
Abstract
Cucumber mosaic virus (CMV) is the one of notorious virus known for its ubiquitous nature and causes substantial yield loss worldwide. The resistance against the Cucumber mosaic virus (CMV) was envisaged in Nicotiana tabacum transgenic lines by introducing viral gene fragments. The chimeric hairpin RNA constructs incorporating 401 bp of coat protein, 411 bp of replicase protein and 361 bp of 2b gene were developed respectively and transformed into N. tabacum. The regenerated transgenic lines introduced with inverted repeats of CMV gene fragments exhibited enhanced resistance against CMV. The preliminary molecular screening and qPCR confirmed the integration of transgene in the transgenic lines. The spectrum of resistance in transgenic lines was evaluated by challenge inoculation with CMV and the resistance was determined through DAC-ELISA. The complete resistance was achieved in the hpRNA-CP transformant with a very low titre (0.029) of CMV followed by hpRNA-REP (0.099) with no symptoms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cucumber mosaic virus (CMV) is one of the well-known destructive viruses causing around 60–100% yield loss on vegetable crops (Palukaitis et al. 1992; Raj et al. 2017). CMV is ubiquitous in nature due to its broad host range and a high degree of diversity. It infects more than 1287 plant species and utmost of the vegetable crops (Kunkalikar et al. 2010). Owing to its inflated adaptive nature and the propensity of rapid evolutionary dynamics, CMV infects wide range host plants (Hong et al. 2003; Ouedraogo et al. 2019). CMV has the highest population diversity due to error-prone replication and short generation (Roossinck 1997; Ouedraogo 2019). CMV undergoes rapid genetic variability and drift through recombination and ressortment than any other RNA virus (Nouri et al. 2014; Pavithra et al. 2019). This often led to the emergence of new strains or variants of CMV and endorses adaptive genes to sustain and broaden host range, infectivity and stability (Ouedraogo et al. 2019; Vinodhini et al. 2020). Moreover, CMV documented to have wide numbers of isolates varies in several molecular and biological properties (Jacquemond 2012). The prevalence of chorosis and mosaic-inducing strains of CMV were also documented in chilli-growing tracts of Tamil Nadu (Vinodhini et al. 2021). There are different strategies were used for the management of CMV. The conventional phytosanitary measures, eradication of associated insect vectors and rouging of infected plants are insufficient to control the virus infection (Hussain et al. 2004). The attempts in developing resistance against CMV using traditional breeding methods were also futile (Watterson 1993). Therefore, induction of a primary line of defense in the host through transgenic manipulation or exogenous application would be stable and reliable for a prolonged period (Rahman et al. 2016). RNA-mediated resistance by the hpRNA expression remains a potent tool in achieving complete resistance against plant viruses (Ntui et al. 2014a). In the present study, resistance derived in transgenic Nicotiana tabatacum by expressing hpRNA of TN chilli isolate of CMV was demonstrated.
Materials and methods
Primers designing
The conserved regions of coat protein (CP), replicase (Rep) and silencing suppressor protein (2b) were determined using comparative multiple sequence alignments of the TN CMV isolates (MT410979; MT422731; MT422729) causing mosaic disease in chilli with other reported CMV isolates available in the NCBI database. The conserved region in the coat protein was determined using Clustal W (www.ebi.ac.uk) from the nucleotide position of 1272 to 1672, replicase protein from 304–714 nt and 2b gene from 121–481 nt proceeded for developing hpRNA construct. The gene-specific RNAi primers were designed using Primer3 software and its specificity was validated further with Gene runner 6.0 (www.generunner.net). The restriction sites in the conserved region were analyzed with the Restriction Mapper version 3 (www.restrictionmapper.org) for primer designing. Therefore, sense primers were designed by including BamHI and ClaI restriction sites and antisense primers were included with KpnI and EcoRI restriction sites (Supplementary Table 1).
hpRNA construct development
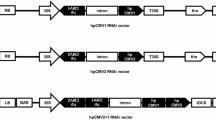
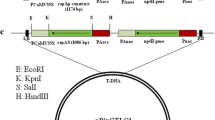
The conserved CP (401 bp), Rep (411 bp) and 2b (361 bp) gene fragment were amplified and cloned into pHANNIBAL vector (CISRO Plant Industry, Australia) in sense orientation between BamHI and ClaI restriction sites and in antisense orientation between KpnI and EcoRI restriction sites. The recombinant pHANNIBAL vector harboring hpRNA cassette with CaMV35S promotor and OCS terminator was sub-cloned into pART27 binary vector (CISRO Plant Industry, Australia) by restricting with NotI enzyme (Fig. 1). Thenceforth, hpRNA cassette was mobilized into Agrobacterium strain LBA4404 using PRK2013 helper strain through triparental mating (Jefferson 1987). The recombinant colonies were selected upon serial dilution (with 0.9% NaCl) and the mobilization of hpRNA cassettes into the A. tumefaciens strain LBA4404 was confirmed through vir gene (F- 5’ ATGTCGCAAGGCAGTAAGCCCA3’; R- 5’GGAGTCTTTCA GCATGGAGCAA3’) and transgene amplification.

HYPERLINK "sps:id::fig1||locator::gr1||MediaObject::0" Vector map of recombinant pHANNIBAL and pART27 plasmid constructs used for plant transformation. The schematic representation of CP hpRNA constructs has been given. The conserved coat protein gene (401 bp) fragments were amplified and cloned in sense (BamHI& ClaI) and antisense (KpnI& EcoRI) orientations. The chimeric gene construct in recombinant pHANNIBAL vector subcloned onto pART27 binary vector by restriction digestion with NotI enzyme. In similar, REP and 2b gene hpRNA constructs also have been developed
Transformation of N. tabacum
The positive conjugants were selected and preceded for transformation in N. tabacum (Kutty et al. 2010). The leaf disc of N. tabacum var. Abirami explants grown under in vitro condition was co-cultivated with Agrobacterium cell suspension culture (OD600-1.0) harboring hpRNA cassette. The transformed ex-plants were regenerated on Murashige Skoog’s (MS) medium supplemented with respective growth hormones and antibiotics (Supplementary Table 2) and incubated at 26 ± 2 °C for 16 h light/8 h dark photoperiod cycle. The selective transfomants were selected upon the kanamycin selective medium and after callus induction transformed to shoot induction medium. The elongated shootlets have been induced for rooting and the putative transgenic lines were transferred to transgenic greenhouse for hardening and maintained.
Molecular screening of transgenic lines
The preliminary confirmation for the integration of hpRNA cassette in the putative transgenic lines was carried out with the amplification of selectable marker and a reporter gene. The transformation efficiency was evaluated by the positive number of transgenic plants/total number of ex-plants used for transformation multiplied by 100. The genomic DNA extracted from the transgenic plant by the CTAB method (Doyle 1990). Further, template DNA was used for PCR amplification of the NPTII gene (F- 5’TTGAGGCGCTAAATGAAACC3’; R- 5’ATTTGCCG ACTACCTTGGTG3’), coat protein (F- 5’TCAACCAGTGCTGGTCGTAA3’; R- 5’GACAAGTCCGAGGAGGCA3’), replicase (F- 5’CCCTCCATCTTACCCAGCAG3’; R- 5’CAAAATGGTGGTTCC GTGA3’) and 2b gene (F-5’GAGCGTTCGATTTCTACTAAGCG3’; R- 5’TTCCGCCCATTCATTACC3’) fragments. The polymerase chain reaction was carried out with conditions of 94 °C for 2 min followed by 94 °C for 30 s of denaturation, 54 °C for 30 s of annealing and 72 °C for 90 s followed by final extension of 72 °C for 10 min with 35 cycles.
Quantification of transgene expression by real-time PCR (qPCR)
The relative expression of the transgene in transgenic lines was determined with qPCR analysis. The total RNA was extracted from the fresh transgenic lines (Chomczynski and Sacchi 1987) and the concentration of total RNA was quantified with a Nanodrop2000 spectrophotometer (Thermo Scientific). RNA samples quantified for a 260/280 ratio over 2.0 were used for analysis and the integrity of RNA was also analyzed by gel electrophoresis. First-strand cDNA was transcribed using CDNA synthesizing kit (Thermo sci.,) and proceeded for the relative expression of the transgene in transgenic lines. Shortly, the reaction mixture contained KAPA SYBR® FAST (Sigma-Aldrich) master mix- 5 µl, forward and reverse primer- 3 µl, water-1 µl and cDNA (1:10 dilution)-1 µl was prepared and preceded with a polymerase chain reaction of 56 °C of annealing for 3 h. The relative expression of CP (F- 5’AACCAGTGC TGGTCGTAACC3’; R- 5’TTGATGAGTCGCGAAAGATG3’), rep (F- 5’AGCAGCTCGAGGAGCAATTA3’; 5’TCGTACTTCCCTCCATACCG3’) and 2b genes (F- 5’TGACAAACGTCGAACTCCAG3’; R- 5’GCTGGG ACTTTTGTGACCTC3’) in transgenic lines was quantified to know copy numbers. The primers were designed for CP, Rep and 2b gene specific to transgene conserved region using Primer 3.0 software. The primer pairs were selected manually based on melting temperature (Tm), GC content and complementarity and validated for amplification efficiency and primer-dimer formation. The house-keeping gene of Elongation factor 1a (EF-1a) was referred from Schmidt and Delaney (2010).
The comparative ∆∆Ct method was used for semi-quantification of transgene expression and the transgene expression was normalized with EF-1a reference gene by subtracting the Ct value of the reference gene from Ct value of transgene which expressed as ∆Ct (∆Ct = Ct transgene–Ct EF-1a). The relative transgene expression to the calibrator was indicated as ∆∆Ct (∆∆Ct = ∆Ct (transgene)–∆Ct (calibrator)). The lower gene expression in each transgene was selected as the calibrator. The amplification plots and fluorescence values were generated in Roche LightCycler ® 480 System. The results were presented as Ct value which is the baseline threshold value and represented as baseline subtracted curve fit. The agarose gel electrophoresis was carried out to validate the qPCR results by the amplification of specific gene products (Mahto et al. 2018).
Challenge inoculations of transgenic plants for virus resistance
The CMV inocula was sap inoculated with 0.1 M of phosphate buffer (pH 7) containing 0.02% mercaptoethanol in 1:2 ratio (infected sample: buffer) by gentle aberration on tobacco transgenic lines and healthy control (Subramanian and Narayanasamy 1973). The inoculated transgenic and control plants were maintained separately at 22 ± 2 °C in an insect-proof greenhouse and observed for symptom development. The level of resistance and qualitative and quantitative measure of CMV was determined through direct antigen coating-ELISA (DAC-ELISA). DAC-ELISA was carried out according to the methodology of Hobbs et al. (1987). The prepared crude extracts (1:10) of inoculated plant samples were tested against CMV-specific polyclonal antiserum (obtained from ICARNRCB, Tiruchirappalli, India) at 1:1000 dilutions. The absorbance was read at 405 nm using a microplate reader (BioTek).
Results
The conserved coat protein (401 bp), replicase (411 bp) and 2b gene (361 bp) fragments homologous to TN chilli isolate of CMV were amplified both in sense and antisense orientations in pHANNIBAL vector. The recombinant pHANNIBAL plasmid harboring hpRNA cassette embodied with CaMV35S promotor, PDK intron, OCS terminator and inverted repeats of transgene such as CP (3765 bp), Rep (3785 bp) and 2b (3687 bp). Thus have been sub cloned into pART27 binary vector to design chimeric gene construct. The recombination was confirmed with restriction digestion of binary vector with NotI enzyme which produced the DNA fragment as exact size of hpRNA cassettes. Subsequently, recombinant plasmid harboring the chimeric gene constructs were selected and mobilized into Agrobacterium strain LBA4404 through triparental mating. The specific transformed colonies were selected and integration of hpRNA cassettes into the Agrobacterium was confirmed with vir gene and transgene-specific primers (Fig. 2).

PCR amplification of vir genes (a) and NPTII (b) marker genes for the confirmation of Agrobacterium-mediated transformation
The leaf disc of Nicotiana was transformed separately with each hpRNA-CP, hpRNA-REP and hpRNA-2b construct by co-cultivation and transferred to a selective medium containing Kanamycin (100 mg l−1). The Kanamycin selection allows the regeneration of ex-plant carrying transgene alone. The calli induction was observed after 10–13 days and the shoot proliferation was observed to be after 17–23 days on basal MS medium with 1 mg/l BAP, 100 mg/l kanamycin and 300 mg/l cephataxamine. After twenty days, 5 cm long shootlets along with leaves were transferred to the rooting medium (Fig. 3). The transgenic plants were transferred onto pots for hardening and substantial growth. The integration of transgene in transgenic lines was evaluated by molecular screening and qPCR analysis. The preliminary confirmation was carried out with PCR analysis using NPTII (marker gene) and transgene0-specific primers (reporter gene). The amplification of ~ 299 bp fragment corresponding to the NPTII gene confirms the integration of hpRNA in the transgenic plants (Fig. 4). In such a way, the concrete CP hpRNA integrated transformants produced amplicon at 401 bp, Rep hpRNA transformants produced 411 bp amplicon and 2b hpRNA transformant produced an amplicon of 361 bp (Fig. 4). The healthy control plants did not show any amplification upon tested for reporter and marker genes.

Phases of regeneration of putative transgenic N. tabacum transformed with RNAi gene construct. a Ex-plants; b Pre-conditioning; c Co-cultivation; d Selection; e, f Calli induction; g, h Shoot induction; i Rooting; j Regeneration of plantlets

Molecular screening transgenic lines with maker and reporter genes: PCR amplification of (a) NPTII gene fragment; (b) coat protein gene; (c) replicase gene; (d) 2b gene expressed from transgenic tobacco lines using specific primers and non-transformant/ healthy control not showing any amplification. The 1 kb molecular marker presented on the left side
The molecular screening results show that five transformants of each hpRNA-CP, hpRNA-REP and hpRNA-2b constructs were positively amplified. The relative expression of transgenes in those selective transgenic lines was analyzed through qPCR for further confirmation of transgene integration. The analysis indicated the stable integration and differential expression of transgenes in the transgenic lines. The semi-quantification of transgene expression was analyzed by the comparative ∆∆Ct method. The differential expression of a transgene in the respective transgenic lines was normalized to the endogenous gene (EF-1a) expressed in Nicotiana. The results showed that the maximum CP gene expression was observed as 230 fold followed by 188.70 fold of REP genes. The level of expression of the 2b gene in the transgenic plants was comparatively low (maximum 95.10 fold). Moreover, 81.26–230 fold of CP transgene expression was observed in hpRNA-CP transformants, 90.01–188.70 fold of REP gene in hpRNA-REP transformants and 10.01–95.10 fold of 2b gene in hpRNA-2b transformants (Table 1). The expression of the 2b gene with CP and REP genes was shown to be relatively lower and further the expression pattern was also validated by gel electrophoresis (Fig. 5).

The expression of transgenes in transgenic lines was determined by semi-quantitative Real-Time PCR. The copy number of CP (a), Rep (b) and 2b (c) expressed in N. tabacum transgenic line determined by qPCR and further validated further by gel electrophoresis
The spectrum of resistance against CMV in the transgenic lines was determined through DAC-ELISA by challenge inoculation. The CMV sap was mechanically inoculated onto twenty-seven transgenic tobacco lines harboring hpRNA cassettes and healthy control plant at 2–3 leaf stage. The inoculated plants were observed regularly for every 24 h for the symptom expression. There was no symptom development was observed in hpRNA-CP and hpRNA-REP transgenic lines even after 20 days of post inoculation. Whereas, mild symptom expression was observed in hpRNA-2b transgenic lines. The healthy control plants produced characteristic mosaic symptom of CMV (Supplementary Fig. 1). The plants were observed for four more weeks at regular intervals and then subjected to DAC-ELISA analysis. The results explicated that the hpRNA-CP transformant was recorded with a very low titre (0.029) of CMV followed by hpRNA-REP (0.099). Besides, the hpRNA-2b transformant was observed with 0.268 titre of CMV (Fig. 6). The healthy control plants with CMV infection recorded 1.784 virus titre (Table 2). Hence, it suggested that hpRNA-CP and hpRNA-REP harboring transgenic lines confer complete resistance against CMV. However, hpRNA-2b harboring transgenic lines produced a partial spectrum of resistance against CMV.

CMV titre in the transgenic N. tabacum lines in DAC-ELISA
Discussion
The abundant expression of hpRNA construct in the plant confer RNA silencing mediated resistance efficiently against virus infections in most cases (Kalantidis et al. 2002; Duan et al. 2008, 2012). Distinctly, transgenic plant expressing hpRNA harboring inverted repeat sequences with intron confer 90% of gene silencing effectively (Smith et al. 2000; Wesley et al. 2001). There are large numbers of transgenic plants developed with the inheritance of post-transcriptional gene silencing (PTGS) through the introduction of dsRNA against invading viruses. The management of severe mosaic caused by CMV is futile. Hence, RNA-mediated resistance is the most effective strategy for developing stable resistance against CMV. The present study demonstrates the spectrum of RNA-mediated resistance against CMV by inverted repeats of conserved gene fragments.
The length of RNAi fragment and sequence similarity is also highly essential for effective RNA silencing mediated resistance (Duan 2012). It is reported that challenging virus with sequence mutation over 10–20% more than of transgene overwhelms resistance mechanism and promote infection. RNAi gene fragment with a length of 300–800 bp effectively induces RNAi silencing in plants (Ritzenthaler 2005). Accordingly, specific hpRNA-CP, hpRNA-Rep and hpRNA-2b gene constructs have been developed to study the efficacy. The developed chimeric gene constructs were mobilized into Agrobacterium strain LBA4404 and preceded by the regeneration of transgenic plants. There are several studies have demonstrated RNA-mediated resistance in different crop plants. But, the frequency of induction of RNA-mediated resistance in transformed plants is more cumbersome. Hence, optimizing the frequency of resistance induction is an essential factor in RNA-mediated resistance.
The integration of transgene in the transgenic line was confirmed by preliminary molecular screening and qPCR analysis. There are around 45–50 percent of the transgenic plants showed that transgene integration implies intact T-DNA integration into transformants. The relative expression frequency of transgenes in the transgenic lines is correlated with virus titre by challenge inoculation with CMV to determine the spectrum of resistance. The transgenic line harboring hpRNA-CP-4 expressed as much as 230 folds in tobacco observed for very low CMV titre (0.029) followed by hpRNA-REP-4 expressed 188 folds with 0.099 virus titre. Kamo et al. (2010) reported that transgenic Gladiolus lines harbor CMV CP gene with lower level expression (24×) in qPCR analysis was apparently not resistance to CMV. It suggests the relative expression of the transgene may correlate with the spectrum of resistance. The higher transgene expression appears to be effective in inducing CP-mediated resistance in transgenic Gladiolus plants (Kamo et al. 2010). Introduction of dsRNA derived from the fragment of the replicase gene produced complete resistance to CMV in tomato, potato and Gladiolus (Kamo et al. 2010; Ntui et al. 2014a). Hu et al., (2011) demonstrated that transgenic tobacco incorporated with a partial fragment of the replicase (1a gene) gene of CMV in an intron-hairpin RNA construct produced complete resistance against CMV.
The constructs designed to express dsRNA in plants in the form of self-complementary hairpin RNA (hpRNA) elicits the high degree and frequency of post-transcriptional gene silencing (PTGS) of invading viruses (Smith et al. 2000; Wang et al. 2010; Chuang and Meyerowitz 2000; Wesley et al. 2001). The complete resistance was achieved in hpRNA-CP and hpRNA-REP harboring transgenic lines with free of symptoms. Indeed, hpRNA-2b harboring transformant with deficient transgene expression showed a partial spectrum of resistance with mild symptoms. The fragmentary or deficient expression of the 2b gene may due to an incomplete suppression of intracellular signaling (Guo and Ding 2002; Ntui et al. 2014b). Apparently, the 2b protein interrupted the silencing mechanism by sequestering siRNAs production by preventing it from entering the RISC complex (Guo and Ding 2002). The mechanism of PTGS is that the synthesis of siRNA from the introduced hpRNA and the expressed siRNA complementary to the target RNA incorporated in the RISC complex. It ultimately led to the degradation of homologous target RNA and confers resistance against invading virus. RNA-mediated resistance involves sequence-specific degradation (Kawazu et al. 2009). It can overcome the potential risk of recombination and heterologous encapsidation occurring in protein/gene-mediated resistance (Hu et al. 2011). The resistance response also seems to considerably vary with different CMV-host combinations (Namba et al. 1992; Kaniewski et al. 1999).
The obtained results from model plants indicate the effectiveness of RNAi in the management of viruses. Further, it can be applied to the development of the available commercial crops to combat the problem that arises from viruses. With further advancement, the successful generation of commercial transgenic plants can be developed by improvising and screening the candidate viral gene constructs. Besides, CMV-2b gene-dependent defense pathway regulation is yet to be studied in detail. The spectrum of resistance and its relation to the ratio of accumulation of each viral RNAs during CMV infection also need to be focused.
Data availability
All the required data has been included within the article.
References
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:156–159
Chuang CF, Meyerowitz EM (2000) Specific and heritable genetic interference by double-stranded RNA in Arabidopsis thaliana. Proc Nat Acad Sci 97(9):4985–4990
Doyle JJ (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Duan CG, Wang CH, Fang RX, Guo HS (2008) Artificial microRNAs highly accessible to targets confer efficient virus resistance in plants. J Virol 82:11084–11095
Duan CG, Wang CH, Guo HS (2012) Application of RNA silencing to plant disease resistance. Silence 3:1–8
Guo HS, Ding SW (2002) A viral protein inhibits the long range signaling activity of the gene silencing signal. EMBO J 21:398–407
Hobbs HA, Reddy DVR, Rajeshwari R, Reddy AS (1987) Use of direct antigen coating and protein a coating ELISA procedures. Plant Dis 71:747–749
Hong JS, Masuta C, Nakano M, Abe J, Uyeda I (2003) Adaptation of Cucumber mosaic virus soybean strains (SSVs) to cultivated and wild soybeans. Theor Appl Genet 107:49–53
Hu Q, Niu Y, Zhang K, Liu Y, Zhou X (2011) Virus-derived transgenes expressing hairpin RNA give immunity to Tobacco mosaic virus and Cucumber mosaic virus. Virology Journal 8:1–11
Hussain M, Mansoor S, Iram S, Zafar Y, Briddon RW (2004) First report of tomato leaf curl New Delhi virus affecting chilli pepper in Pakistan. Plant Pathol 53:94
Jacquemond M (2012) Cucumber mosaic virus. Adv Virus Res 84:439–504
Jefferson RA (1987) Assaying chimeric genes in plants: the GUS gene fusion system. Plant Mol Biol Report 5:387–405
Kalantidis K, Psaradakis S, Tabler M, Tsagris M (2002) The occurrence of CMV-specific short RNAs in transgenic tobacco expressing virus-derived double-stranded RNA is indicative of resistance to the virus. Mol Plant Microbe Interact 15(8):826–833
Kamo K, Jordan R, Guaragna MA, Hsu H, Ueng P (2010) Resistance to Cucumber mosaic virus in Gladiolus plants transformed with either a defective replicase or coat protein subgroup II gene from Cucumber mosaic virus. Plant Cell Rep 29(7):695–704
Kaniewski W, Ilardi V, Tomassoli L, Mitsky T, Layton J, Barba M (1999) Extreme resistance to cucumber mosaic virus (CMV) in transgenic tomato expressing one or two viral coat proteins. Mol Breed 5:111–119
Kawazu Y, Fujiyama R, Noguchi Y (2009) Transgenic resistance to Mirafiori lettuce virus in lettuce carrying inverted repeats of the viral coat protein gene. Transgenic Res 18(1):113–120
Kunkalikar SR, Sudarsana P, Rajagopalan P, Zehr UB, Ravi KS (2010) Biological and molecular characterization of Capsicum chlorosis virus infecting chilli and tomato in India. Adv Virol 155(7):1047–1057
Kutty PC, Parveez GKA, Huyop F (2010) An easy method for Agrobacterium tumefaciens-mediated gene transfer to Nicotiana tabacum cv. TAPM26. J Biol Sci 10(6):480–489
Mahto BK, Sharma P, Rajam MV, Reddy PM, Dhar-Ray S (2018) An efficient method for Agrobacterium-mediated genetic transformation of chilli pepper (Capsicum annuum L.). Indian J Plant Physiol 23(3):573–581
Namba S, Ling K, Gonsalves C, Slightom JL, Gonsalves D (1992) Protection of transgenic plants expressing the coat protein gene of watermelon mosaic virus ii or zucchini yellow mosaic virus against six potyviruses. Phytopathology 82:940–946
Nouri S, Arevalo R, Falk BW, Groves RL (2014) Genetic structure and molecular variability of Cucumber mosaic virus isolates in the United States. PLoS ONE 9(5):e96582
Ntui VO, Kong K, Azadi P, Khan RS, Chin DP, Igawa T, Mii M, Nakamura I (2014a) RNAi-mediated resistance to Cucumber mosaic virus (CMV) in genetically engineered tomato. Am J Plant Sci 2:1–12
Ntui VO, Kong K, Azadi P, Khan RS, Ohara M, Goto Y, Watanabe M, Fukami M, Nakamura I, Mii M (2014b) Transgenic tobacco lines expressing defective CMV replicase-derived dsRNA are resistant to CMV-O and CMV-Y. Mol Biotechnol 56(1):50–63
Ouedraogo RS, Pita JS, Somda IP, Traore O, Roossinck MJ (2019) Impact of cultivated hosts on the recombination of Cucumber mosaic virus. J Virol 93(7):1–9
Palukaitis P, Roossinck MJ, Dietzgen RG, Francki RIB (1992) Cucumber mosaic virus. Adv Virus Res 41:281–348
Pavithra BS, Govin K, Renuka HM, Krishnareddy M, Jalali S, Samuel DK, Himabindu K (2019) Characterization of Cucumber mosaic virus infecting coleus (Plectranthus barbatus) in Karnataka. Virus Disease 30(3):403–412
Rahman MS, Akanda AM, Mian IH, Bhuiyan MKA, Hossain MM (2016) New sources of resistance to Cucumber mosaic virus in Capsicum annuum. J Crop Sci Biotechnol 19(3):249–258
Raj SK, Kumar S, Gautam KK, Kaur C, Samad A, Zaim M, Hallan V, Singh R (2017) The progress of research on Cucumoviruses in India. In: A century of plant virology in India, pp 217–253
Ritzenthaler C (2005) Resistance to plant viruses: old issue, news answers? Curr Opin Biotechnol 16(2):118–122
Roossinck MJ (1997) Mechanisms of plant virus evolution. Annu Rev Phytopathol 35(1):191–209
Schmidt GW, Delaney SK (2010) Stable internal reference genes for normalization of real-time RT-PCR in tobacco (Nicotiana tabacum) during development and abiotic stress. Mol Genet Genomics 283(3):233–241
Smith NA, Singh SP, Wang MB, Stoutjesdijk PA, Green AG, Waterhouse PM (2000) Total silencing by intron-spliced hairpin RNAs. Nature 407(6802):319–320
Subramanian KS, Narayanasamy P (1973) Mechanical transmission of whitefly borne Yellow mosaic virus of Lablab niger. Midikus Curr Sci 47:92–93
Vinodhini J, Rajendran L, Raveendran M, Rajasree V, Karthikeyan G (2020) Characterization of Cucumber mosaic virus (CMV) subgroup IB infecting chilli in Tamil Nadu, India. 3 Biotech 10(11):1–10
Vinodhini J, Rajendran L, Abirami R, Karthikeyan G (2021) Co-existence of chlorosis inducing strain of Cucumber mosaic virus with tospoviruses on hot pepper (Capsicum annuum) in India. Sci Rep 11(1):1–9
Wang Z, Kraft JJ, Hui AY, Miller WA (2010) Structural plasticity of Barley yellow dwarf virus-like cap-independent translation elements in four genera of plant viral RNAs. Virology 402(1):177–186
Watterson JC (1993) Development and breeding of resistance to pepper and tomato viruses. In: Kyle MM (ed) Resistance to viral disease of vegetables. Timber Press, Portland, pp 80–110
Wesley SV, Helliwell CA, Smith NA, Wang M, Rouse DT, Liu Q, Gooding PS, Singh SP, Abbott D, Stoutjesdijk PA (2001) Construct design for efficient, effective and high-throughput gene silencing in plants. Plant J 27(6):581–590
Acknowledgements
We are thankful to Department of Plant Pathology, TNAU, Coimbatore for providing facilities during the course of study. We would like to extend our gratitude to CISRO Plant Industry, Australia and Dr. Varanavasiappan, Asst. Professor, Department of Biotechnology, TNAU for their immense help and support.
Author information
Authors and Affiliations
Contributions
GK designed the project, supervised, revised and approved the manuscript for submission; JV drafted the experiments and prepared the manuscript; LR gave proposition to conduct the experiment. All authors reviewed and approved the submission.
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflict of interest.
Research involving human and animal participants
This manuscript does not contain any experiments involving human or animals participants.
Consent for publication
Authors have agreed to submit in its current form for publication in this journal.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vinodhini, J., Rajendran, L. & Karthikeyan, G. Engineering resistance against Cucumber mosaic virus in Nicotiana tabacum through virus derived transgene expressing hairpin RNA. 3 Biotech 13, 143 (2023). https://doi.org/10.1007/s13205-023-03576-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-023-03576-1