
Abstract
The non-structural protein 5 (NS5) is the most conserved protein among flaviviruses, a family that includes the dengue virus. It functions both as an RNA-dependent RNA polymerase and an RNA-methyltransferase and is therefore essential for the replication of viral RNA. The discovery that dengue virus NS5 protein (DENV-NS5) can also localize to the nucleus has resulted in renewed interest in its potential roles at the host-virus interface. In this study, we have used two complementary computational approaches in parallel – one based on linear motifs (ELM) and another based on tertiary structure of the protein (DALI) – to predict the host proteins that DENV-NS5 might interact with. Of the 42 human proteins predicted by both these methods, 34 are novel. Pathway analysis of these 42 human proteins shows that they are involved in key host cellular processes related to cell cycle regulation, proliferation, protein degradation, apoptosis, and immune responses. A focused analysis of transcription factors that directly interact with the predicted DENV-NS5 interacting proteins was performed, followed by the identification of downstream genes that are differentially expressed after dengue infection using previously published RNA-seq data. Our study provides unique insights into the DENV-NS5 interaction network and delineates mechanisms whereby DENV-NS5 could impact the host-virus interface. The novel interactors identified in this study could be potentially targeted by NS5 to modulate the host cellular environment in general, and the immune response in particular, thereby extending the role of DENV-NS5 beyond its known enzymatic functions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dengue is a global public health concern. It is estimated that over 390 million people are infected by the dengue virus each year (Guzman et al. 2010; Bhatt et al. 2013; WHO 2022, Jan 10). Dengue is caused by any of the four genetically related serotypes of the dengue virus (DENV-1, DENV-2, DENV-3, DENV-4) that belong to the genus Flavivirus which includes other human-infecting arthropod-borne viruses such as Zika, Japanese encephalitis virus (JEV), tick-borne encephalitis (TBE), West Nile virus (WNV), and yellow fever virus (YFV). DENV is a positive-strand RNA virus with an approximately 11-kb long genome comprising 10 genes – three encoding structural proteins (Env, PreM, Capsid) and seven encoding non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5), all of which are initially translated as a single polyprotein. NS5, the largest and the most conserved of non-structural proteins is the replicase comprising of two functional domains—an RNA-dependent RNA polymerase (RdRp) domain at its C-terminus, required for replication and RNA-methyltransferase (MTase) domain at the N-terminus. The RNA MTase activity ensures the capping of the viral RNA, enhancing its stability and facilitating polyprotein translation (Liu et al. 2010; Klema et al. 2016; El Sahili and Lescar 2017). DENV-NS5 elicits a strong T cell response indicating its immunogenicity, making it an important target for both drug discovery and vaccine design (Duangchinda et al. 2010). Furthermore, the NS5 proteins of other flaviviruses such as Japanese encephalitis virus (JEV), tick-borne encephalitis (TBE) and West Nile virus (WNV) have been shown to inhibit type I interferon (IFN) signaling by interfering with the activation of the transcription factor STAT1 by Janus kinase (JAK) (Laurent-Rolle et al. 2010). One study demonstrated that DENV-NS5 could bind to STAT2 and target it for proteasomal degradation (Morrison et al. 2013) suggesting a role in immune-modulation. These reports suggest that the flaviviral NS5 protein is capable of manipulating host cells and the immune environment to promote viral replication and survival justifying efforts to better understand DENV NS5’s ability to interact with, and probably influence host cellular processes.
Previous studies using either computational or experimental approaches have shown that NS5 of DENV (and Zika virus) interacts with numerous host proteins. DenvInt (https://denvint.000webhostapp.com/) is a dengue-specific database of serotype-related experimental evidence of protein–protein interactions (PPIs) (Dey and Mukhopadhyay 2017) that curates’ data from yeast-two-hybrid (Y2H), bacterial two-hybrid, pull-down, and co-localization experiments (Khadka et al. 2011, Le Breton et al. 2011; Mairiang et al. 2013). Another database, HVPPI (Human-virus PPI) provides a comprehensively annotated database of human-virus PPIs as well as online tools for their functional analysis (Li et al. 2022). Some pull-down studies using human cell lines such as HEK293T and Huh7 cells either by infecting or transfecting the cells with DENV-2/DENV-2 NS5 (Carpp et al. 2014; De Maio et al. 2016; Poyomtip et al. 2016, Shah et al. 2018) have identified important proteins such as those of the spliceosome complex and Golgi network as interactors of NS5. Much of this data has been compiled in a recent review (Bhatnagar et al. 2021).
Our interest in re-visiting the protein–protein interaction (PPI) landscape stemmed from the finding that DENV-NS5 encoded a nuclear localization signal (NLS) (Brooks et al. 2002) and that it accumulates in the nucleus of DENV-infected cells in a serotype-specific manner (Hannemann et al. 2013). This information implied that NS5 protein functions were more complex and encompassed both the cytoplasmic and nuclear compartments. There is a formal possibility that nuclear NS5 could modulate host gene expression by directly binding to the upstream regulatory regions of host genes or via interactions with nuclear transcriptional complexes and/or factors. Thus, in addition to its primary function as the viral replicase and the RNA capping enzyme, DENV-NS5 is a pleiotropic effector that may interact with host proteins participating in a variety of biological pathways. Therefore, identifying NS5-interactors among host proteins can greatly enhance our understanding of the mechanistic bases of the co-option or subversion of host cellular processes by DENV.
While the list of physiological processes likely influenced by NS5 is both large and diverse based on available experimental and computational interactomes, it is of interest to determine the effect of NS5 on molecular processes within specific host cellular compartments. In this study, we have used a combination of both linear motif-based and three-dimensional structure-based bioinformatic tools to identify a common, core set of host proteins that DENV-NS5 could potentially interact with. Based on these findings, we have further identified the transcription factors (TFs) that these DENV-NS5 interacting proteins could directly interact with. Finally, using publicly available transcriptomics data obtained from naïve and dengue-infected patient samples, we have identified certain downstream genes that are differentially expressed, indicating that these are very likely modulated by DENV-NS5 in accordance with our prediction of interactors. Our study, therefore, identifies novel interactors that could be targeted by NS5 to potentially modulate the host cellular environment in general and the immune response in particular. Identification of these interactors also provides likely and testable mechanisms underlying the observed effects of dengue infection on host cells.
Materials and methods
Data sources
NS5 protein sequences for all four Dengue serotypes (DENV-1, 2, 3, 4) were retrieved from NCBI (DENV-1 WP74, DENV-2 S16803, DENV-3 CH53489, DENV-4 TVP360). Protein sequences of the dengue NS5 protein for all the sequenced isolates were downloaded from the VIPR database (https://www.viprbrc.org) (Pickett et al. 2012). We used only those sequences for which the complete genome sequences were available to ensure the surety of the serotype. Serotype-wise multiple sequence alignment was performed using Clustal-ω (https://www.ebi.ac.uk/Tools/msa/clustalo/) (Sievers and Higgins 2014).
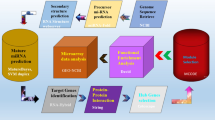
The crystal structure of full-length DENV-3 NS5 protein complexed with S-adenosyl-L-homocysteine (SAH) is available in the worldwide Protein Data Bank (http://www.wwpdb.org/ (Berman et al. 2003); PDB ID: 4V0Q; Resolution 2.30 Å (Zhao et al. 2015). Using this as a reference, three-dimensional (3D) structures of NS5 protein for serotypes 1,3 and 4 were modelled using i-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) (Zhang 2008). The crystal structure of DENV-2 NS5 is also available (PDB ID 5ZQK; Resolution 2.30 Å) (El Sahili et al. 2019). It was used to model the structure of the NS5 protein of serotype 2. Interaction networks were created and visualized using Cytoscape (version 3.9.0) (Shannon et al. 2003). Pathway analysis was done using WebGestalt (WEB-based Gene SeT AnaLysis Toolkit) (Liao et al. 2019). The bioinformatics pipeline for the analysis performed in this work is depicted in Fig. 1.

Bioinformatics pipeline for prediction of human interacting partners of DENV-NS5 protein and downstream analysis for identification of differentially expressed genes: Both sequence-based (ELM) and structure-based (DALI) analysis was done to predict the human proteins that are potential interacting partners of DENV-NS5 protein. A total of 42 proteins were common to predictions made by both approaches. Transcription factors that could directly interact with these 42 proteins were identified using JASPAR and The Human Transcription factors database. The downstream target genes regulated by these transcription factors were identified using the DoRothEA database, and their differential expression during dengue infection was verified using published RNA-seq data (described in materials and methods)
Eukaryotic linear motifs (ELM) analysis
Eukaryotic Linear Motifs (ELMs) of the DENV-NS5 were predicted using the Eukaryotic Linear Motif resource server (ELM) (http://elm.eu.org) (Dinkel et al. 2016). The motif probability cut-off was taken as 100 (default value) and Homo sapiens was selected as the preferred species to predict the conserved human peptide linear motifs in DENV-NS5 protein for all four serotypes separately. Since the NS5 protein shuttles between the nucleus and cytoplasm, the motifs were not filtered based on cell compartmentalization. After the multiple sequence alignment step (performed on the isolated sequences downloaded from the VIPR database) using Clustal-ω (Sievers and Higgins 2014), only those ELMs that were > 95% conserved in all the available strains and had a p-value of ≤ 0.05 were considered for further analysis.
NetSurfP 2.0 predicts the surface accessibility of amino acids (https://services.healthtech.dtu.dk/service.php?NetSurfP-2.0) (Klausen et al. 2019). This was used to identify buried and exposed residues in DENV-NS5 protein (all serotypes). The motifs that were found to lie entirely in buried stretches were removed from further analysis (ELMs selected for further analysis are listed in Supplementary table 1). Using the list of all domains annotated as interacting with one or more ELM class, available on the ELM server, protein domains interacting with the predicted ELMs were identified. Protein domain/ superfamily data from NCBI-CDD was used to extract the human proteins that could be possible interacting partners of DENV-NS5 proteins (Marchler-Bauer et al. 2015).
Tertiary protein structure prediction using i-TASSER
3D protein structures of NS5 were obtained from i-TASSER (Zhang 2008) for all four dengue serotypes. I-TASSER is a widely used, freely available tool used for protein 3D structure prediction that was ranked as the no.1 server for protein structure prediction in recent community-wide CASP15 experiments (https://predictioncenter.org/casp15/zscores_interdomain.cgi). The crystal structure of DENV3-NS5 protein (PDB ID 4V0Q) (Zhao et al. 2015) was used as a reference for modelling the structure of NS5 from serotype 1, 3 and 4. DENV-2 NS5 crystal structure (PDB ID 5ZQK) (El Sahili et al. 2019) was used as a reference to model DENV-2 NS5. (Supplementary Fig. 1 contains high-resolution 3D protein structures modelled using i-TASSER along with the C-scores, TM-score and estimated RMSD values). The modelled structure selected for further analysis have C-score > 1.5, TM-score > 0.9 and Z-score alignment > 1. Ramachandran plots were plotted for all four modelled NS5 structures using PROCHECK (Laskowski et al. 1993). A few amino acids were found to lie in the disallowed regions. Therefore, we undertook energy minimization using GROMACS (Pronk et al. 2013). Thereafter, Ramachandran plots were plotted again to determine if most amino acids fell within the acceptance region (Supplementary Fig. 2).
Structural similarity prediction of host proteins by DENV-NS5
We investigated protein mimicry using structural similarities detected by the DALI server (Holm 2020). DALI compares the 3D structural coordinates of two PDB entries by alignment of alpha carbon distance matrices, allowing for differences in domain order, and produces a structural similarity score. For this study, we ran the modelled DENV-NS5 protein structures from each serotype through an exhaustive PDB25 search that compares one query structure against a representative subset of the Protein Data Bank for structurally similar proteins. A z-score above 2.0 and default settings of a score cut-off of 40 bits and sequence overlap cut-off of 50% were used. Out of these results, only protein structures from Homo sapiens were retained. The human interacting partners of these structurally similar proteins so identified were extracted from BioGRID (Stark et al. 2006), a database that curates protein–protein interactions based on experimental evidence.
Pathway analysis for the protein interacting partners of DENV-NS5 predicted commonly through ELM and DALI
Only those human proteins predicted as DENV-NS5 interactors by both ELM and DALI were considered for further analysis. The 42 proteins are common to both predictions were fed into the STRING database, with the selected organism as Homo sapiens, to identify the degree of interaction of these proteins within the reconstructed network (Mering et al. 2003). Pathway analysis was done using WebGestalt (WEB-based Gene SeT AnaLysis Toolkit (Liao et al. 2019). The bubble plot depicting the different enriched pathways was made using R studio (Allaire 2012) (Fig. 2).
Identification of downstream transcription factors and differentially expressed genes potentially modulated by DENV-NS5
Transcription factors (TFs) were identified from the set of proteins belonging to the first interactors of the 42 DENV-NS5 interacting proteins predicted by both ELM and DALI (first interactors were identified using BioGRID). Each TF was then queried in the DoRothEA database (Garcia-Alonso et al. 2019) to identify their downstream gene targets. Only interactions with the highest number of supporting evidence (wherein evidence ranged from A (high quality) to E (low quality)) were selected. Subsequently, the gene expression of each gene of interest was examined in previously published gene expression data sets from dengue patients presenting with different degrees of disease severity (dengue fever – DF or dengue haemorrhagic fever – DHF) (Banerjee et al. 2017, Poonpanichakul et al. 2021). Only those studies were chosen for which raw read counts were available. The genes that showed significant differential expression between DF/DHF and naïve were selected and further evaluated using a manual literature search on PubMed. The list of differentially expressed genes (DEGs) obtained is shown in Supplementary table 7. Pathway enrichment of these DEGs was performed using pathfindR (https://cran.r-project.org/package=pathfindR) (Ulgen et al. 2019).
Results
Inferring the NS5-human protein interactome
In the present study, both ELM and DALI were used for the prediction of human proteins that could interact with DENV-NS5. In our work, we have restricted our analysis to a set of 42 human protein interactors predicted in both analyses (Fig. 1, Fig. 3A, Supplementary table 2). ELM which detects linear motifs yielded 153 interactors that were common to all four serotypes (Supplementary table 3) whereas DALI which detects 3D structures yielded 2630 protein interactors (Supplementary table 4). Thus, ours is a stringent approach focused on potential protein interactors that are predicted by both linear motif and 3D structural analyses. We explored the level of overlap between our approach and previously published yeast 2-hybrid screen (compiled in (Dey and Mukhopadhyay 2017)) and bioinformatic analysis (Lasso et al. 2019) (total 177 proteins were identified from these studies and are listed in Supplementary material of (Bhatnagar et al. 2021)), and found an overlap of 8 proteins (p-value 0.0038, Fisher’s exact test) validating our approach. These eight proteins are WW domain-containing E3 ubiquitin protein ligase 1 (WWP1); three tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation proteins– YWHAB, YWHAE and YWHAZ; exportin1 (XPO1), the DNA repair-associated protein BRCA1; catenin beta 1/β-catenin (CTNNB1) and growth factor receptor bound protein 2 (GRB2). These eight proteins are highlighted in Supplementary table 2. The remaining 34 proteins are novel that have been identified in this study. Interestingly, we did not identify any significant overlap between proteins predicted by our approach and those identified by previous pull-down studies (Carpp et al. 2014; De Maio et al. 2016; Poyomtip et al. 2016, Shah et al. 2018). This difference is probably likely because, while Y2H and PPI prediction studies take into account only the direct interaction, pull-down studies can result in the isolation of entire protein complexes even if only a subset of components interact directly with the protein of interest.
Our analysis showed that amongst the 42 proteins we identified there were some proteins that are known to be directly involved in immune responses indicating the ability of DENV-NS5 to influence host anti-viral responses. Noteworthy amongst these are proteins such as CISH and SOCS5, that form a part of classical negative feedback of cytokine signaling (Yoshimura et al. 2018); NEDD4L, a ubiquitin ligase that inhibits TGF-β signaling through proteasomal degradation of its receptor TGFBR1 and a downstream adaptor SMAD2/3 (Gao et al. 2009); XPO-1, which is known to regulate NFAT-AP1 transcriptional complex (Gruffaz et al. 2019) that has a pivotal role in a T cell activation during a productive immune response (Macian et al. 2001). GRB2 provides a critical link between the RAS and the EGFR signaling pathway (Rozakis-Adcock et al. 1993), and thus is potentially impeded by the binding of NS5. STAT5A is a very well-established downstream effector for cytokines IL-2, IL-7 and GM-CSF, and also many growth hormones. STAT5A also induces anti-apoptotic proteins such as BCL2L1/BCLXL (Socolovsky et al. 1999; Lin and Leonard 2000). TRAF5 is a key molecule in the tumor necrosis factor receptor family that is central to both the innate and adaptive immune response (Hildebrand et al. 2011). Another protein, AP4M1, a part of the endosomal-lysosomal system involved in the recognition and sorting of cargo proteins with tyrosine-based motifs (Hirst et al. 1999) needed for cytotoxic responses was also identified as an NS5 interactor (Repnik et al. 2013). AP4M1 RNA was earlier found to be upregulated in DENV-2-infected ECV304 endothelial-like cells suggesting utilization of the AP-4 complex by the virus for its release from the infected cells (Liew and Chow 2006).
Other notable interactors identified by our analysis were proteins such as WWP1, WWP2, NEDD4, USP7, SMURF1, COP1 that ubiquitinate to degrade proteins involved in key cellular processes. Ubiquitin-mediated proteolysis is known to regulate multiple events such as viral entry, viral replication, cell cycle and apoptosis, essential for viral infection and survival (extensively reviewed in references Isaacson and Ploegh 2009; Gustin et al. 2011)). Proteins involved in cell cycle regulation (CDC20, CCNB1, CCNE1), apoptosis (YAP1), and metabolism (RAPTOR) were also identified. Finally, three NS5 interactors were vital components in the WNT signaling pathway – FBXW11 and APC that ubiquitinate beta-catenin, a key downstream component of WNT signaling (Rao and Kühl 2010) and CTNNB1 were also identified. Thus, NS5 possibly interacts with multiple host proteins that are involved in the key cellular processes of proliferation, apoptosis, protein degradation, and immune responses. Key proteins along with the pathways they regulate are summarized in Table 1 (with references).
All the predicted DENV-NS5 interactors were analyzed using the STRING database to identify the degree of interaction based on the central rule that the higher the degree, the more central the protein is in its interaction with other proteins. We found that proteins CTNNB1, XPO1, CCNB1, USP7 and YWHAB were among the top five with the highest node degree, implying that there is a high probability that DENV-NS5 on interaction with these proteins would perhaps lead to greater control over associated cellular pathways. Supplementary table 5 lists the 42 DENV-NS5 interactors along with the degree of interaction as obtained from STRING.
Pathway analysis for the 42 identified DENV-NS5 interactors
Pathway-based enrichment analysis using WebGestalt (WEB-based Gene SeT AnaLysis Toolkit) (Liao et al. 2019) revealed 12 pathways within which these 42 host proteins participated (Fig. 2). Supplementary table 6 lists the results of the KEGG pathway enrichment along with the enrichment scores, p-values and false discovery rates (FDRs). Notable amongst these was the PI3K-Akt signaling pathway that regulates fundamental functions such as cellular proliferation, growth, transcription, and protein translation – all of which are heightened during an anti-viral response. The cell cycle and the Hippo signaling pathway which contain many genes that support cellular proliferation and growth were found to be significantly enriched. Prolactin acts both as a hormone as well as a cytokine (Harvey et al. 2015), stimulating the secretion of other cytokines and expression of cytokine receptors including IFN-γ, IL-1, and IL-10 (Enninga et al. 2014). In our analysis, it was notable that the prolactin signaling pathway that activates many other signaling cascades containing Src, PI3K-Akt, and MAPK kinases, was also significantly enriched. In summary, our analysis revealed that DENV-NS5 is capable of interacting with host proteins that participate in key cellular processes that are vital to a successful anti-viral immune response.

Pathway enrichment of human interacting partners of DENV-NS5 protein: KEGG pathway enrichment of interacting proteins of DENV-NS5. The Y-axis shows all enriched pathways with FDR < 0.05 and the X-axis shows enrichment ratios representing the number of proteins present in the reference gene set. The size of the dot shows the size of the gene set. The intensity of the color of the nodes is inversely proportional to the p-value i.e., the more intense the color, the lower the p-value and the greater the significance
Identification of downstream transcription factors and differentially expressed genes (DEGs) potentially modulated by DENV-NS5
Cellular pathways are defined as sequential events that ultimately result in a phenotypic or functional consequence. Therefore, we were curious to understand the ripple effects of these 42 direct interactions to truly appreciate the overall impact of predicted DENV-NS5- host protein interactions. We, therefore, mapped the interaction network of DENV-NS5 protein and the 42 human proteins (orange circles in Fig. 3A) along with all potential downstream interactions. We then focused on transcription factors among the downstream interactors (pink circles in Fig. 3A) in order to identify key transcriptional programs that would be potentially affected by NS5-host protein interactions and found five well-studied transcription factors. These were – STAT1 (directly downstream of SOCS5), ETS1 (downstream of BRCA1), RelA (downstream of CTNNB1), E2F1 (downstream of CCNE1) and SP1 (downstream of BRCA1).


Predicted DENV-NS5 interactome: A Schematic representing the proteins predicted to be interacting with DENV-NS5 and their first interactors. Direct DENV-NS5 interactors are shown in orange circles. Transcription factors (TF) present in the first interactors of DENV-NS5 interacting proteins are shown in pink and the genes regulated by them are shown as a TF network. The genes identified to be differentially expressed in previously published dengue RNA-seq data (described in methods) have been shown in light green. Genes in dark green are DEGs additionally reported in other studies relevant to dengue. Some of the edges between nodes have been removed to improve readability. B Transcriptional profile of genes shown in dark green in A. Normalized read counts of selected DEGs have been shown in dengue vs naïve subsets. B-H (Benjamini-Hochberg) adjusted p-values are shown and indicated by *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
One of the 42 interactors of DENV-NS5 was USP7 (orange circle with a blue ring at around 4 o’clock in Fig. 3A). This protein has been reported to directly ubiquitinate, and thereby target for degradation, proteins such as (a) FOXO4 (part of the PI3K-Akt pathway) (b) p53 (an important tumor suppressor that ensures genome stability) (c) MDM2 (a p53 antagonist whose gene is transcriptionally inhibited by p53 and which, in turn, is a p53-specific ubiquitinase that targets p53 for degradation) (d) ERCC6, a chromatin remodelling factor, (e) DNMT1, a DNA methyltransferase responsible for de novo DNA methylation (f) UHRF1, an E3 ubiquitin-protein ligase regulating the G1/S transition (g) PTEN, a tumor suppressor and a key modulator of the mTOR signaling pathway and (h) DAXX, a pro-apoptotic protein that often functions with Fas (reviewed in (Van der Horst et al. 2006)). Therefore, it is likely that the ability of DENV-NS5 to cause pleiotropic effects may be attributed to its interaction with host proteins situated at hubs that are common to several physiological pathways.
Transcription factors affected by DENV-NS5 that have immunological relevance (pink circles in Fig. 3A) are briefly discussed below. A few of the key interactors downstream of these TFs have been highlighted in light green if they were identified previously through RNA-seq studies, or in dark green if they were also validated in dengue infection. The DEGs additionally reported in other studies relevant to dengue (dark green circles in Fig. 3A) have been shown in Fig. 3B. Pathway enrichment analysis of these DEGs showed that there were genes identified that are in involved in cellular processes like apoptosis and cell death (like BCL2L12, TLR3, CDK5), protein metabolism (like PAX6, FGFR1) and protein phosphorylation (like AURKA, DKK1), indicating that DENV-NS5 can potentially modulate several cellular pathways via its interaction with upstream proteins that are vital for its own propagation (Supplementary table 8). A list of key downstream effectors potentially impacted by DENV-NS5 have been tabulated in Table 1 and briefly discussed below.
STAT1 (pink circle at around 4 o’clock in Fig. 3A) is a part of the family of signal transducer and activator of transcription (STAT) proteins that mediate cellular responses downstream of interferon signaling pathways. After Type I IFN signaling, STAT1 is phosphorylated, translocate to the nucleus as part of a larger complex to induce the transcription of interferon-stimulated genes (ISGs) such as Interferon gamma (IFN-γ) to drive an anti-viral immune response (Ramana et al. 2002). Dengue infections are often accompanied by cytokine storms (Srikiatkhachorn et al. 2017), and the binding of NS5 to SOCS5, a suppressor of cytokine signaling probably enhances interferon related events besides resulting in runaway cytokine expression.
ETS1 is known to induce the expression of some cytokines (IFN-γ, IL-5, TFN-α) and repress the expression of others (IL-2, IL-4, IL-10)(Russell and Garrett-Sinha 2010). It is tempting to hypothesize that ETS1 downregulation via direct interaction of BRAC1 with NS5 results in poor Th1 cytokine production, thereby inhibiting the process of viral clearance, and increased production of cytokines such as IL-10 that have been associated with severe dengue manifestations. Further, ETS1 is also known to directly control the expression of certain chemokines (CXCL4, CCL2, and CXCL8) either by direct gene regulation, cooperative gene regulation or regulation of calcium flux (Russell and Garrett-Sinha 2010). Thus, NS5, through BRCA1 and ETS1, could be responsible for disrupting the cell-mediated immune response (Th1) that is key in antiviral response.
RelA is a subunit of NF-κB that, along with other NF-κB subunits such as p52, p50, p105 or RelB, can act as a transcriptional activator or repressor (Hiscott et al. 2001). Since its presence is probably regulated via ubiquitin degradation directed through CTNNB1, binding of NS5 to CTNNB1 could affect this regulation. There is then a possibility that RelA upregulates genes such as EGR1 that in turn can increase the expression of cytokines and chemokines such as IL-1β and CXCL2. It is interesting to note that EGR1 transcription is upregulated during dengue infection (Fig. 3B, top row, middle panel), and so is its downstream target IL-1β during dengue disease. Another protein of interest that we identified was TNFRSF10b (alternative name – DR5, encircled with a red circle at approximately 9 o’clock in Fig. 3A), which is a receptor for TRAIL that results in caspase-8-mediated apoptosis.
E2F1 is a transcription factor that plays a crucial role in processes such as cell cycle progression, proliferation, and microtubule formation (Wu et al. 2001) (pink circle at approximately 11 o’clock in Fig. 3A). We identified eleven E2F1-regulated downstream effectors whose genes are upregulated when analysed by RNA-seq in dengue disease (see Fig. 3A).
SP1 is a transcription factor downstream of MAPK and PI3K-Akt signaling pathways and controls many cellular processes that involve differentiation, apoptosis, immune responses and chromatin remodelling (Black et al. 2001) (pink circle at approximately 1 o’clock in Fig. 3A). Genes regulated by SP1 encode proteins such as Pax6 (an NFATC1 repressor), LDLR (receptor for the endocytosis of cholesterol-rich LDL), LRP5 (associated with Wnt signaling) and FASLG (mediator of apoptosis). Transcriptional upregulation of LCAT and LDLR is confirmed by prior RNA-seq studies and dengue infection-related studies as well (LCAT – row 2, column 1 and LDLR – row 3, column 3 in Fig. 3B). Thus, the cell-to-cell spread of DENV may be enhanced by the regulation of LDLR that has been found to bind LDLR ligand-like motifs in the dengue envelope and capsid proteins (Guevara et al. 2015).
Thus, the potential interaction of DENV-NS5 with host proteins most likely results in a cascade of downstream effects, some of which can be directly associated with the immune response or with dengue disease. A recent single-cell RNA-seq analysis study performed on naïve vs dengue patients revealed differential expression of many genes that were identified as downstream targets of DENV-NS5 in our study including TFs like STAT1 and SP1, and genes such as CXCL8, EGR1, PFKL and LDLR, in one or more immune cells(Xu et al. 2022).
Discussion
Viruses have evolved ways to modulate host cellular processes for their own survival, be it by increased viral production, evasion of the host immune system or increasing the long-term survival of latently infected cells. One of the ways of taking over cellular processes is by mimicking the binding surfaces of host proteins, enabling physical interactions with a large set of host proteins (reviewed in (Guven-Maiorov et al. 2016, Fishburn et al. 2022)).
The p-HIPSTER database predicts human-viral PPIs of all human-infecting viruses (Lasso et al. 2019). Its approach combines both sequence and structural similarity prediction as is the case with our study. However, the tools we used for these predictions are different. p-HIPSTER uses PrePPI (http://bhapp.c2b2.columbia.edu/PrePPI), a database that combines predicted and experimentally determined protein–protein interactions (PPIs) using a Bayesian framework. By contrast, we have modelled our protein tertiary structures using i-TASSER, determined the structural similarity using DALI and identified the NS5-human interacting partners using BioGRID, which is based on experimentally validated data, and combined it with ELM-based predictions. Our study specifically focused on our protein of interest, DENV-NS5 protein. In addition to identifying interactors of NS5, we performed a downstream analysis to identify transcription factors and human proteins, and identified the cellular processes directly or indirectly influenced by it. Another deep structural analysis study has been done on Zika-NS5 protein and STAT2 to identify the amino acid residues important for their interaction and IFN antagonism (Dar et al. 2017). Future studies like these involving deep structural analysis could be performed on these identified interacting partners of DENV-NS5 to determine the amino acid residues involved in these interactions (Kumar et al. 2022, Singh et al. 2022a, b).
Our simultaneous and rigorous two-pronged approach to deciphering PPI’s of DENV-NS5 resulted in the identification of 42 host proteins whose functions spanned both the cytoplasm and the nucleus. Further analysis of the downstream interactors of these 42 proteins revealed proteins that, in publicly available transcriptomic studies, exhibit significant fold-change during DENV infection. Our focus on downstream transcription factors has uncovered known as well as previously unsuspected ramifications of DENV-NS5 interactions with host proteins involved in the host immune response.
Our study identified eight previously validated interactors of DENV-NS5 and identified 34 novel interactors, many of which play key roles in anti-viral immune responses or in dengue pathophysiology due to their involvement in the processes of cell cycle regulation, proliferation, protein degradation, apoptosis, and cytokine production among others. It enables the formulation of testable hypotheses regarding the influence of DENV-NS5 on the mediators of the host responses involved in the entire pathogenic pathway starting with a viral effector through host intermediaries and eventually to terminal host effectors. Thus, this study provides insights into how DENV (and other Flaviviruses, owing to high sequence conservation of NS5 protein across all Flaviviruses), might hijack the human host proteome by means of molecular mimicry to facilitate their replication and/or contribute to pathogenesis, an understanding of which can open novel lines of inquiry into both prophylactic and therapeutic options.
Conclusions
Our stringent approach of using both sequence and tertiary structure-based information for studying DENV-NS5 protein that is capable of interacting with human proteins led to the identification of a common, minimal set of novel human interactors. Further identification of downstream interacting partners identified TFs that could be targeted by DENV-NS5 for the ease of establishing infection. Our analysis of previously published RNA-seq data from naïve and dengue-infected individuals has identified certain differentially expressed genes which could be indirectly modulated by DENV-NS5 via interaction with host proteins, some of which interact with transcription factors. This study has led to the generation of the testable hypothesis that requires further experimental validation. However, our analysis clearly indicates that the potential role of DENV-NS5 during viral infection extends well beyond its known enzymatic functions. Further validation of these predicted interactors is required to understand the cumulative effects of these interactions in dengue infection.
Dengue has been a particularly difficult disease to control. With no universal vaccine or antivirals currently available to tackle the disease, other strategies such as behavioral changes (e.g., using mosquito nets) and vector control remain the only options. Under these circumstances, it is important to explore therapeutic options that could help patients by controlling viral replication and modulating immune responses, particularly those that could potentially damage the host tissues. Our study provides mechanistic insights into human-dengue virus interactions at the molecular level, some of which may be used to design and test therapeutic interventions at key pathways to prevent viral multiplication and spread as well as mitigate or prevent adverse clinical outcomes.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information.
References
Allaire J (2012) RStudio: integrated development environment for R. Boston, MA 770(394):165–171
Ashour J, Laurent-Rolle M, Shi P-Y, García-Sastre A (2009) NS5 of dengue virus mediates STAT2 binding and degradation. J Virol 83(11):5408–5418
Banerjee A, Shukla S, Pandey AD, Goswami S, Bandyopadhyay B, Ramachandran V, Das S, Malhotra A, Agarwal A, Adhikari S (2017) RNA-seq analysis of peripheral blood mononuclear cells reveals unique transcriptional signatures associated with disease progression in dengue patients. Transl Res 186(62–78):e69
Berman H, Henrick K, Nakamura H (2003) Announcing the worldwide protein data bank. Nat Struct Mol Biol 10(12):980–980
Bhatnagar P, Sreekanth GP, Murali-Krishna K, Chandele A, Sitaraman R (2021) Dengue virus non-structural protein 5 as a versatile, multi-functional effector in host–pathogen interactions. Front Cell Infect Microbiol. https://doi.org/10.3389/fcimb.2021.574067
Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O (2013) The global distribution and burden of dengue. Nature 496(7446):504–507
Black AR, Black JD, Azizkhan-Clifford J (2001) Sp1 and krüppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol 188(2):143–160
Brooks AJ, Johansson M, John AV, Xu Y, Jans DA, Vasudevan SG (2002) The interdomain region of dengue NS5 protein that binds to the viral helicase NS3 contains independently functional importin beta 1 and importin alpha/beta-recognized nuclear localization signals. J Biol Chem 277(39):36399–36407
Byk LA, Iglesias NG, De Maio FA, Gebhard LG, Rossi M, Gamarnik AV (2016) Dengue virus genome uncoating requires ubiquitination. MBio. https://doi.org/10.1128/mBio.00804-16
Carpp LN, Rogers RS, Moritz RL, Aitchison JD (2014) Quantitative proteomic analysis of host-virus interactions reveals a role for Golgi brefeldin A resistance factor 1 (GBF1) in dengue infection. Mol Cell Proteomics 13(11):2836–2854
Chen J, Ng MM-L, Chu JJH (2015) Activation of TLR2 and TLR6 by dengue NS1 protein and its implications in the immunopathogenesis of dengue virus infection. PLoS Pathog 11(7):e1005053
Cortese M, Kumar A, Matula P, Kaderali L, Scaturro P, Erfle H, Acosta EG, Buehler S, Ruggieri A, Chatel-Chaix L (2019) Reciprocal effects of fibroblast growth factor receptor signaling on dengue virus replication and virion production. Cell Rep 27(9):2579-2592.e2576
Cui L, Lee YH, Kumar Y, Xu F, Lu K, Ooi EE, Tannenbaum SR, Ong CN (2013) Serum metabolome and lipidome changes in adult patients with primary dengue infection. PLoS Negl Trop Dis 7(8):e2373
Dar HA, Zaheer T, Paracha RZ, Ali A (2017) Structural analysis and insight into Zika virus NS5 mediated interferon inhibition. Infect Genet Evol 51:143–152
De Maio FA, Risso G, Iglesias NG, Shah P, Pozzi B, Gebhard LG, Mammi P, Mancini E, Yanovsky MJ, Andino R (2016) The dengue virus NS5 protein intrudes in the cellular spliceosome and modulates splicing. PLoS Pathog 12(8):e1005841
Dey L, Mukhopadhyay A (2017) DenvInt: a database of protein–protein interactions between dengue virus and its hosts. PLoS Negl Trop Dis 11(10):e0005879
Dinkel H, Van Roey K, Michael S, Kumar M, Uyar B, Altenberg B, Milchevskaya V, Schneider M, Kühn H, Behrendt A (2016) ELM 2016—data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res 44(D1):D294–D300
Duangchinda T, Dejnirattisai W, Vasanawathana S, Limpitikul W, Tangthawornchaikul N, Malasit P, Mongkolsapaya J, Screaton G (2010) Immunodominant T-cell responses to dengue virus NS3 are associated with DHF. Proc Natl Acad Sci 107(39):16922–16927
Durán A, Carrero R, Parra B, González A, Delgado L, Mosquera J, Valero N (2015) Association of lipid profile alterations with severe forms of dengue in humans. Adv Virol 160(7):1687–1692
El Sahili A, Lescar J (2017) Dengue virus non-structural protein 5. Viruses 9(4):91
El Sahili A, Soh TS, Schiltz J, Gharbi-Ayachi A, Seh CC, Shi PY, Lim SP, Lescar J (2019) NS5 from dengue virus serotype 2 can adopt a conformation analogous to that of its Zika virus and Japanese encephalitis virus homologues. J Virol. https://doi.org/10.1128/JVI.01294-19
Enninga EAL, Holtan SG, Creedon DJ, Dronca RS, Nevala WK, Ognjanovic S, Markovic SN (2014) Immunomodulatory effects of sex hormones: requirements for pregnancy and relevance in melanoma. Mayo Clin Proc. https://doi.org/10.1016/j.mayocp.2014.01.006
Fishburn AT, Pham OH, Kenaston MW, Beesabathuni NS, Shah PS (2022) Let’s get physical: flavivirus-host protein-protein interactions in replication and pathogenesis. Front Microbiol. https://doi.org/10.3389/fmicb.2022.847588
Fontaine KA, Sanchez EL, Camarda R, Lagunoff M (2015) Dengue virus induces and requires glycolysis for optimal replication. J Virol 89(4):2358–2366
Gao S, Alarcón C, Sapkota G, Rahman S, Chen P-Y, Goerner N, Macias MJ, Erdjument-Bromage H, Tempst P, Massagué J (2009) Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-β signaling. Mol Cell 36(3):457–468
Garcia-Alonso L, Holland CH, Ibrahim MM, Turei D, Saez-Rodriguez J (2019) Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res 29(8):1363–1375
Giraldo MI, Xia H, Aguilera-Aguirre L, Hage A, van Tol S, Shan C, Xie X, Sturdevant GL, Robertson SJ, McNally KL (2020) Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature 585(7825):414–419
Grifoni A, Tian Y, Sette A, Weiskopf D (2020) Transcriptomic immune profiles of human flavivirus-specific T-cell responses. Immunology 160(1):3–9
Gruffaz M, Yuan H, Meng W, Liu H, Bae S, Kim J-S, Lu C, Huang Y, Gao S-J (2019) CRISPR-Cas9 screening of Kaposi’s sarcoma-associated herpesvirus-transformed cells identifies XPO1 as a vulnerable target of cancer cells. Mbio 10(3):e00866-e819
Guevara J, Romo J, McWhorter T, Guevara NV (2015) Analogs of LDL receptor ligand motifs in dengue envelope and capsid proteins as potential codes for cell entry. J Viruses 2015:1–15
Gustin J, Moses A, Fruh K, Douglas J (2011) Viral takeover of the host ubiquitin system. Front Microbiol 2:161
Guven-Maiorov E, Tsai C-J, Nussinov R (2016) Pathogen mimicry of host protein-protein interfaces modulates immunity. Semin Cell Dev Biol. https://doi.org/10.1016/j.semcdb.2016.06.004
Guzman MG, Halstead SB, Artsob H, Buchy P, Farrar J, Gubler DJ, Hunsperger E, Kroeger A, Margolis HS, Martínez E (2010) Dengue: a continuing global threat. Nat Rev Microbiol 8(12):S7–S16
Hannemann H, Sung P-Y, Chiu H-C, Yousuf A, Bird J, Lim SP, Davidson AD (2013) Serotype-specific differences in dengue virus non-structural protein 5 nuclear localization. J Biol Chem 288(31):22621–22635
Harvey S, Martínez-Moreno CG, Luna M, Arámburo C (2015) Autocrine/paracrine roles of extrapituitary growth hormone and prolactin in health and disease: an overview. Gen Comp Endocrinol 220:103–111
Hildebrand JM, Yi Z, Buchta CM, Poovassery J, Stunz LL, Bishop GA (2011) Roles of tumor necrosis factor receptor associated factor 3 (TRAF3) and TRAF5 in immune cell functions. Immunol Rev 244(1):55–74
Hirst J, Bright NA, Rous B, Robinson MS (1999) Characterization of a fourth adaptor-related protein complex. Mol Biol Cell 10(8):2787–2802
Hiscott J, Kwon H, Génin P (2001) Hostile takeovers: viral appropriation of the NF-kB pathway. J Clin Investig 107(2):143–151
Holm L (2020) Using Dali for protein structure comparison. Structural bioinformatics. Springer, pp 29–42
Isaacson MK, Ploegh HL (2009) Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 5(6):559–570
Kanlaya R, Pattanakitsakul S-N, Sinchaikul S, Chen S-T, Thongboonkerd V (2010) The ubiquitin− proteasome pathway is important for dengue virus infection in primary human endothelial cells. J Proteome Res 9(10):4960–4971
Khadka S, Vangeloff AD, Zhang C, Siddavatam P, Heaton NS, Wang L, Sengupta R, Sahasrabudhe S, Randall G, Gribskov M (2011) A physical interaction network of dengue virus and human proteins. Mol Cell Proteomics. https://doi.org/10.1074/mcp.M111.012187
Klausen MS, Jespersen MC, Nielsen H, Jensen KK, Jurtz VI, Soenderby CK, Sommer MOA, Winther O, Nielsen M, Petersen B (2019) NetSurfP-2.0: improved prediction of protein structural features by integrated deep learning. Proteins: Struct, Funct, Bioinform 87(6):520–527
Klema VJ, Ye M, Hindupur A, Teramoto T, Gottipati K, Padmanabhan R, Choi KH (2016) Dengue virus nonstructural protein 5 (NS5) assembles into a dimer with a unique methyltransferase and polymerase interface. PLoS Pathog 12(2):e1005451
Kumar S, Bhardwaj VK, Singh R, Das P, Purohit R (2022) Identification of acridinedione scaffolds as potential inhibitor of DENV-2 C protein: an in silico strategy to combat dengue. J Cell Biochem. https://doi.org/10.1002/jcb.30237
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26(2):283–291
Lasso G, Mayer SV, Winkelmann ER, Chu T, Elliot O, Patino-Galindo JA, Park K, Rabadan R, Honig B, Shapira SD (2019) A structure-informed atlas of human-virus interactions. Cell 178(6):1526-1541.e1516
Laurent-Rolle M, Boer EF, Lubick KJ, Wolfinbarger JB, Carmody AB, Rockx B, Liu W, Ashour J, Shupert WL, Holbrook MR (2010) The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J Virol 84(7):3503–3515
Le Breton M, Meyniel-Schicklin L, Deloire A, Coutard B, Canard B, De Lamballerie X, Andre P, Rabourdin-Combe C, Lotteau V, Davoust N (2011) Flavivirus NS3 and NS5 proteins interaction network: a high-throughput yeast two-hybrid screen. BMC Microbiol 11(1):1–11
Li S, Zhou W, Li D, Pan T, Guo J, Zou H, Tian Z, Li K, Xu J, Li X (2022) Comprehensive characterization of human–virus protein-protein interactions reveals disease comorbidities and potential antiviral drugs. Comput Struct Biotechnol J. https://doi.org/10.1016/j.csbj.2022.03.002
Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B (2019) WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 47(W1):W199–W205
Liew KJ, Chow VT (2006) Microarray and real-time RT-PCR analyses of a novel set of differentially expressed human genes in ECV304 endothelial-like cells infected with dengue virus type 2. J Virol Methods 131(1):47–57
Lin J-X, Leonard WJ (2000) The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene 19(21):2566–2576
Liu L, Dong H, Chen H, Zhang J, Ling H, Li Z, Shi P-Y, Li H (2010) Flavivirus RNA cap methyltransferase: structure, function, and inhibition. Front Biol 5(4):286–303
Liu J, Li Q, Li X, Qiu Z, Li A, Liang W, Chen H, Cai X, Chen X, Duan X (2018) Zika virus envelope protein induces G2/M cell cycle arrest and apoptosis via an intrinsic cell death signaling pathway in neuroendocrine PC12 cells. Int J Biol Sci 14(9):1099
Macian F, López-Rodríguez C, Rao A (2001) Partners in transcription: NFAT and AP-1. Oncogene 20(19):2476–2489
Mairiang D, Zhang H, Sodja A, Murali T, Suriyaphol P, Malasit P, Limjindaporn T, Finley RL Jr (2013) Identification of new protein interactions between dengue fever virus and its hosts, human and mosquito. PLoS One 8(1):e53535
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI (2015) CDD: NCBI’s conserved domain database. Nucleic Acids Res 43(D1):D222–D226
McMahon SB, Monroe JG (1996) The role of early growth response gene 1 (egr-1) in regulation of the immune response. J Leukoc Biol 60(2):159–166
Medin CL, Fitzgerald KA, Rothman AL (2005) Dengue virus nonstructural protein NS5 induces interleukin-8 transcription and secretion. J Virol 79(17):11053–11061
Mering CV, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B (2003) STRING: a database of predicted functional associations between proteins. Nucleic Acids Res 31(1):258–261
Morrison J, Laurent-Rolle M, Maestre AM, Rajsbaum R, Pisanelli G, Simon V, Mulder LC, Fernandez-Sesma A, García-Sastre A (2013) Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog 9(3):e1003265
Nainggolan L, Tahapary DL, Dewi BE, Harbuwono DS, Soewondo P (2017) The association of lipoprotein changes and the development of plasma leakage in dengue infection. eJournal Kedokt Indones 5(1):6–50
Ohno M, Sekiya T, Nomura N, Ji Daito T, Shingai M, Kida H (2020) Influenza virus infection affects insulin signaling, fatty acid-metabolizing enzyme expressions, and the tricarboxylic acid cycle in mice. Sci Rep 10(1):1–12
Pandey AD, Goswami S, Shukla S, Das S, Ghosal S, Pal M, Bandyopadhyay B, Ramachandran V, Basu N, Sood V (2017) Correlation of altered expression of a long non-coding RNA, NEAT1, in peripheral blood mononuclear cells with dengue disease progression. J Infect 75(6):541–554
Park T, Kang M-G, Baek S-H, Lee CH, Park D (2020) Zika virus infection differentially affects genome-wide transcription in neuronal cells and myeloid dendritic cells. PLoS One 15(4):e0231049
Pickett BE, Sadat EL, Zhang Y, Noronha JM, Squires RB, Hunt V, Liu M, Kumar S, Zaremba S, Gu Z (2012) ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res 40(D1):D593–D598
Poonpanichakul T, Chan-In W, Opasawatchai A, Loison F, Matangkasombut O, Charoensawan V, Matangkasombut P (2021) Innate lymphoid cells activation and transcriptomic changes in response to human dengue infection. Front Immunol. https://doi.org/10.3389/fimmu.2021.599805
Poyomtip T, Hodge K, Matangkasombut P, Sakuntabhai A, Pisitkun T, Jirawatnotai S, Chimnaronk S (2016) Development of viable TAP-tagged dengue virus for investigation of host–virus interactions in viral replication. J Gen Virol 97(3):646–658
Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, Van Der Spoel D (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29(7):845–854
Raghupathy R, Chaturvedi U, Al-Sayer H, Elbishbishi E, Agarwal R, Nagar R, Kapoor S, Misra A, Mathur A, Nusrat H (1998) Elevated levels of IL-8 in dengue hemorrhagic fever. J Med Virol 56(3):280–285
Ramana CV, Gil MP, Schreiber RD, Stark GR (2002) Stat1-dependent and-independent pathways in IFN-γ-dependent signaling. Trends Immunol 23(2):96–101
Rao TP, Kühl M (2010) An updated overview on Wnt signaling pathways: a prelude for more. Circ Res 106(12):1798–1806
Ravi K, Keerthi Kumar N, Aishwarya DM (2021) The lipid profile as a marker for predicting the severity of dengue fever. Int J Adv Med 8(5):691
Rawlinson SM, Pryor MJ, Wright PJ, Jans DA (2009) CRM1-mediated nuclear export of dengue virus RNA polymerase NS5 modulates interleukin-8 induction and virus production. J Biol Chem 284(23):15589–15597
Repnik U, Česen MH, Turk B (2013) The endolysosomal system in cell death and survival. Cold Spring Harb Perspect Biol 5(1):a008755
Rozakis-Adcock M, Fernley R, Wade J, Pawson T, Bowtell D (1993) The SH2 and SH3 domains of mammalian Grb2 couple the EGF receptor to the Ras activator mSos1. Nature 363(6424):83–85
Russell L, Garrett-Sinha LA (2010) Transcription factor Ets-1 in cytokine and chemokine gene regulation. Cytokine 51(3):217–226
Sessions OM, Tan Y, Goh KC, Liu Y, Tan P, Rozen S, Ooi EE (2013) Host cell transcriptome profile during wild-type and attenuated dengue virus infection. PLoS Negl Trop Dis 7(3):e2107
Shah PS, Link N, Jang GM, Sharp PP, Zhu T, Swaney DL, Johnson JR, Von Dollen J, Ramage HR, Satkamp L (2018) Comparative flavivirus-host protein interaction mapping reveals mechanisms of dengue and Zika virus pathogenesis. Cell 175(7):1931-1945.e1918
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504
Sievers F, Higgins DG (2014) Clustal omega, accurate alignment of very large numbers of sequences. Multiple sequence alignment methods. Springer, pp 105–116
Singh R, Bhardwaj VK, Purohit R (2022a) Inhibition of nonstructural protein 15 of SARS-CoV-2 by golden spice: a computational insight. Cell Biochem Funct. https://doi.org/10.1002/cbf.3753
Singh R, Kumar S, Bhardwaj VK, Purohit R (2022b) Screening and reckoning of potential therapeutic agents against DprE1 protein of Mycobacterium tuberculosis. J Mol Liq 358:119101
Socolovsky M, Fallon AE, Wang S, Brugnara C, Lodish HF (1999) Fetal anemia and apoptosis of red cell progenitors in Stat5a−/− 5b−/− mice: a direct role for Stat5 in Bcl-XL induction. Cell 98(2):181–191
Soto-Acosta R, Mosso C, Cervantes-Salazar M, Puerta-Guardo H, Medina F, Favari L, Ludert JE, Del Angel RM (2013) The increase in cholesterol levels at early stages after dengue virus infection correlates with an augment in LDL particle uptake and HMG-CoA reductase activity. Virology 442(2):132–147
Srikiatkhachorn A, Mathew A, Rothman AL (2017) Immune-mediated cytokine storm and its role in severe dengue. Semin Immunopathol. https://doi.org/10.1007/s00281-017-0625-1
Stark C, Breitkreutz B-J, Reguly T, Boucher L, Breitkreutz A, Tyers M (2006) BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34(1):535–539
Tian Y, Seumois G, De-Oliveira-Pinto LM, Mateus J, Herrera-de la Mata S, Kim C, Hinz D, Goonawardhana NS, de Silva AD, Premawansa S (2019) Molecular signatures of dengue virus-specific IL-10/IFN-γ Co-producing CD4 T cells and their association with dengue disease. Cell Rep 29(13):4482–4495
Ulgen E, Ozisik O, Sezerman OU (2019) pathfindR: an R package for comprehensive identification of enriched pathways in omics data through active subnetworks. Front Genet 10:858
Van der Horst A, de Vries-Smits AM, Brenkman AB, van Triest MH, van den Broek N, Colland F, Maurice MM, Burgering BM (2006) FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol 8(10):1064–1073
van Gorp EC, Suharti C, Mairuhu AT, Dolmans WM, van Der Ven J, Demacker PN, van Der Meer JW (2002) Changes in the plasma lipid profile as a potential predictor of clinical outcome in dengue hemorrhagic fever. Clin Infect Dis 34(8):1150–1153
WHO (2022) Dengue and severe dengue. From https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue#:~:text=The%20number%20of%20dengue%20cases,affecting%20mostly%20younger%20age%20group.
Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, Nuckolls F, Giangrande P, Wright FA, Field SJ (2001) The E2F1–3 transcription factors are essential for cellular proliferation. Nature 414(6862):457–462
Xu G, Gao Y, Pan T, Li S, Zhang Y, Guo J, Tian Z, Xu J, Li Y, Li X (2022) Dynamic immune ecosystem of dengue infection revealed by single-cell sequencing. J Leukoc Biol. https://doi.org/10.1002/JLB.6MA0622-738RR
Yoshimura A, Ito M, Chikuma S, Akanuma T, Nakatsukasa H (2018) Negative regulation of cytokine signaling in immunity. Cold Spring Harb Perspect Biol 10(7):a028571
Yu H, Huang X, Ma Y, Gao M, Wang O, Gao T, Shen Y, Liu X (2013) Interleukin-8 regulates endothelial permeability by down-regulation of tight junction but not dependent on integrins induced focal adhesions. Int J Biol Sci 9(9):966
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinform 9(1):1–8
Zhao Y, Soh TS, Zheng J, Chan KWK, Phoo WW, Lee CC, Tay MY, Swaminathan K, Cornvik TC, Lim SP (2015) A crystal structure of the dengue virus NS5 protein reveals a novel inter-domain interface essential for protein flexibility and virus replication. PLoS Pathog 11(3):e1004682
Acknowledgements
We would like to thank Dr. Sakshi Chaudhary (ICGEB, New Delhi) and Love Panchariya (ICGEB, New Delhi) for their valuable inputs at the time of manuscript revision.
Funding
The authors did not receive support from any organization for the submitted work.
Author information
Authors and Affiliations
Contributions
Conceptualization and design of pipeline was done by JS and PB. Data curation and analysis was done by PB. RNA-seq data curation and analysis was done by PB. Figures were made by PB and PB. PB wrote the first draft. Rewriting, reviewing and editing were done by PB, MKK, AC and RS.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Human and animal participants
This research does not involve human participants and/or animals.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bhatnagar, P., Bajpai, P., Shrinet, J. et al. Prediction of human protein interactome of dengue virus non-structural protein 5 (NS5) and its downstream immunological implications. 3 Biotech 13, 180 (2023). https://doi.org/10.1007/s13205-023-03569-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-023-03569-0