
Abstract
Protein aggregation, their mechanisms and trends in the field of neurodegenerative diseases is still far from completely being decoded. It is mainly attributed to the complexity surrounding the interaction between proteins which includes various regulatory mechanisms involved with the presentation of abnormal conditions. Although most proteins are functional in their soluble form, they have also been reported to convert themselves into insoluble aggregates under certain conditions naturally. Misfolded protein forms aggregates which are mostly unwanted by the cellular system and are mostly involved in various pathophysiologies including Alzheimer’s, Type II Diabetes mellitus, Kurus’s etc. Challenges lie in understanding the complex mechanism of protein misfolding and its correlation with clinical evidence. It is often understood that due to the slowness of the process and its association with ageing, timely intervention with drugs or preventive measures will play an essential role in lowering the rate of dementia causing diseases and associated ailments in the future. Today approximately more than 35 proteins have been identified capable of forming amyloids under defined conditions, and nearly all of them have been associated with disease outcomes. This review incorporates a major understanding from the history of diseases associated with protein misfolding, to the current state of neurodegenerative diseases globally, highlighting challenges in drug development and current state of research in a comprehensive manner in the field of protein misfolding diseases. There is increasing clinical association of protein misfolding with regards to amyloids compelling us to thread questions solved and further helping us design possible solutions by generating a pathway-based research on which future work in this field could be driven.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The clinical significance of amyloids and amyloidosis have been more prevalent since nineteenth century although there have been reports of specific significant abnormalities in both liver and spleen (“white stone” in spleen) related to protein misfolding as early as 1639 which was later reviewed by Cohen et al. (1986). The term “amyloid” was first introduced by Rudolf Virchow when he examined large tissue abnormalities in cerebral corpora amylacea and wax-like deposits from spleen, kidney and liver (Virchow 1854; Kumar et al. 2009). The first association of amyloids with a definitive role in diseased conditions was investigated when Alois Alzheimer first reported the presence of senile plaques along with neurofibrillary tangles in the brain of a middle-aged woman. She was suffering from a loss of memory and progressive loss of cognitive function. It was indeed the first reported evidence of Alzheimer’s disease (AD) (Forman et al. 2004). The pathological hallmarks of other similar neurodegenerative conditions like Parkinson’s disease, Lewy bodies (LB) and Lewy neuritis (LN) were further reported by Friedreich Lewy (Lewy et al. 1912).
The primary structure of proteins which, is an assembly of a series of amino acids joined by peptide bonds is stable in terms of thermodynamics and its own intracellular and extracellular conditions. It is well dependent on the cellular microenvironment related to pH, temperature and cellular contents. Its primary structure governs the proper folding of the protein into its native state, as concluded by Afinsen’s experiment in 1973 (Afinsen 1973; Kumar et al. 2009). A shift in the standard conditions of the cellular environment in terms of pH encounters with destabilizing agents, and in vivo changes like mutation can often disturb the primary sequence of the protein which ultimately leads to improper folding of the entire protein or parts of the protein which drives the loss in native structure and leads to aggregation and misfolding of proteins (Dobson 2003; Kumar et al. 2009). Various factors such as the nature of protein being synthesised, the concentration of the protein, the in vivo action of multiple molecular chaperones and proteases and the molecular crowding effect, all have been identified as essential factors that influence the improper folding of proteins followed by aggregation (Minton 2001). Since the native structure of the protein is of primary importance, it is evident that any disturbance leading to the formation of the native state will be fatal in terms of the functioning of the protein (Goldberg et al. 2003; Konar et al. 2019). Misfolding of proteins often leads to awkward transitions in the structure which might result in the exposure of the deeply buried hydrophobic residues from the core of the protein to the solvent side. This, in turn, acts as an epicentre for the formation of well-ordered and insoluble larger fibrillar aggregates known as amyloid fibrils.
In vivo mechanisms for combating protein misfolding
The molecular mechanisms underlying protein misfolding and aggregation is still mysterious to the majority of biophysicists (Dubey et al. 2005; Nallamsetty et al. 2007). Majority of the proteins synthesised undergo the folding process in the cytoplasm once they cross the quality control checkpoints of the ribosomes and begins to interact with heat shock proteins and molecular chaperones. Recent studies also prove that chaperones are not only essential in correctly folding improperly folded proteins but also try and prevent proteins from misfolding by rescuing misfolded proteins, giving them an apparent second chance to gain their native structure. This process is energetically dependent, and hence it is now understood that why ATP is required in high amounts or is present in high amounts in regions associated with cellular stress (Dobson et al. 2003; Agbas et al. 2018). There are many heat shock proteins like Hsp 70, Hsp 90, Hsp 40, along with various catalysts like protein disulphide isomerase and prolyl peptidyl isomerase which helps in the folding of the protein in vivo (Sarkar and Dubey 2010). An active presence of protein quality control in the living cell often recognizes and degrades proteins which are misfolded to prevent further aggregation. If the quality control system of the present cell is compromised, it might lead to the deposition of misfolded proteins in various tissues and organs (Kaganovich et al. 2008; Sarkar and Dubey 2010).
The Endoplasmic reticulum serves as excellent quality control equipment for monitoring proper folding of proteins and also is a reservoir of the variety of chaperones (Dobson 2004). The endoplasmic reticulum is home to a large number of folding catalysts which makes it the go-to cell organelle for protein folding and hence is also closely associated with protein misfolding diseases (Kaufman et al. 2002; Agbas et al. 2018). Aggregation of proteins also leads to impairment of the ubiquitin–proteasome system, which even aggravates the collection of proteins (Kumar et al. 2009). The mechanism of aggregation of protein is in itself a multi-complex phenomenon involving many intermediates. The intermediates are responsible for the formation of the aggregates. The insoluble aggregates have been well characterised which are now called as amyloid fibrils (Dobson et al. 1999; Chiti et al. 1999).
The intermediate states have been inferred to be as an essential stage in the process governing the folding of proteins to reach the native state. This folding pathway is often referred to as Non-cooperative or multistate folding. In many cases, these states may lie away from the productive path called “off-pathway intermediates”. Irrespective of when these intermediates are formed or populated, they end up interacting with each other driving the formation of aggregates and hence shifting the equilibrium towards protein aggregation. At this state, intermolecular interactions are greater than intramolecular interactions. These aggregates are classified as either amorphous or ordered, of which “amyloid fibril” is the most commonly found ordered state. Also, the formation of amyloids is highly dependent on factors like pH, temperature, peptide, protein concentration and ionic strength (Calamai et al. 2005).
Amyloids not associated with diseases
Most significant research in the field of amyloidosis has been emphasised on the pathogenic form of amyloids. There have been numerous studies which reported amyloids are formed by proteins not necessarily associated with disease but in general. There are also many proteins which are known to form amyloid-like features under in vitro conditions but are not associated with any diseased conditions, which indicate that the formation of amyloid structures is an inherent property of any polypeptide chain and this type of association has been found in nature as well (Stefani and Dobson 2003; Fink et al. 2004; Chiti and Dobson 2006). This statement is further strengthened when functional amyloid formation in Escherichia coli was reported formed from the self-assembly of the protein Curlin, which is utilised by E. coli to colonise inert surfaces and mediate host binding proteins. The Curlin amyloid was characterised and was found to be consistent with other amyloid structures, with the diameter ranging from 6–12 nm also containing extensive β sheet structure (Chapman et al. 2002; Fowler et al. 2007). Streptomyces coelicolor, a filamentous bacterium which is known to produce aerial hyphae, is mainly due to a secreted protein called Chaplin. Chaplins have been found in the hyphae of this bacterium and have the ability to form amyloid fibrils which can act cooperatively bringing significant changes in aerial development (Mackay et al. 2001; Chiti and Dobson 2006).Other than these examples from bacteria, functional amyloids formation have recently been highlighted in mammals. Melanosomes, which are lysosome-related organelles differentiate into melanocytes thus permitting epidermal production of melanin have also been characterised by intraluminal fibrous striations on which melanin granules are formed (Berson et al. 2003; Chiti and Dobson 2006). Among more recent studies, another example of such functional amyloid is the RNA- binding protein-Cytoplasmic polyadenylation element-binding protein (CPEB). Other than the structural part which binds with the RNA regulating it to translate a subset of cellular mRNAs, it also contains an alternative amyloid-forming domain. The protein also has a similar domain reported in fruit fly as Orb2. Several studies in the fruit fly Drosophila, sea slug Aplysia, and in mice, all have shown that the amyloid formed by Orb2/CPEB in these organisms has a role in the consolidation of the memory which is independent of its RNA binding motif (Roberts 2016). Studies on Orb2/CPEB had already revealed that the ones found in the fruit fly and the ones found in sea slug are available in two isoforms: Orb2a and Orb2b having a common Prion-like domain (PLD). It is important to note that the Orb2a isoform is a synaptic protein and has an octapeptide (YNKFVNFI) sequence before the prion-like domain, which is important for both efficiencies, as well as the amyloid-like oligomers, formed. The Orb2b form has a longer 162 amino acid portion before the PLD which is canonical to CPEB in its RNA binding functions. The PLD of Orb2a acts in long term memory consolidation independent of its RNA binding domain (Khan et al. 2012) (Fig. 1).

Schematic representation of some of the states which can be adopted by polypeptide chains during folding, and pathways by which they can obtain various intermediate states and form aggregates ultimately leading to beta pleated sheets and fibrillar aggregates
Elucidation on the structure of amyloids
During the initial phase, protein molecules were thought not to interact with each other and exist as an independent entity. The molecular structures of amyloid fibrils have always been important as the proper representation of molecular structure help us understand the interaction between monomers of the protein which drive the formation of amyloids. The importance of structural studies lies in the fact that these will help us study the mechanism of formation of amyloids; the kinetic studies associated with the formation, the interactions which will help us design and develop inhibitors against the fibrillization. In 1997 the first study was carried out to elucidate the structure with the help of synchrotron X-ray diffraction method which revealed the common core structure of the amyloid aggregates which consists of the helical array of beta-sheets parallel to the long fibre and the strands at a right angle to the axis (Blake 1997). Other than the structural characterisation, in vitro and in vivo detection techniques were also developed. Usually, the detection of both amyloids and protein aggregates poses a unique challenge. In vitro detection techniques would include turbidity measurements, light scattering, Thioflavin T assay and various imaging techniques including AFM, SEM and TEM (Khan et al. 2012). During the initial period, there were low-resolution methods for structural studies on proteins before the advent of better and advanced techniques. For a long time, only methods like TEM followed by Atomic Force Microscopy and X-ray crystallography (Blake 1997; Serpell et al. 2000; Chiti and Dobson 2006). Until the early 2000s, one of the major problems as stated in literature was the difficulty in a detailed molecular level characterisation of amyloid fibrils mainly due to its inability to form crystals and also to be too large for NMR spectroscopic studies (Chiti and Dobson 2006). In vivo detection of amyloids includes correlated fluorescence spectroscopy, magnetic resonance spectroscopy and current methods also include single photon emission computed tomography and multi-photon microscopy etc. (Khan et al. 2017). Though Thioflavin T is commonly used to detect the amyloid fibrils but not amorphous aggregates both in vivo and in vitro, its activity is largely dependent on pH, changes in pH might cause Thioflavin T to form micelles and decrease the fluorescence by several folds (Khurana et al. 2005; Khan et al. 2017).The situation changed with the advent of solid-state NMR (SS-NMR) spectroscopy for structural studies on amyloid fibrils. It also helped in being able to generate nano and microcrystals peptide fragments having amyloidogenic fibrillar characteristics (Petkova et al. 2002; Ritter et al. 2005; Chiti and Dobson 2006). With the use of SS-NMR along with computational energy minimization processes, Tycko and co-workers had for the first time put forward a structure of amyloid β fibrils formed from the first 40 residues of amyloid-β peptide, pH maintained was 7.4 and temperature was 24 °C under quiescent conditions (Petkova et al. 2002; Chiti and Dobson 2006). The structure of Aβ1-40 elucidated arrangement in form of a pair of β-strands, involving regions approximately from 12th to 24th residue and 30th to 40th residue in the core region of the fibrils. The interesting feature in this arrangement is that there a loop from residues 25–29 which connects these two β strands which do not belong to the same β sheet but instead form parts of two distinct β sheets within the same protofilament. The different Aβ molecules are stacked upon each other in a parallel arrangement (Petkova et al. 2002; Chiti and Dobson 2006). Rigorous structural studies with TEM, Scanning Transmission Electron Microscope (STEM) have shown that the single protofilament consists of four β-sheets which are separated by distances of approximately 10 Å (Chiti and Dobson 2006). The key features highlighted in this structure was further validated by Site-directed spin labelling couple to electron paramagnetic resonance (SDSL-EPR) (Torok et al. 2002; Chiti and Dobson 2006). With advancements in SSNMR, a technique which gives us the liberty to measure specific internuclear distances and torsional angles have allowed researchers to carefully decipher the structure of an 11-residue fragment of the protein transthyretin within amyloid-like fibril. This study revealed that the peptide fragment adopted an extended β strand within the fibrils. This study was also important from the point that it revealed that the molecules within there are a certain degree of uniformity within the fibrils in terms of torsion angles associated with the side chain which had previously been associated with crystal structures. It laid the foundation for further analysis through X-ray crystallography (2002; Jaroneic et al. 2004; Chiti and Dobson 2006). One of the highs in the structural study of amyloids came when a Sup35 derived small hexapeptide sequence (GNNQQNY) which was incubated to generate a three-dimensional crystal possessing the critical features of amyloids. This study helped to gain information on the structure of the peptides, and in the way, the molecules are packed themselves with unprecedented resolution using high-resolution X-ray Crystallography. The crystals consisted of a pair of β sheets in parallel orientation in which each molecule contributed to a single β strand. The β strands were stacked parallel to each other in both the sheets. The sheets interacted mainly via the side-chain interactions of Aspargine at second and sixth position and Glutamine at fourth position. The interactions were such that there was no water in the region formed in between them. Other side chains which were external had more hydration and were at a considerable distance from the next β sheet pair which suggested for a less intimate interaction specifically about crystal contact instead of representing a characteristic of amyloid fibrillar state (Chiti and Dobson 2006) (Fig. 2).

Diagram showing two proteolytic pathways of the amyloidogenic precursor protein. ED and ID are extracellular and intracellular domain, respectively. APP is a membrane bound type-1 protein which can be processed by two pathways, the amyloidogenic and non amyloidogenic pathway. In the non amyloidogenic pathway, there is proteolysis of the APP in the ectodomain of the protein which produces bigger soluble Aβ fragments. This step is catalysed by the α Secretase. In addition to this, the C-terminal domain of the potein is attached to the membrane is processed by γ Secretase which produces smaller fragments with very less cytotoxicity called p3 and the intracellular domain of APP which is known to show certain neuroprotective activity. In the amyloidogenic pathway, the enzyme that causes proteolysis of the APP in the N terminal, APPβ and CTF β. CTFβ is further processed by γ secreatse which produces Aβ fragments insoluble (Carrillo-Mora et al. 2014)
Studies on ss NMR, cryo-electron microscopy (cryo-EM) and X-ray micro crystallography have been able to generate a large amount of data which has increased our knowledge about the structure of amyloid fibrils in a detailed manner by drastically comparing a vast array of peptides and proteins. The details of the arrangement of Aβ40protofilaments and two of their models helped us understand both the similarities and the differences in particular among the different polymorphic forms of this vital system (Paravastu et al. 2008; Petkova et al. 2006; Chiti and Dobson 2017).
The structural models were developed based on the constraints modelled for ssNMR, TEM imaging and mass per length measurements obtained with STEM. In one of the models, the protofilaments are 6 nm in width and have lateral association to form a striated ribbon-like structure (Petkova et al. 2006; Chiti and Dobson 2017) although Paravastu et al. reported that the protofilaments are 7 nm in width and has no association and hence does not form striated ribbon appearance (Paravastu et al. 2008). The similarity between the two models includes that each polypeptide chain has a flexible N- terminus and residues from the 10–22nd position and 30–40th position forms pair of β strands which is linked by a loop. Similarities also include that the strands are stacked into β sheet, lying along the axis of the protofilaments, strands being arranged in parallel having a few intermolecular contacts via a side chain. Despite these standard features between the two models, the polymorphism in the Aβ40 with striated ribbons, each protofilament is reported to consist of four β sheets having overall twofold symmetry with the second polymorph has six β sheets are present and has threefold symmetry. The differences between the two polymorphs also include their respective side-chain interactions. Other structures which have been reported of Aβ40 and Aβ42 in recent times using ssNMR have indicated that there are more possible polymorphs which could be obtained with the same peptide (Colvin et al. 2016; Chiti and Dobson 2017).
Studies involving other proteins which have modelled are human IAPP, the median of whose protofilaments have been deduced using similar techniques. Specific other proteins from other diseased systems have also been worked upon. However, the detailed protofilaments structure have not been worked out yet although the regions or sequences which can form β strands in the fibrils have been identified like transthyretin, Human PrP, human insulin, calcitonin and β-microglobulin (Helmus et al. 2011; Yang et al. 2010; Bateman et al. 2011; Daebel et al 2012; Chiti and Dobson 2017). More recently, the fibril structure of the prion domain (218–289) of HET-s from P. anserina have been worked out using ss-NMR. This structure revealed that it is a single protofilament fibril consisting of a left-handed β-solenoid or β helix structure in which each molecule contributes to two windings of β solenoid and eight β-strands, where the first four strands add to the primary winding and the last four strands contribute to the second winding resulting in four parallel β sheets running along the fibril axis (Van Melekebeke et al. 2010; Vasquez-Fernandez et al. 2016; Chiti and Dobson 2017). A similar structure has also been elucidated for human prion protein based on the X-ray fibre diffraction data collected, although each molecule in this structure contributes to four windings in the β-helix (Vasquez-Fernandez et al. 2016). Transthyretin 11 residue peptide corresponding to the sequence of amino acids from 106–115 have also been carried out using high-resolution structural analysis using a series of experimental techniques. The high-end atomic resolution structures at 5 nm of a three fibril polymorph having pairs of two, three and four protofilaments were solved (Fitzpatrick et al. 2017).
The X-ray diffraction studies have also helped us increase our knowledge about the various types of interactions which are most likely to be present in amyloid fibrils. These studies have been made on microcrystals of short peptides generally fewer than ten amino acids. This system has been already used to study model conditions related to approximately thirty states revealing series of standard features among all these conditions. Participation of each peptide molecule in formation of β-strand and in-register alignment of β-strands within β-sheet which helps in optimizing intermolecular interactions, the presence of pairs of β-sheets parallel to each other drives formation of aggregation along the fibril axis along with right interactions of the side chain within these pairs. The various structures have been classified based on specific characteristics: (a) whether the strands in the sheet are parallel or antiparallel (b) whether the -adjacent sheets interact with face-to-face orientation or face-to-back orientation (c) whether the corresponding strand termini from different sheets point the same or opposite directions (Eisenberg and Jucker 2012; Chiti and Dobson 2017). A novel micro electron diffraction technique has been applied to determine an 11 residue peptide structure which was assembled into nanocrystals at a 1.4 Å resolution which further strengthened our ability to solve formation of amyloids up to atomic resolution (Rodriguez et al. 2015; Chiti and Dobson 2017). Electron microscopic studies can be used to differentiate polymorphic fibrils and help us understand the structural differences among amyloids. Characterisations of amyloids are surrounded on the degree of twist, the number of filaments in each fibril as well as the diameter and mass per unit length of the fibril (Riek 2017).
Solid-state NMR spectra can also observe structural polymorphism, can be as separate peak signals due to differential chemical shift resonance or as extensive lines which indicate the actual macroscopically observed polymorphisms at atomic levels. When high-resolution X-ray crystallography is performed to study short amyloid peptides, they tend to form microcrystals of different confirmations of steric zippers (Colletier et al. 2011; Riek 2017). Different types of polymorphisms are observed-(a) the packing polymorphism is dependent upon the differences in packing between β strands when they are arranged either in parallel or antiparallel orientation. (b) The segmental polymorphism is where particular segments of the amyloid protein or peptide are involved in the cross β sheet formation; for instance, in the Aβ42 protein, there are various segments which are identified as regions which form cross-linked sheets individually and also are part of β sheet core. (c) There are also assembly polymorphisms between different amyloid proteins which are mainly due to the interactions of the individual protofilaments forming a fibril although the atomic structure could show the same arrangement. The probable cause behind the strong interactions of the protofilaments forming the fibril is the repeated interactions of the weak forces, which in summation increases their potency. (d) Lastly, there is also a possibility of the polymorphism occurring due to the side chains of the individual amino acids.
The transition of β structured aggregates into amyloid fibrils occurs by monomer or protofibrils addition varying from protein to protein which forms β- aggregates. The different conformational states, interconversions between them are well-governed and monitored by the local cellular environment. The various states of proteins are all utilised by the cells for various biological functions, including the unfolded proteins and amyloid fibrils, and conformational diseases occur when the regulation within the cells fail similarly to an occurrence of metabolic disorders (Fig. 3).

Graphical representation of various classes of drugs in Phase III clinical trials as of January, 2018 (Cummin et al. 2018). Majority of the drugs currently in the market are targeted towards anti amyloidogenic activity with very few drugs are targeted towards being neuroprotective or acting on the metabolic activity. Neurotransmitters (NT) are second to Anti amyloidogenic drugs in the pipeline
The role of amyloids and amyloid-like structures in human disease biology
Some diseased conditions in humans arise due to the failure of a specific protein peptide to adapt to its functional state. Even if the protein’s primary structure is formed with the correct amino acid sequence, improper folding of the protein makes it attain a different structure other than the native state which might often cause pathological conditions to form which are referred to as protein misfolding or protein conformational diseases. Improper folding of the protein not only reduces its actual biological role but also affect its production at the natural level. This reduction might also be a result of various posttranslational modifications like the increased probability of the protein being degraded by the quality control system involving the Golgi-endoplasmic reticulum system. There is a large chunk of protein misfolding diseases is associated with the conversion of parent proteins or specific peptide sequences from their soluble state to highly ordered fibrillar aggregates (Chiti and Dobson 2006). These structures are also called amyloid fibrils or plaques when they accumulate extracellularly and form intracellular inclusions, which is a more appropriate term when the morphological comparison of amyloid fibrils built intracellularly, and extracellular plaques are compared (Westermark et al. 2005; Chiti and Dobson 2006; Ashraf et al. 2014). The other group of protein folding diseases is due to improper protein folding due to mutations which shifts the amino acid blueprint (Ashraf et al. 2014) (Fig. 4).

Graphical representation of various mechanisms of actions of drugs in Phase II clinical trials as of January, 2018 (Cummin et al. 2018). 1. Monoclonal Antibody, 2. Polyclonal Antibody, 3. Mast Cell Stabilizer, 4. NMDA Receptor, 5. BACE1 Inhibitor, 6. Nor Epinephrine Reuptake Inhibitor, 7. Insulin Signalling Modulator, 8. Anti-Viral therapy 9. 5-HT6 inhibitors, 10. AchE Inhibitors, 11. Agitation therapy 12. Anti- Psychosis 13. Anti-Inflammatory 14. Phosphodiesterase Inhibitors 15. RAGE antagonist, 16. Anti- Tau aggregator, 17. Serotonin Reuptake Inhibitor, 18. Dopamine Receptor Modulator, 19. Dual-Orexin Receptor Antagonist. Based on the mechanism, most of the drugs are targeted as BACE1 inhibitors and the second highest being the use of monoclonal antibodies. This figure marks the areas in which future drug research in the field of neurodegenerative diseases could be targeted
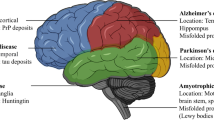
Precisely 37 peptides or proteins have already been found to be associated with amyloid deposits causing pathological conditions in humans. The proteins involved in these conditions are both secretory and cytosolic. Secretory proteins tend to form extracellular deposits and the proteins which are cytosolic form intracellular deposits having amyloid-like characteristics. An interesting fact is that the polypeptides which form amyloids are mostly very short in size having lesser than 100 amino acid residues and only four has greater than 400 residues, and none has more than 700 residues (Chiti and Dobson 2017). Some of the proteins associated with amyloidosis forms deposits in the central nervous system, which causes the vast array of neurodegenerative diseases such as Alzheimer and Parkinson. In contrast, other proteins form deposits in different types of tissues which results in amyloids in non-neuropathic diseases. More than half of the amyloid-related diseases are sporadic in nature which usually have a history of late-onset which also suggests that the sporadic group of disorders is more so because of a result of progressive loss in regulatory control with ageing although some have been reported to be hereditary (Dobson 2003; Powers et al. 2009; Knowles et al. 2014; Chiti and Dobson 2017) (Table 1).
Interestingly a small number of amyloidosis could also be as a result of medical treatments including dialysis-related amyloidosis, haemodialysis, injection localized amyloidosis associated with treatment of type 1 diabetes and iatrogenic Creutzfeldt–Jakob disease which results from organ transplants or from biological derivatives which might be contaminated with prion proteins (Chiti and Dobson 2017). Creutzfeldt–Jakob disease is also known to be caused by the consumption of meat from carcasses of cattle infected with the bovine form of the disease (Chiti and Dobson 2017). The factors which cause a high rate of transmission of prions diseases in humans are now are strict regulation which has resulted in the diagnosis of no new diseases in the recent few years (Chiti and Dobson 2017). Neurodegenerative conditions where aggregations occur in the brain, like Alzheimer's and Parkinson's are mostly sporadic, but hereditary forms are also reported. Both AA amyloidosis and AL amyloidosis are sporadic, whereas the transthyretin peptide associated amyloidosis is reported to be both sporadic as well hereditary. The Apo A proteins I and IV mainly affect multiple organs, although Apo proteins II and III mainly cause renal amyloidosis. Lysozyme, gelsolin and cystatin associated amyloidosis are also systematic and hereditary (Chiti and Dobson 2017).
Molecular mechanism of beta-amyloid formation
Amyloids are formed by sequential cleavage of APP protein. APP is a significant Type I integral membrane protein of 695–770 amino acids which is sequentially cleaved by BACE 1 or α-Secretase to CTFβ-99 amino acids and CTF α-83 amino acids, respectively. Overexpression of APP protein causes a higher amount of Amyloidβ formation. It is generally accepted that APP protein is first transported to the cell surface, where it is processed by αSecretase to CTF α followed by processing by γSecretase which resides in multiples locations in Golgi bodies and cell surface. APP which is not handled by αSecretase is internalised by endocytic vesicles and is processed by BACE 1 (Beta-site APP Cleaving Enzyme), an aspartic protease with acidic pH cleaving it to generate CTFβ, further processed by γSecretase to Aβ40 and Aβ42 which is then transported to cell surface and secreted via recycling vesicles (Carillo Mora et al. 2014; Ashraf et al. 2014). The elongated Aβ variant as compared to the Aβ40 has more aggregation, and it is assumed that the length of Aβ is an essential feature for both neurotoxicity and pathogenesis of AD (Table 2).
Strangely the normal function of Aβ peptide is still yet to be fully explored. It is known to have functions related to activation of kinases, protection from damages caused by metal oxidation, help in the transport of cholesterol and damages to ion channels (Malnoney and Lahiri 2011; Yang et al. 2014). The amyloid precursor protein is a common protein which is expressed almost in all mammalian tissues. It is highly expressed in kidneys and brains, and similar proteins are found in organisms like Drosophila and Caenorhabditis elegans, which suggests that the proteins also function in developmental biology and during the development of organisms. In vitro studies also suggests cleavage of APP products also play a role in cell adhesion and growth of neurons (Yang et al. 2014). Over the past few decades, there have been enough studies on understanding the molecular mechanisms underlying the development of the of various neurodegenerative diseases with special emphasis on Alzheimer’s disease. Pathological remarks include plaques formed extracellularly and intracellular neurofibrillary tangles in brain comprises mainly of Aβ protein and tau protein in its hyperphosphorylated state. The hyperphosphorylated tau protein also forms extracellular deposits which disrupt synaptically dense regions. The Aβ deposits in both extracellular and intracellular regions also show synaptotoxic effects existing in a highly dynamic equilibrium between the small, soluble forms and larger, insoluble fibrillar aggregates. Recent studies have also revealed locations in other portions of the central nervous system outside the brain like deposits in the retina of cataract patients. Clinical data from various researches converges ultimately linking Aβ accumulation and neuroinflammation, synaptic loss, impaired neuronal function and loss in cognition and memory (Selkoe 2001, 2002; Querfurth and LaFerla 2010; Jucker and Walker 2013; Meliet al. 2014; Zuroff et al. 2017) (Table 3).
Mechanism of formation
The formation of amyloids is a cooperative process that in general adheres to the nucleation-dependent polymerisation model-2 phases: the lag phase, where peptide self-associates into small oligopeptides transiently which is followed by the formation of a thermodynamically unfavourable nucleus which is used as a template in elongation phase for fibril growth. Increased B-sheet formation, confirmed via CD and FTIR (De Toma et al. 2012). Research indicates there is a high propensity of formation of beta-sheet motifs in proteins involved in AD and T2DM. The transition from alpha to β sheet exposes a lot of hydrophobic amino acids on the surface of the protein, which causes them to form aggregates. These proteins are also responsible for interacting with other proteins in their native state and influencing them to undergo changes turning them into abnormal proteins and hence are also called infective proteins (Ashraf et al. 2014) (Table 4).
9–10 nm microfilaments called neurofilaments are usually localised within neuronal cell body, axon and dendrites which forms a significant portion of neuronal cytoskeleton thus providing structural support. Neurofibrillary Tangles (NFTs) occurring in the hippocampal pyramidal cells are signature features associated with AD and similar changes have been reported to occur in Down syndrome's patient causing mental retardation and also happen in lesser intellectual people. Paired Helical Twisted Filaments (PHF) is usually the building components of NFTs which are either neurofilaments or microtubules and is majorly formed by Tau protein’s C terminal hyperphosphorylation intracellularly. Conformational changes in tau protein and the formation of NFTs impair any communication and transport between the cell body and synapses which is prime importance in the context of neuronal function and survival. In normal conditions, neuronal microtubules are usually bound by tau proteins and stabilise them, but when hyperphosphorylated tau fails in binding with microtubules which destabilises them, and hence aggregation of hyperphosphorylated tau proteins cause the formation of NFTs which with time change into filamentous form interfering with various neuronal functions.
Signatures of AD
NFTs, Cerebral Amyloid Angiopathy (CAA), neuroinflammation and synaptic loss are the primary drivers of the disease towards clinical diagnosis (Ashraf et al. 2014). This is along with miscompartmentalization of metal like Cu, Fe and Zn (De Toma et al. 2012) which might also contribute to AD onset and progress.
Oxidative stress hypothesis
Reactive Oxygen and Nitrogen Species are often produced by various cellular processes in humans which often play a dual role as beneficial in cellular signalling pathways as well as can be deleterious which might lead to damage of cellular structures including lipid membrane, nucleic acids et cetera. Since there is a higher consumption of oxygen in the brain cells as compared to cells in other tissues, it implies that the cells in brain are more prone to oxidative stress. Since neurons contain a high amount of polyunsaturated fatty acids which can interact with ROS and lipid peroxidation can occur along with molecular apoptosis. Less proportion of glutathione in neurons also makes them more prone to oxidative damage (Nunomura et al. 2006; Mohsenzadegan and Mishrafiey 2012; Pocernish and Butterfield 2012; Liu et al. 2017).
Cholinergic hypothesis
Bartus et al. first proposed that dysfunction in the cholinergic activity of brain in healthy and early dementia patients has a role to play in causing dementia and relative cognitive impairments. Repairing damages to cholinergic pathways may lead to reduce the effect of serious loss in cognitive functions. Studies have shown that Acetylcholine (Ach) which is an important neurotransmitter in the brain, mainly regulating processes dealing with memory and function is greatly reduced owing to loss of its both muscarinic and nicotinic receptors. Along with Acetylcholinesterase (AchE) and Choline acetyltransferase (ChAT) in the brain of patients suffering from AD, Acetylcholine shows a continuous decline. Acetylcholine is made from acetyl CoA, and choline and the process is catalysed by Choline acetyltransferase and the acetylcholine receptor along with transfer of nerve impulses (Auld et al. 2003; Kihara and Shimohama 2004, Schliebs and Arendt 2006; Schliebs and Arendt 2011; Liu et al. 2017).
Inflammatory response
Chronic inflammatory response in the brain is another pathological hallmark of AD. In comparison to normal patients, there is overexpression of acute-phase proteins and pro-inflammatory cytokines. Microglia and astrocytes are the main causes of the inflammatory response. The activated cells produce pro-inflammatory mediators like interleukin 1, interleukin 6, tumour necrosis factor, interleukin 8, prostaglandin, leukotriene, coagulation factors, protease and protease inhibitors et cetera. The production of these substances actually kills the neighbouring neurons (Griffith and Mrak et al. 2002; Tuppo and Arias et al. 2005; Town et al. 2005; Finch and Morgan 2007).
Metal ion hypothesis
There is an important role played by metals ions in maintaining homeostasis. The relationship between metal ions and neurodegeneration has recently been an attractive area of study (Farina et al. 2013; Savelieff et al. 2014; Liu et al. 2017). Metal ions present in the brain are usually a part of the various metalloprotein enzymes as cofactors. The concentration of the various metal ions in the brain are regulated directly by the blood–brain barrier and when the system of this homeostasis between the metal ions and the blood–brain barrier breakdown, it begins to affect the oxidative stress response of the mitochondria and causes wrong folding of proteins ultimately leading to neurodegeneration (Jomova et al. 2010; Popescu and Nichol 2011; Zheng and Monnot 2012; Muhoverac and Vidal 2013; Liu et al. 2017). Studies have shown that aluminium, zinc, copper and iron leads to changes in the conformation of the amyloid β protein. Aluminium leads to accumulation of Aβ and tau protein; also aluminium and copper lead to the development of nerve inflammation (Kawahara 2005; Walton 2013; Liu et al. 2017).
Prevalence of disease
The global prevalence of dementia has increased recently accounting for some 35.6 million around 2010 and projected to be almost double by 2030 and expected to reach up to 65.7 million by 2030 inclusive of both industrialised and developing countries (Ashraf et al. 2014). It was also reported that the annual incidence of AD was 4.6 million cases (Carillo Mora et al. 2014).
Factors governing the amyloid formation
It is well known that amyloid formation is primarily a result of properties which are fairly common to all peptides and proteins. Protein biochemistry also suggests that the sequence of amino acids in a particular protein which might be accessible which majorly influences the relative stability of the protein in its native state via the entire folding process. This plays a key role in a particular sequence of a protein having more tendencies to develop amyloids. Polypeptides with different sequences can form amyloids at various rates although they have a common starting point of a partially folded structure or an improperly folded native state.
Hydrophobicity, charge and secondary structure
One of the common factors among various amyloids formed is the relative abundance of hydrophobic amino acid residues. Substitutions of amino acids in sequences which play critical role in governing the structure of the protein or the folding process by hydrophobic residues or replacing hydrophobic amino acids with hydrophilic residues can increase the chances of the sequence to develop amyloids or amyloid-like properties or decrease it in case of the latter (Chiti et al. 2002; Wurth et al. 2002; Chiti and Dobson 2006). Also, it has been reported that naturally occurring proteins tend to avoid major hydrophobic clusters as part of evolution (Chiti and Dobson 2006). The net charge of a peptide or overall sequence locally, also affects the formation of amyloids. In an experiment with AcP, propensity of the protein to aggregate was tested by single amino acid substitution. It was reported that increasing the net positive charge of the protein decreased its propensity to aggregate and decreasing the net positive charge increased the aggregate formation which was verified by its binding with Congo red and Thioflavin T. It was also reported that the protein aggregation could be accelerated in the presence of macromolecules which would exhibit an overall positive charge on the protein (Konno 2001; Chiti and Dobson 2006). On the contrary, large studies comparing a vast majority of proteins in its native, as well as non-native state, states that non-native states of protein have a lower amount of hydrophobic residues and have a lower overall charge as compared to the native state. This also confirms that the non-native state of proteins has a low probability of forming amyloid-like aggregates under normal physiological conditions (Chiti and Dobson 2006). Also, one other factor that governs the amyloid formation is the presence of the β sheet. Evolution of proteins naturally has assured that there is the elimination of sequences containing alternating hydrophobic and hydrophilic sequences which favour the formation of the β sheet (Chiti and Dobson 2006).
Amino acid sequence
In the experiment related to AcP, various amino acid substitutions were performed to study the effect of sequences on the rate of formation of aggregates which also helped in the development of an equation which could also validate the formation of aggregates for various other proteins. This study also helped us understand the similarity in principles and the mechanisms which govern the mechanism of formation of polypeptide molecules. A number of other factors like the number of aromatic side chains, exposed surface area of the amino acids, net dipole moment also play an important role in determining aggregate forming capability. The relationship between these simple factors and the rate of formation of amyloids is mostly related to the cluster of amino acids forming a simple polymer on the contrary to the proper molten globule state during the normal process of folding which consists of the major secondary structures present in the native state. Increasing understanding of the factors governing the formation of amyloids by increasing the probability of the β sheet formation has helped bioinformatics to develop newer software which could predict the amyloidogenic precursor regions in a protein in unstructured peptides or proteins. This software has been validated for correctly predicting the amyloid-forming regions of the amyloid β protein and α synuclein protein. The sequences predicted by the software to form amyloids was found to be true when the sequences were determined experimentally to form a stable fibrillar core or play a role in the formation of the fibrils (Pawar et al. 2005; Chiti and Dobson 2006).
Unfolded regions within the protein
Although the key regions that associate with the formation of the fibrils can now be easily identified based on various physicochemical parameters of amino acids, actual aggregation of polypeptides exhibiting various levels of secondary structure and wide-range of interactions are also influenced by additional factors. Studies on AcP with limited proteolysis revealed that in the presence of trifluoroethanol in moderate amount, the regions or the sequences which were found to promote amyloid aggregation were flexible and solvent-exposed along with having a high propensity to develop aggregates. Regions that are not involved in aggregation although having high propensities to aggregate were found to be partially buried in the residual structure and other solvent-exposed regions which are not involved in the aggregation process have a low propensity for the formation of amyloid fibrils (Chiti and Dobson 2006).
Role of nucleotide imbalance and neurodegeneration
There is a particular requirement of maintaining a constant level of nucleotides in both dividing cells and quiescent cells. It is equally essential for neural tissues as it requires a high amount of ATP produced by mitochondria. Mitochondria are prone to oxidative stress considerably along with associated damages to DNA, since an imbalance in nucleotide ratio results in mitochondrial depletion because of improper DNA replication. These sorts of genetic defects could often lead to infantile death, although there is a right amount of variability among the manifestations of clinical symptoms in this form of diseases.
Maintenance of the right ratio of nucleotides is critical for proper DNA integrity and prevention of neurodegeneration. Nucleotide levels are kept proper in eukaryotes by two pathways, namely: de novo and salvage pathway (Micheli et al. 2011; Fasullo and Endres 2015). DNA damage results in increased synthesis of dNTP levels which is required for DNA replication and repair (Chabes et al. 2003; Fasullo and Endres 2015). Single gene defects which result in deficiencies of critical enzymes in the pathways have often led to conditions resulting in pathological conditions related to neurology. Purine nucleotides salvage resulting in conditions like gout, neurodegeneration and odd behavioural problems (Fu et al. 2014; Fasullo and Endres 2015). To have a comprehensive understanding of the various pathologies, it is essential to understand the role of dNTPs in neurological physiology, basic biochemistry of the pathways by which the purine and pyrimidine nucleotides are balanced within normal conditions and the effects on cellular conditions when they are imbalanced. Understanding these concepts is also crucial for designing novel drug targets and develops strategies for applications of gene therapy in future.
Nucleotide metabolism needs to be regulated properly in the nervous system, because although the matured, and differentiated cells do not undergo cell division, but there is still a high requirement and metabolism of ATP. Astrocytes and glycolytic pathway do produce lactate, but the neurons still use oxidative phosphorylation as their main source to derive energy and the lactate produced by the astrocytes is consumed. To maintain the high level of ATP, there is the need of maintenance of high copy number of the mitochondrial DNA without any errors (Belanger et al. 2011; Federico et al. 2012; Fasullo and Endres 2015). Other than acting as a source of energy source, also as co-factors and building blocks of nucleic acids, nucleotides and bases also have a significant role in maintaining cell physiology and signalling (Abbaracchio et al. 2009; Fasullo and Endres 2015). cAMP is important for the regulation and development of neuronal connectivity. ATP is also an important co- stimulator motor- sensory-motor, hypothalamus, parasympathetic and sympathetic nerves and also has neuroprotective functions and helps in regeneration of neurons from stem cells (Averaimo et al. 2014; Fasullo and Endres 2015). Guanosine is also known to modulate glutamatergic neurotransmission by facilitating the uptake of L-glutamate by glial cells (Fasullo et al. 2015). Since our cellular system constantly requires dNTPs hence, it becomes imperative that it is both generated as well as destroyed. Sometimes the dNTPs can also be formed from the freely circulating bases, and for this reason, there is a greater dependence on the salvage pathway than the de novo pathway for the generation of the purines and pyrimidines. In de novo pathway the constituents for purine synthesis are- phosphoribosyl pyrophosphate (PRPP), glycine and glutamine and for pyrimidine synthesis constituents include carbamoyl phosphate, aspartate and PRPP. Main regulatory enzyme for purine synthesis is glutamine PRPP amidotransferase which can be inhibited feedback mechanism upon the synthesis of ribonucleotide monophosphates (Fu et al. 2014; Fasullo and Endres 2015). Pyrimidine de novo pathway is regulated at the step of carbamoyl phosphate synthetase (Nelson et al. 2008). In neural tissues, salvage pathway is one of the major pathways for the generation of purines and pyrimidines. The purine bases in free form like hypoxanthine and guanine can be salvaged by hypoxanthine–guanine phosphoribosyl transferase (HGPRT). HGPRT phosphoribosyltransferase is located in the nervous system and is found in high amounts in the brain. DNA repair and nucleotide salvage mechanisms are of particular importance in neuroprotection. It can be concluded that nucleotide imbalance might often lead to wide varieties of neurodegenerative diseases; as a result, there are multiple changes in neural physiology. Diseases like Lesch–Nyhan syndrome and Mitochondrial depletion syndrome both show the importance of salvage of guanine hence illustrates the importance of guanine nucleotides in maintaining physiology and integrity of the nervous system. Recent studies on various uracil derivatives have been shown to cause inhibition of the enzymes acetylcholinesterase (AchE) and butyrylcholinesterase (BuChE) which are often targeted for the treatment of Alzheimer’s disease. Studies have also shown that that dCTP are actually deficient in certain neurodegeneration cases and hence can be a viable factor for neurodegenerative diseases (Gonzalez-Vioque et al. 2011; Fasullo and Endres 2015).
Effect of amyloids on physiology
Effect on cells
It is well reported in various studies that the intermediate stages in amyloid formation, the oligomers have cytotoxic effects on cells. However, there is lack of consensus among the researches as to what exactly is the underlying mechanism (Gharibayan et al. 2007; Sayed et al. 2015). Another school of thought that also exists is that the precursor stages to the formation of the amyloids are actually cytotoxic which later manifests to the point of amyloidosis although the actual aggregates are inert in nature (Gharibayan et al. 2007; Yang et al. 2014). Several works conclude that the soluble oligomeric intermediates are the toxic species which are responsible for the cytotoxic effects (Glabe and Kyed 2006; Yang et al. 2014). Amyloid fibrils are already known to disrupt various ion channels in the cell membrane, specially the Calcium ion channels (Glabe and Kayed 2006). Also, another possible mechanism is that the oligomers interact directly with the cell membrane surface and tissue surface virtue of their exposed hydrophobic surfaces which cause the cytotoxic effects (Pepys 2006; Ghabriyan et al. 2007; Yang et al. 2014). Cellular damages due to oligomers also happen due to interaction of the oligomers interacting with various cellular receptors (Yang et al. 2014). It is also believed that if the oligomers get too large, it can disrupt the plasma membrane by further interactions (Lee et al. 2012; Yang et al. 2014). The ultimate lack of consensus in the exact mechanisms underlying the various effects of the oligomers on the cells is mainly because of the high amount of polymorphism among the beta sheet oligomers (Ghabriyan et al. 2007; Yang et al. 2014). The cytotoxic pathways of the amyloidogenic oligomers have pretty general pathways of exerting their effect although there are also some specific pathways reported for some proteins. The most cytotoxic effect is seen by the soluble oligomeric state which precedes the actual amyloidogenic state. They are so because of their solubility properties which can easily penetrate the cell membrane which causes the influx of calcium ions from the surrounding endoplasmic reticulum ultimately causing the apoptosis of the cells (Glabe and Kayed 2006; Hamley 2012; Yang et al. 2014). Other than the solubilising effect of the oligomers, they also cause thinning of membranes, a carpeting effect and detergent effect, where the amphipathic amyloid fibrils interact with the phospholipid bilayer membrane resulting in leakage across membranes (Yang et al. 2014). Since the concentration of the naturally forming amyloid β peptide within the cells is at a much lower concentration than what is required for the formation of the amyloids, it is believed that the preceding soluble oligomeric states interact with certain portions within the cell membrane which accelerates the formation of amyloid fibrils in vivo. Hence the formation of amyloid fibrils within the cells proves that the oligomeric states do interact with certain specific regions within the cells as a step towards the formation of amyloid fibrils in vivo (Yang et al. 2014). Lastly, amyloid fibrils have been known to cause cell death via both necrotic and apoptotic pathways, but it is difficult to differentiate between the two as both the necrotic and apoptotic pathway markers are present in the affected cells (Gharibayan et al. 2007; Yang et al. 2014). Under specific interactions that the amyloidogenic proteins show, primary examples are Aβ peptide which interacts with microglial receptors causing inflammation. Another example is the transthyretin protein and its amyloid fibrils interacting with the receptors for advanced glycation end products (RAGE) causing receptors to undergo oxidative stress and inflammation and other cytotoxic effects (Sousa et al. 2001; Butterfield et al. 2001; Yang et al. 2014).
Effect on tissues and organs
Amyloidosis is the condition of deposition of amyloids in various organs within the body. Cellular death due to deposition of amyloids or due to their other oligomeric states often affects the organs and tissues at large, which ultimately leads to the death of the tissues and organ failure at large. There are also reports suggesting that the actual death of the tissues and organs have taken place before the actual formation of the amyloids at large, suggesting the role of the soluble oligomers and protofilaments showing cytotoxic effects. Hence it also becomes important to differentiate between amyloidosis conditions that are actually due to the amyloidogenic deposits or due to the oligomeric action of oligomers and protofilaments of certain species of protein (Dember 2006; Yang et al. 2014). The effect of amyloid-beta fibril is more pronounced in organs such as heart, where large deposit amyloid fibrils can cause significant changes in the gross physiological and morphological properties of the heart which can often lead to cardiac dysfunction. The major underlying mechanism is because of the loss of elastic property of the heart muscles, which reduces overall ventricular filling causing heart failure. Also, the infiltration of the amyloid fibrils causes necrosis of the cells and tissues of the heart (Roberts and Waller 1983; Pepys 2006; Yang et al. 2014).
Current trends in Amyloid disease treatment and research into new therapeutics
There are many approaches for the treatment of amyloid depositing diseases. This review shall include a brief of the current therapies for various amyloid-forming diseases and strategies for development of new drugs for the treatment of diseases due to amyloidogenic diseases.
There have been extensive researches which have focused majorly on identifying the underlying mechanisms of Aβ neurotoxicity which as a result have yielded identification and association of various pathways. Among the various pathways involved most of them have identified axonal degeneration and oxidative stress (Butterfield et al. 2013; Salvadore et al. 2017), mitochondrial dysfunction (Kerr et al. 2017; Salvadore et al. 2017) and abnormal calcium signalling (Salvadore et al. 2017). Till recent times drug development strategies against AD have focused on the amyloid hypothesis and most of the randomized clinical trials have been designed to target this protein (Salvadore et al. 2017). The outcomes of the clinical trials have had an overall failure rate of 99.6%, and a major reason for this has been the late diagnosis of the disease, where there are extensive pathology and neurodegeneration (Cummings et al. 2014; Salvadore et al. 2017). Now that it has been identified that axonal degeneration is one of the early signs of AD disease progression, understanding the mechanisms which trigger this phenomenon can be an interesting point for future drug development (Salvadore et al. 2017). Current researches have revealed that actual accumulation of Aβ peptide starts occurring 15 years before the onset of the disease which beckons future research to be focused on the development of methods to identify the disease at early stages (Grimaldi et al. 2018). Current prognosis of the disease is based on the cognitive assessments of the patients who present significant cognitive and behavioural changes. Computed Tomography scan (CT) and Magnetic Resonance Imaging (MRI) scan are used for initial diagnosis of cases in patients with cognitive AD signs (McKhann et al. 2011; Dubois et al. 2014; Grimaldi et al. 2018). Also less frequently used is Positron Emission Tomography (PET) to visualise the amyloid plaques mainly in cases, where confirmation is needed. Since the PET scan comes in costly, it cannot be used for widespread diagnosis of the disease. Besides the PET scanning of amyloid images, some other pathophysiological markers inclusive of cerebrospinal fluid tests looking for the presence of Aβ1-42, total tau, ptau et cetera have also been used for diagnosis. It is shown that lower levels of Aβ1-42 in CSF along with higher levels of ptau are more strongly correlated with the AD disease progression (Johnson et al. 2012; Frisoni et al. 2017; Grimaldi et al. 2018). It is noteworthy that therapeutically, clinical trials are hugely hindered, partly because of the lack in availability of biomarkers and reliable methods to monitor and understand the proper development and progress of the disease. In this regard, the currently available biomarkers are expensive and invasive and have majorly focused on brain and CSF (Grimaldi et al. 2018). A recent study by Grimaldi et al. has concluded that retina is used as a model for the brain as the pathological changes associated with the brain during AD can be seen in the retina, and hence newer therapeutic strategies could be developed keeping the retina in mind. The research has successfully identified retinal biomarkers which could be used for understanding the progress of AD and hence anticipates future treatments. Grimaldi and colleagues identified the presence of amyloid plaques, tau tangles, neurodegeneration and astrogliosis at the pre-symptomatic stage in the retinal ganglion cell layer in mice models. They also report that retinal microglia at the pre-symptomatic stage were anti-inflammatory which had changed its phenotype to pro-inflammatory during disease progression and hence becoming neurotoxic (Grimaldi et al. 2018).
Role of A1 and A2A receptors in neurodegenerative diseases
At present neurological pharmacology mainly deals with symptomatic interventions and trying to lower the symptomatic effects on the patients. There is a lack of drug development which can inhibit disease from happening. In recent times the ribonucleoside adenosine has come into light with its neuromodulatory activity in case of neurodegenerative diseases. Research also suggests that adenosine receptors play an important role in the modulation of cognitive function (Rahman 2009). Adenosine is a very common nucleoside which is found in all cells including glia and neurons and plays a role in synaptic transmission, neuronal excitability in the CNS. It generally exerts its effect by four (A1, A2A, A2B and A3) G protein-coupled receptors ultimately regulating vital processes such as sleep and arousal, cognition, memory, neurodegeneration et cetera. The powerful pharmacological effect of adenosine is mediated by A1 and A2A receptors. There have been studies which have investigated the expression of A1, and A2A receptors and studies have been done on developing antagonists for these receptors experiments have shown positive results in elevating cognitive effects by down regulation of A1 and A2A receptors (Cunha 2001; de Mendonca and Ribeiro 2001; Angulo et al. 2003; Rahman 2009). Pieces of evidence from post mortem analysis of AD patients also suggest that adenosine receptors change their pattern of localization and density from various regions in the brain in case of normal and diseased brains (Albasanz et al. 2008).It is important to note that both A1 and A2R receptors and caffeine can all regulate the brain metabolism although it is unclear whether there is any relation between the modulatory effects of adenosine and the effects of the receptors and caffeine on the brain tissue. In general, adenosine has a homeostatic role in the majority of eukaryotic cells (Joo et al. 2007; Peart et al. 2007; Wendler et al. 2007; Gomes et al. 2011). Observations suggest that extracellular levels of adenosine are modified in conditions of brain damage. This is probably due to the increased requirements of ATP, which is required for maintenance of cell viability which leads to disproportionally higher levels of adenosine (Gomes et al. 2011). The sources and clarification of extracellular adenosine is though unclear. Although the literature suggests that obnoxious stimuli in brain causes an increase in extracellular levels of adenosine (Cunha 2001; Fredholm et al. 2005; Gomes et al. 2011).
Challenges in the development of neuroprotective drugs
At various stages of translational research, scientific reports fail to differentiate between the exact mechanisms of the drug. A drug could be working upon the improvement of symptoms, or it could help reduce the side effects of the disease. Any drug that does either of the work may be considered as neuroprotective. When a drug goes for clinical trials, the promoter has to decide the exact mechanism of the drug for measuring the trial’s outcome. There are several reasons why clinical trials for neurodegenerative drugs take improvement of symptoms as the major parameter for a successful clinical trial (Franco and Navarro 2018). Since for ethical reasons, patients continue taking the already prescribed medicine; it becomes difficult to understand the effect of any new drug used via clinical trials. In cases of Parkinson’s clinical trial, patients continue taking levodopa, and to understand the effect of any new drugs, there should be long terms effects of the new drug which must be visible even after leaving the use of levodopa. There are lacking consensual rules for proper evaluation and approval of drugs which can function via prevention of neuronal death; in other words, drugs which modify the progression of neurodegenerative disease. The current scientific community is also fast working towards developing reliable biomarkers which could be used to measure the extent and progress of neurodegeneration in humans in vivo via Positron emission tomography. Regulatory bodies should also look for the approval of safe drugs to be used in large population for cohort studies on long term effects on neuroprotection with a general thumb rule that the drug should delay clinical symptoms or disease progression (Franco and Navarro 2018).
Drug discovery strategies used against amyloidosis
According to population studies, it is estimated that by 2050 one in every eighty, five people will be suffering from AD. Eightfold of this probability do end up showing early signs of AD and dementia and are at risk of progressing to manifest off the disease. Disease-modifying therapies (DMT) which will prevent the onset or delay the progression of the disease are urgently needed. A 1-year delay in onset of the disease by 2020 will reduce the number of affected patients by 9.2 million people worldwide by 2050 (Brookmeyer et al. 2007, 2017; Cummings et al. 2018). Multiple therapeutic strategies are in place to identify disease ameliorating compounds and their effects on the diseased state. Natural compound based identification includes ethnobotanical knowledge from earlier days which had effects of improving cognition and memory. Reports are suggesting that the Mediterranean diet which is rich in vegetables, fruits, cereals and olive oil have reduced risk for development of Alzheimer’s in cohort studies carried out in small population from New York. Diets containing a high rate of flavonoids and polyphenolic compounds, consumption of red wine which contains resveratrol also have a protective effect against the development of dementia and associated conditions. Curcumin, a compound found in the turmeric spice which is commonly used in Southeast Asian diets along with Epigallocatechin-3-gallate (EGCG), myricetin and certain polyphenolic compounds found in green tea are associated with lowering cognitive disabilities (Luchsinger and Mayeux 2004; Arntzen et al. 2010; Eisele et al. 2015; Yamada et al. 2015).
Transthyretin Amyloidosis is caused by Transthyretin a homotetrameric protein which is mainly involved in the transport of Vitamin A and thyroxine in its wild type form, and it is the wild type form which is associated wild type transthyretin amyloidosis (wtTTR) which was formerly known as senile systematic amyloidosis. The conversion of normal transthyretin into amyloidogenic form requires the dissociation of the homotetramer into individual monomers, specific conformational changes and assembly of fibrils. The propensity to form the amyloids is majorly influenced by specific amino acid substitutions and micro environmental factors such as pH and oxidative stress (Sayed et al. 2015).
Systematic AA amyloidosis results in organ dysfunction because of extracellular deposition of N-terminal fragments of SAA in the form of amyloid fibrils insoluble in nature. Glycosaminoglycans like heparin sulphate are known to act as promoters of amyloid fibrils formation in its early phase by acting as chaperones. Eprodisate is negatively charged sulphonated molecule which is structurally analogous to heparin sulphate and hence undergoes competitive binding inhibition between SAA and glycosaminoglycans. It has reduced AA amyloid development in experimental mouse models (Dember et al. 2007; Sayed et al. 2015).
Inhibitors of amyloid fibril formation
One of the current strategies for development of drugs against amyloidosis includes identification of lead compounds which work against (1) toxic amyloid formation or stabilisation of the native form and prevention of the aggregation, (2) remodelling or degradation of already formed insoluble oligomers or fibrils.
There have been multiple approaches to develop inhibitors at various stages of amyloid fibril formation. One of the major attractive potential therapeutic approaches was, paving the way for the development of metal ion chelators, especially iron and copper. They have been shown to play an important role in the amyloid β formation and has now been investigated as a treatment option to a great extent (Regland et al. 2001; Hardy and Selkoe 2002; Hamley 2012; Yang et al. 2014). There are various approaches which have been used including those of in vitro cell-based approaches or platforms which are used widely to screen various small molecules that inhibit the formation of fibril focusing on overall effect rather than focussing on one particular mechanism (Kim et al. 2006; Chen et al. 2010; Velander et al. 2017).
Tafamidis
Tafamidis is a drug that is orally administered, which preferentially bind to the homotetrameric form of transthyretin and stabilises it and does not carry the risk of non-steroidal anti-inflammatory drugs use. The most common side effects included urinary tract infections (UTI) and diarrhoea. There have been no reports of Tafamidis, causing any thyroid-related hormonal problems (Sayed et al. 2015). Tafamidis is known to bind to the thyroxine-binding site of the Transthyretin tetramer and inhibits its dissociation into monomers, thus blocking the rate-limiting step in Transthyretin amyloidosis (Castano and Maurer 2020).
Oligonucleotide based therapies
Oligonucleotide based therapies which include small interfering RNAs (siRNA) and antisense RNAs can cause changes at the translational level without integrating themselves within the genome. Antisense oligonucleotides are small 13–25 nucleotides long single-stranded DNA molecules that bind to mRNAs and degrade them hence affecting the transcription of the same. Oligonucleotide therapies for transthyretin inclusive of that of siRNA and antisense oligonucleotides are already in clinical trials and are being regularly investigated in clinical studies (Sayed et al. 2015).
Peptide-based Amyloid inhibition strategies
Until a major part of the twentieth century, peptides were usually not seen as good candidates belonging to lead like molecules as compared to other small molecules. Major weaknesses were attributed to proteolytic degradation, low bioavailability, high clearance rates and poor physical or chemical instability, though they are particularly suitable as drugs because of their high specificity, selectivity and potency (Henninot et al. 2018; Armiento et al. 2019). Peptides are an attractive alternative to small molecules as anti-amyloid drugs. Small molecules lack large surfaces which are required for proper interaction between an amyloidogenic region of a protein and the anti-amyloid molecule. Other than this, antibodies which are also another alternative have very high costs of production owing to its purity and sensitivity, might have a low cell membrane and/blood–brain barrier permeability. Keeping all these factors in mind, developing small peptides can be a better alternative which can combine several factors to its advantage like low cost of production, better selectivity, lesser immunogenicity and so on (Craik et al. 2013; Armiento et al. 2019). Current strategies for development of peptide-based amyloid inhibitors are mainly based on (a) recognition of amyloid self-assembly core regions, (b) cross- amyloid interactions, (c) interactions with chaperones and other non-amyloidogenic peptides and the fourth strategy includes designing inhibitors using combinatorial libraries optimized using peptide chemistry tools (Armiento et al. 2019).
Natural product-based amyloid inhibitors
Majority of the polyphenol based compounds identified till date is chiefly associated reducing the effects of the late and progressive disease pathology. Major examples include oleuropein and oleocanthal found in olive oil, resveratrol found commonly in fruits and red wine, curcumin found in turmeric, EGCG and catechin found in green tea, rosamarinic acid found in certain culinary herbs.
Epigallocatechin-3-gallate
Recent in vitro studies suggest that concentrations in the range of micromole/litre which is commonly found in green tea efficiently inhibits fibril formation in amyloid formation related to β-protein, α-synuclein and TTR converting existing fibrils into non-fibril conformers. Transthyretin tetramerization is observed when epigallocatechin-3-gallate binds to wild type and variant TTRs at three different sites other than the thyroxine-binding sites (Sayed et al. 2015). EGCG has progressed to clinical trials for the treatment of Alzheimer’s. Also, the multiple effects on the inflammatory pathway, anti-oxidant activity and preventing metal-chelating pathways, polyphenols is a class of compound that often exhibits the structural backbone for the rational drug design process for the development of anti-amyloid compounds (Velander et al. 2017).
Taxifolin
In a recent study by Mahdvamhier et al. (2017), it was reported that taxifolin which is a novel ubiquitous compound, commonly found in natural herbs and foods had been shown to bind to the amyloid fibrils and reduce their related toxicity. Taxifolin binds to amyloid and causes it to form a very large globular chain like fibrils. Various amyloid specific indicators were used to track the entire process. Taxifolin was seen to bind to prefibrillar species of amyloids rather than stabilising its native state. It was also stated that that binding of taxifolin diverts the amyloid-forming pathway from the beta-sheet aggregates to a more globular chain which has much lower beta-sheet content as well as much lower exposed hydrophobic patches. Taxifolin binding capacity is much reduced when the aggregates are formed at large by the end of the growth phase. ThT fluorescence monitored this specific test. These results do indicate that natural products like these could be used as amyloid fibril inhibitors (Mahdaviemhier et al. 2017).
Curcumin
Curcumin, have been reported in multiple cases to modulate various amyloid-forming activities in various protein systems. Curcumin has been identified as a pan- assay interference compound implying that it tends to give false-positive results in high throughput screening and hence there is a requirement of multiple validations by various orthogonal assays. Nevertheless, curcumin has been reported in a wide source of literature to have anti-amyloidogenic activity by inhibiting the formation of amyloids, amyloid induced cytotoxicity and providing beneficial assistance and effects against amyloid structures and reducing the plaque burden in vivo. Possible mechanisms include (1) inhibition of amyloid aggregation in proteins like amylin and amyloid β and (2) acceleration of α- synuclein aggregation activity to skip the slow pre fibrillar forming stages hence skipping the more cytotoxic intermediates. Computational studies including curcumin has helped us understand that it makes new non-covalent interactions with the typical cross beta spinal structure called the steric zipper within amyloid fibrils (Ono et al. 2004; Yang et al. 2005; Guerro-Munoz et al. 2015; Jha et al. 2016; Nedumpully-Goovindan et al. 2016; Nelson et al. 2017; Valender et al. 2017). A study by Sarkar et al. a molten globule like intermediate was identified in 2,5-diketo-d-gluconate reductase A (DKGR) at pH 2.5 having a considerable β-sheet structure. The molten globule state amyloidogenic property at protein concentrations greater than 50 µM. A 1:1 molar ratio of the protein along with curcumin prevents amyloid formation based on results of Thioflavin T assay and AFM imaging (Sarkar et al. 2009). The computational studies also helped us realize curcumin is basically intercalated between hydrophobic amino acid residues in the beta-sheets irrespective of the amyloid structure and its parent protein. This study also helped in understanding that curcumin would also be able to bind to the alpha amyloid structures formed during the earliest stage of the generation of amyloids (Knight et al. 2006; Abedini et al. 2009).
Non- catechol
Non-catechol derived compounds- In recent studies which included cinnamomum tree extract showed anti-amyloidogenic and anti-fibril formation activity in tau and hen egg-white lysozyme proteins. It was also reported that cinnamaldehyde, the major constituent cinnamon bark oil acts against Tau 187 amyloid-forming without affecting the tau- associated microtubule assembly (Peterson et al. 2009; George et al. 2013; Ramshini et al. 2015; Velander et al. 2017).
Oleocanthal
Derived from olive oil also prevents tau amyloid formation by preventing its change in conformation to beta sheet secondary structure in a dose dependent manner. Further studies with the help of Mass spectrometry had revealed an adduct formation full length with the lysine residue in a small hexa peptide sequence via formation of Schiff’s base in the tau protein. The complete mechanism was not been able to deciphered because of the lack of clarity in the exact way oleocanthal binds with the lysine residue in tau protein and also due to high signal:noise ratio in the MS reports (Li et al. 2009; Velander et al. 2017).
Recent studies also showed another Nordihydroguaiaretic (NDGA), resveratrol and myricetin were successful in remodelling the preformed amyloid oligomers and fibrils towards SDS stable aggregates along with exhibiting lowered cytotoxicity compared to controls which contained untreated preformed oligomers. Other assays also revealed various other naturally derived polyphenol derivatives like rufigallol, trihydroxybenzophenone and exifone as well as porphyrin and hematin were shown to inhibit the amyloid formation by tau protein indicated by the presence of a higher amount of soluble tau protein fraction compared to the insoluble portion between treated and untreated samples. These results were also validated by orthogonal TEM and ThT assays (Taniguchi et al. 2005; Velander et al. 2017).
Challenges and future perspectives
One of the significant issues with natural inhibitors which have already been tested so far have moderate inhibition potential. Various validation studies such as synthesis of structural analogue and structural and functional relationship are often required based on medicinal chemistry studies. Another common issue is also their solubility and bioavailability in the human system (Valender et al. 2017). There is a significant number of animal studies which are based on similar physicochemical properties and generally covers the majority of polyphenolic compounds and flavonoids. Only a few animal studies are present which have tested compounds which are structurally different. We realise that a limited amount of structural diversity among various compounds will be a limiting factor in the discovery of new inhibitors for neurodegenerative diseases. To identify a large number of compounds, large scale platforms for virtual screening are required, which needs a large platform. Some work has already been done by Lee et al. when they had implemented a quantitative high throughput screening method for screening of a large number of compounds for the discovery of inhibitors for tau assembly. Valender et al. had also developed a semi high throughput fluorescent 384 well plate which can be readily expanded up to 1596 well plate mainly to screen inhibitors for various amyloidogenic proteins.
We are yet to fully discover the exact mechanistic pathways by which various amyloidogenic proteins become amyloidogenic in the first place. Apart from the various biochemical factors for a protein which triggers the process development of amyloidosis, there are certain factors which are yet to be discovered or understood. Currently, there is a significant gap in knowledge in our understanding of the exact mechanism of an inhibitor in preventing the formation of amyloids or how do they decrease the formation of preformed amyloids. There is also a lack of proper biophysical characterization of the state, where the inhibitor and the protein is interacting and how they are interacting. Advanced techniques related to mass spectrometry might help us understand the effect of inhibitors on the various post-translational changes associated with proteins in the amyloidogenic state. They will also lead us to understand what sort of covalent and non-covalent interactions are majorly responsible for the interaction and inhibition of amyloids in vivo. This knowledge will ultimately help us for shifting our strategy towards more structure-based drug design. Due to lack of knowledge in development of new drugs by factors involving money, time involved in clinical trials, and dearth of knowledge in the subject, it is of prime importance to focus our strategies on natural inhibitors despite its shortcoming along with the idea of drug re-prescribing which implies the use of already approved FDA drugs available in the market for nervous system diseases so as to reduce the cost and the time investment on their pharmacological studies involving ADMET and various other assays.
References
Abedini A, Raleigh DP (2009) A critical assessment of the role of helical intermediates in amyloid formation by natively unfolded proteins and polypeptides. Protein Eng Des Sel. 22(8):453–459
Agbas A (2018) Trends in protein aggregation in neurodegenerative diseases. Open Access Peer Rev. https://doi.org/10.5772/intechopen.81224
Albasanz JL, Perez S, Barrachina M, Ferrer I, Martín M (2008) Up-regulation of adenosine receptors in the frontal cortex in Alzheimer's disease. Brain Pathol 18:211–219
Angulo E, Casado V, Mallol J, Canela EI, Vinals F, Ferrer I, Lluis C, Franco R (2003) A1 adenosine receptors accumulate in neurogenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol 13:440–451
Armiento V, Spanopoulou A, Kapurniotu A (2019) Peptide-based molecular strategies to interfere with protein misfolding, aggregation, and cell degeneration. Angew Chem Int Ed 2019(58):2–15
Arntzen KA, Schirmer H, Wilsgaard T, Mathiesen EB (2010) Moderate wine consumption is associated with better cognitive test results: a 7 year follow up of 5033 subjects in the Tromso Study. Acta NeurolScand Suppl 190:23–29
Ashraf G, Greig NH, Khan AT, Hassan I, Tabrez S, Shakil S, Sheikh AI, Zaidi KS, Wali AM, Jabi RM, Firz KC, Naeem A, Alhazza MI, Damanhouri AG, Kamal AM (2014) Protein misfolding and aggregation in Alzheimer’s disease and type 2 diabetes mellitus. CNS NeurolDisord Drug Targets 13(7):1280–1293
Auld DS, Kornecook TJ, Bastianetto S, Quirion R (2003) Prog Neurobiol 3:209–245
Averaimo S, Nicol X (2014) Intermingled cAMP, cGMP and calcium spatiotemporal dynamics in developing neuronal circuits. Front Cell Neurosci 8:376
Bateman DA, Tycko R, Wickner RB (2011) Experimentally derived structural constraints for amyloid fibrils of wild-type transthyretin. Biophys J 101:2485–2492
Bélanger M, Allaman I, Magistretti PJ (2011) Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14:724–738
Berson JF, Theos AC, Harper DC, Tenza D, Raposo G, Marks MS (2003) Proprotein con-vertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J Cell Biol 161:521–533
Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM (2007) Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement 3:186–191
Brookmeyer R, Abdalla N, Kawas CH, Corrada MM (2017) Forecasting the prevalence of preclinical and clinical Alzheimer’s disease in the United States. Alzheimers Dement 14:121–129
Butterfield DA, Drake J, Pocernich C, Castegna A (2001) Evidence of oxidative damage in Alzheimer’sdisease brain: central role for amyloid b-peptide. Trends Mol Med 7:548–554
Butterfield DA, Swomley AM, Sultana R (2013) Amyloid b- peptide (1–42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid Redox Signal 19:823–835
Calamai M, Chiti F, Dobson MC (2005) Amyloid fibril formation can proceed from different conformations of a partially unfolded protein. Biophys J 89:4201–4210
Carrillo-Mora P, Luna R, Barneque-Colin L (2014) Amyloid beta: multiple mechanisms of toxicity and only some protective effects? Oxid Med Cell Longev 2014:795375
Castano A, Maurer SM (2020) Cardiac amyloidosis-heart failure: companion to Braunwald's heart disease (Fourth edition). 301–310.e3.
Chabes A, Bilyana G, Domkin V, Zhao X, Rothstein R, Thelander L (2003) Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 112:391–401
Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J (2002) Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295:851–855
Chen J, Armstrong AH, Koehler AN, Hecht MH (2010) Small molecule microarrays enable the discovery of compounds that bind the Alzheimer’s Abeta peptide and reduce its cytotoxicity. J Am Chem Soc 132(47):17015–17022
Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75:333–366
Chiti F, Dobson CM (2017) Protein misfolding, amyloid formation and human disease: a summary of progress over the last decade. Annu Rev Biochem 86:351–354
Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, Dobson CM (1999) Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc Natl Acad Sci USA 96:3590–3594
Chiti F, Taddei N, Baroni F, Capanni C, Stefani M et al (2002) Kinetic partitioning of protein folding and aggregation. Nat Struct Biol 9:137–143
Colletier JP, Laganowsky A, Landau M, Zhao M, Soriaga AB, Goldschmidt L, Flot D, Cascio D, Sawaya MR, Eisenberg D (2011) Molecular basis for amyloid-b polymorphism. 158. Proc Natl Acad Sci 108:16938–16943
Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I (2016) Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J Am Chem Soc 138:9663–9674
Craik DJ, Fairlie DP, Liras S, Price D (2013) The future of peptide-based drugs. Chem Biol Drug Des 81(1):136–147
Cummings JL, Morstorf T, Zhong K (2014) Alzheimer’s disease drug development pipeline: few candidates, frequent failures. Alzheimers Res Ther 6:37–43
Cummins J, Lee G, Ritter A, Zhong K (2018) Alzheimer’s disease drug development pipeline. Alzheimer’s Dementia 4:195–214
Cunha RA (2001) Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: different roles, different sources and different receptors. Neurochem Int 38:107–125
Daebel V, Chinnathambi S, Biernat J, Schwalbe M, Habenstein B (2012) β-Sheet core of tau paired helical filaments revealed by solid-state NMR. J Am Chem Soc 134:13982–13989
De Toma A, Salamekh S, Ramamoorthy A, Lim HM (2012) Misfolded proteins in Alzheimer’s disease and type II diabetes. Chem Soc Rev 41(2):608–621
Dember LM (2006) Amyloidosis - associated kidney disease. J Am Soc Nephrol 17:3458–3471
Dember LM et al (2007) Eprodisate for the treatment of renal disease in AA amyloidosis. N Engl J Med 356(23):2349–23260
Dobson CM (1999) Protein misfolding, evolution and disease. Trends Biochem Sci 24:329–332
Dobson CM (2003) Protein folding and misfolding. Nature 426:884–890
Dobson CM (2004) Principles of protein folding misfolding and aggregates. Semin Cell Biol 15(1):3–16
Dubey VK, Lee J, Blaber M (2005) Redesigning symmetry-related “mini-core” regions of FGF-1 to increase primary structure symmetry: thermodynamic and functional consequences of structural symmetry. Prot Sci 14:2315–2323
Dubois B (2014) Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol 13:614–629
Eisele YS, Monteiro C, Fearns C, Encalada SE, Wiseman RL, Powers ET (2015) Targeting protein aggregation for the treatment of degenerative diseases. Nat Rev Drug Discov 14(11):759–780
Eisenberg D, Jucker M (2012) The amyloid state of proteins in human diseases. Cell 148:1188–1203
Farina M, Avila DS, da Rocha JB, Aschner M (2013) Metals, oxidative stress and neurodegeneration. Neurochem Int 62(5):575–594
Fasullo M, Endres L (2015) Nucleotide salvage deficiencies, DNA damage and neurodegeneration. Int J Mol Sci 16:9431–9449
Federico A, Cardaioli E, da Pozzo P, Formichi P, Gallus GN, Radi E (2012) Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci 322:254–262
Finch CE, Morgan TE (2007) Systemic inflammation, infection, ApoE alleles, and Alzheimer disease: a position paper. Curr Alzheimer Res 4:185–189
Fink AL, Uversky VN (2004) Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim Biophys Acta 1698:131–153
Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SHW (2017) Cryo-EM structures of tau filaments from Alzheimer's disease. Nature 547:185–190
Forman MS, Trojanowski JQ, Lee VM (2004) Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. 2004. Nat Med 10:1055–1063
Fowler DM, Koulov AV, Balch WE, Kelly JW (2007) Functional amyloid from bacteria to humans. Trends Biochem Sci 32:217–224
Franco R, Navarro G (2018) Adenosine A2A receptor antagonists in neurodegenerative diseases: huge potential and huge challenges. Front Psychiatry 9:68
Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM (2005) Adenosine and brain function Int. Rev Neurobiol 63:191–270
Frisoni GB (2017) Strategic roadmap for an early diagnosis of Alzheimer’s disease based on biomarkers. Lancet Neurol 16:661–676
Fu R, Ceballos-Picot I, Torres RJ, Larovere LE, Yamada Y, Nguyen KV, Hegde M, Visser JE, Schretlen DJ, Nyhan WL (2014) Lesch-Nyhan disease international study group. Brain. Genotype–phenotype correlations in neurogenetics: Lesch-Nyhan disease as a model disorder. Brain 137:1282–1303
George RC, Lew J, Graves DJ (2013) Interaction of cinnamaldehyde and epicatechin with tau: implications of beneficial effects in modulating Alzheimer’s disease pathogenesis. J Alzheimer’s Dis 36(1):21–40
Gharibyan AL, Zamotin V, Yanamandra K, Moskaleva OS, Margulis BA, Kostanyan IA, Morozova-Roche LA (2007) Lysozyme amyloid oligomers and fibrils induce cellular death via different apoptotic/necrotic pathways. J Mol Biol 365:1337–1349
Glabe CG, Kayed R (2006) Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neuorology 66:74–S78
Goldberg AL (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426(6968):895–899
Gomes C, Kaster M, Tome A, Agastinho P, Cunha R (2011) Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochim Biophys Acta 1808:1380–1399
González-Vioque E, Torres-Torronteras J, Andreu AL, Martí R (2011) Limited dCTP availability accounts for mitochondrial DNA depletion in mitochondrial neurogastrointestinalencephalomyopathy (MNGIE). PLoS Genet 7:e1002035
Griffin WS, Mrak RE (2002) Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer's disease. J Leukocyte Biol 72(2):233–238
Grimaldi A, Brighi C, Peruzzi G, Rogozzino D, Bonanni V, Limatola C, Ruocco G, Angelantonio GS (2018) Inflammation, neurodegeneration and protein aggregation in the retina as ocular biomarkers for Alzheimer’s disease in the 3xTg-AD mouse model. Cell Death Disease 9:685
Guerrero-Munoz MJ, Gerson J, Castillo-Carranza DL (2015) Tau oligomers: the toxic player at synapses in Alzheimer’s disease. Front Cell Neurosci 9:464
Hamley IW (2012) The amyloid beta peptide: a chemist’s perspective. Role in Alzheimer’s and fibrillization. Chem Rev 112:5147–5192
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356
Helmus JJ, Surewicz K, Apostol MI, Surewicz WK, Jaroniec CP (2011) Intermolecular alignment in Y145Stop human prion protein amyloid fibrils probed by solid-state NMR spectroscopy. J Am Chem Soc 133:13934–13937
Henninot A, Collins JC, Nuss MJ (2018) The current state of peptide drug discovery: back to the future? J Med Chem 2018(61):1382–1414
Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM, Griffin RG (2004) High-resolution molecular structure of a peptide in an amyloid fibril determined by magic angle spinning NMR spectroscopy. Proc Natl Acad Sci USA 101:711–716
Jha NN, Ghosh D, Das S, Anoop A, Jacob RS, Singh PK (2016) Effect of curcumin analogs on alpha-synuclein aggregation and cytotoxicity. Sci Rep 6:28511
Johnson KA, Fox NC, Sperling RA, Klunk WE (2012) Brain imaging in Alzheimer disease. Cold Spring Harb Perspect Med 2:a006213
Jomova K, Vondrakova D, Lawson M, Valko M (2010) Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem 345(12):91–104
Joo JD, Kim M, Horst P, Kim J, D'Agati V, Emala CW, Lee HT (2007) Acute and delayed renal protection against renal ischemia and reperfusion injury with A1 adenosine receptors. Am J Physiol Ren Physiol 293:F1847–F1857
Kaganovich D, Kopito R, Frydman J (2008) Misfolded proteins partition between two distinct quality control compartments. Nature 454:1088–1095
Kaufman RJ (2002) The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol 3(6):411–421
Kawahara M (2005) Effects of aluminium on nervous system and its possible link with neurodegenerative diseases. J Alzheimer's Dis 8(2):171–182
Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA (2017) Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci 40:151–166
Khan JM, Qadeer A, Chaturvedi SK, Ahmad E, Rehman SA, Gourinath S, Khan RH (2012) SDS can be utilized as an amyloid inducer: a case study on diverse proteins. PLoS ONE 7:e29694
Khan JM, Siddiqi MK, Alam P, Khan RH (2017) Comparative insight into surfactants mediated amyloidogenesis of lysozyme. Int J Biol Macromol 83:315–325
Khurana R, Coleman C, Ionescu-Zanetti C, Carter SA, Krishna V, Grover RK, Roy R, Singh S (2005) Mechanism of thioflavin T binding to amyloid fibrils. J Struct Biol 151:229–238
Kihara T, Shimohama S (2004) Alzheimer’s disease and acetylcholine receptors. Acta Neurobiol Exp (Wars) 64(1):99–105
Kim W, Kim Y, Min J, Kim DJ, Chang YT, Hecht MH (2006) A high-throughput screen for compounds that inhibit aggregation of the Alzheimer’s peptide. ACS Chem Biol 1(7):461–469
Knight JD, Hebda JA, Miranker AD (2006) Conserved and cooperative assembly of membrane-bound alpha-helical states of islet amyloid polypeptide. Biochemistry 45(31):9496–9508
Konar M, Mathew A, Dasgupta S (2019) Effect of silica nanoparticles on the amyloid fibrillation of lysozyme. ACS Omega 4:1015–1026.
Konno T (2001) Amyloid-induced aggregation and precipitation of soluble proteins: an electrostatic contribution of the Alzheimer's beta (25–35) amyloid fibril. Biochemistry 40(7):2148–2154
Knowles TPJ, Vendruscolo M, Dobson CM (2014) The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15(6):384–396
Kumar S, Ravi VK, Swaminathan R (2009) Suppression of lysozyme aggregation at alkaline pH by tri- N-acetylchitotriose.Biochem. Biophysics Acta 1794:913–920
Lee CC, Sun Y, Huang HW (2012) How type II diabetes-related islet amyloid polypeptide damages lipid bilayers. Biophys J 102:1059–1068
Li W, Sperry JB, Crowe A, Trojanowski JQ, Smith AB, Lee VM (2009) Inhibition of tau fibrillization by oleocanthal via reaction with the amino groups of tau. J Neurochem 110(4):1339–1351
Liu Z, Zhang A, Sun H, Han Y, Kong L, Wang X (2017) Two decades of new drug discovery and development for Alzheimer's disease. RSC Adv 7:6046
Luchsinger JA, Mayeux R (2004) Dietary factors and Alzheimer’s disease. Lancet Neurol 3(10):579–587
Mackay JP, Matthews JM, Winefield RD, Mackay LG, Haverkamp RG et al (2001) The hydrophobin EAS is largely unstructured in solution and functions by forming amyloid-like structures. Structure 9(2):83–91
Mahdavimehr M, Meratan AA, Ghobeh M, Ghasemi A, Saboury AA, Nemat-Gorgani M (2017) Inhibition of HEWL fibril formation by taxifolin: mechanism of action. PLoS ONE 12(11):e0187841
Maloney B, Lahiri DK (2011) The Alzheimer’s amyloid b-peptide (Ab) binds a specific DNA Ab-interacting domain (AbID) in the APP, BACE1 and APOE promoters in a sequence-specific manner: characterizing a new regulatory motif. Gene 488:1–12
McKhann GM (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7:263–269
Meli G, Lecci A, Manca A, Krako N, Albertini V, Benussi L, Ghidoni R, Cattaneo A (2014) Conformational targeting of intracellular Abeta oligomers demonstrates their pathological oligomerization inside the endoplasmic reticulum. Nat Commun 5:3867
Micheli V, Camici M, Tozzi MG, Ipata PL, Sestini S, Bertelli M, Pompucci G (2011) Neurological disorders of purine and pyrimidine metabolism. Curr Top Med Chem 11:923–947
Minton AP (2001) The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J Biol Chem 276(14):10577–10580
Muhoberac BB, Vidal R (2013) Abnormal iron homeostasis and neurodegeneration. Front Aging Neurosci 5:32
Nallamsetty S, Dubey VK, Pande M, Ambasht PK, Jagannadham MV (2007) Accumulation of partly folded states in the equilibrium unfolding of ervatamin A: spectroscopic description of the native, intermediate, and unfolded states. Biochimie 89:1416–1424
Nedumpully-Govindan P, Kakinen A, Pilkington EH, Davis TP, Ke PC, Ding F (2016) Stabilizing off-pathway oligomers by polyphenol nanoassemblies for IAPP aggregation inhibition. Sci Rep 6:19463
Nelson DL, Lehninger AL, Cox MM (2008) Lehninger principles of biochemistry, 5th edn. WH Freeman and Company, New York, pp 882–900
Nelson KM, Dahlin JL, Bisson J, Graham J, Pauli GF, Walters MA (2017) The essential medicinal chemistry of curcumin. J Med Chem 60(5):1620–1637
Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA (2006) J Neuropathol Exp Neurol 65:631–641
Ono K, Hasegawa K, Naiki H, Yamada M (2004) Curcumin has potent anti-amyloidogenic effects for Alzheimer's beta-amyloid fibrils in vitro. J Neurosci Res 75:742–750
Paravastu AK, Leapman RD, Yau WM, Tycko R (2008) Molecular structural basis for polymorphism in Alzheimer’s β-amyloid fibrils. PNAS 105:18349–18354
Pawar AP, DuBay KF, Zurdo J, Chiti F, Vendruscolo M, Dobson CM (2005) Prediction of "aggregation-prone" and "aggregation-susceptible" regions in proteins associated with neurodegenerative diseases. J Mol Biol 350:379–392
Peart JN, Headrick JP (2007) Adenosinergic cardio protection: multiple receptors, multiple pathways. Pharmacol Ther 114:208–221
Pepys MB (2006) Amyloidosis. Annu Rev Med 57:223–241
Peterson DW, George RC, Scaramozzino F, LaPointe NE, Anderson RA, Graves DJ (2009) Cinnamon extract inhibits tau aggregation associated with Alzheimer’s disease in vitro. J Alzheimer’s Dis 17(3):585–597
Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD (2002) A structural model for Alzheimer's beta -amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA 99:16742–16747
Petkova AT, Yau WM, Tycko R (2006) Experimental constraints on quaternary structure in Alzheimer’s β-amyloid fibrils. Biochemistry 45:498–512
Pocernich C, Butterfield D (2012) Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim Biophys Acta 1822(5):625–630
Popescu BF, Nichol H (2011) Mapping brain metals to evaluate therapies for neurodegenerative disease. CNS Neurosci Ther 17:256–268
Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE (2009) Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 78:959–991
Querfurth HW, Laferla FM (2010) Alzheimer’s disease. N Engl J Med 362(4):329–344
Rahman A (2009) The role of adenosine in Alzheimer’s disease. Curr Neuropharmacol 7:207–216
Ramshini H, Ebrahim-Habibi A, Aryanejad S, Rad A (2015) Effect of Cinnamomum verum extract on the amyloid formation of hen egg-white lysozyme and study of its possible role in Alzheimer’s disease. Basic ClinNeurosci 6(1):29–37
Regland B, Lehmann W, Abedini I, Blennow K, Jonsson M, Karlsson I, Sjogren M, Wallin A, Xilinas M, Gottfries CG (2001) Treatment of Alzheimer’s disease with clioquinol. Dement Geriatric Cognit Disord 12:408–414
Riek R (2017) The three-dimensional structures of amyloids. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a023572
Ritter C, Maddelein ML, Siemer AB, Luhrs T, Ernst M et al (2005) Correlation of structural elements and infectivity of HETs prion. Nature 435:844–848
Roberts RG (2016) Good amyloid, bad amyloid—What’s the difference? PLoS Biol 14(1):e1002362
Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE (2015) Structure of the toxic core of α-synuclein from invisible crystals. Nature 525:486–490
Salavadores N, Sanhueza M, Manque P, Court F (2017) Axonal degeneration during aging and its functional role in neurodegenerative disorders. Front Neurosci 11:451
Sarkar N, Singh AN, Dubey VK (2009) Effect of curcumin on amyloidogenic property of molten globule like intermediate state of 2,5-diketo-D-gluconate reductase A. Biol Chem 390:1057–1061
Sarkar N, Dubey VK (2010) Protein nano-fibrilar structure and associated diseases. Curr Proteom 7:116–120
Savelieff MG, DeToma AS, Derrick JS, Lim MH (2014) The ongoing search for small molecules to study metal-associated amyloid beta species in Alzheimer’s disease. AccChem Res 47(8):2475–2482
Sayed HR, Hawkins NP, Lachmann JH (2015) Emerging treatments for amyloidosis. Kidney Int 87:516–526
Schliebs R, Arendt T (2006) The significance of the cholinergic system in the brain during aging and in Alzheimer's disease. Neural Transm 113:1625–1644
Schliebs R, Arendt T (2011) The cholinergic system in aging and neurodegeneration. Behav Brain Res 221:555–563
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81(2):741–766
Selkoe DJ (2002) Alzheimer’s disease is a synaptic failure. Science 298:789–791
Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA (2000) Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-β conformation. Proc Natl Acad Sci USA 97(9):4897–4902
Sousa MM, Yan SD, Fernandas R, Guimaraes A, Stern D, Saraiva MJ (2001) Familial amyloid polyneuropathy: receptor for advanced glycation end products-dependent triggering of neuronal inflammatory and apoptotic pathways. J Neurol 21:7576–7586
Stefani M, Dobson CM (2003) Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med 81:678–699
Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M (2005) Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J Biol Chem 280(9):7614–7623
Torok M, Milton S, Kayed R, Wu P, McIntire T (2002) Structural and dynamic features of Alzheimer’s Aβpeptide in amyloid fibrils studied by site-directed spin labeling. J Biol Chem 27:40810–40815
Town T, Nikolic V, Tan J (2005) The microglial "activation" continuum: from innate to adaptive responses. J Neuroinflam 2:24
Tuppo EE, Arias HR (2005) The role of inflammation in Alzheimer’s disease. Cell Biol 37:289–305
Valender P, Wu L, Henderson F, Zhang S, Bevan D, Xu B (2017) Natural product-based amyloid inhibitors. Biochem Pharmacol 139:40
Van Melckebeke H, Wasmer C, Lange A, Ab E, Loquet A (2010) Atomic-resolution three dimensional structures of HET-s (218–289) amyloid fibrils by solid-state NMR spectroscopy. J Am Chem Soc 132:13765–13775
Vazquez-Fernandez E, Vos MR, Afanasyev P, Cebey L, Sevillano AM (2016) The structural architecture of an infectious mammalian prion using electron cryomicroscopy. PLOS Pathog. 12:e1005835
Virchow R (1854) Zurcellulosefrage virchows. Arch Pathol Anat Physiol 6:416–426
Walton JR (2013) Aluminum involvement in the progression of Alzheimer's disease. J Alzheimer's Dis 35(1):7–43
Wendler CC, Amatya S, McClaskey C, Ghatpande S, Fredholm BB, Rivkees SA (2007) A1 adenosine receptors play an essential role in protecting the embryo against hypoxia. Proc Natl Acad Sci USA 104:9697–9702
Wurth C, Guimard NK, Hecht MH (2002) Mutations that reduce aggregation of the Alzheimer's Abeta42 peptide: an unbiased search for the sequence determinants of Abeta amyloidogenesis. J Mol Biol 319(5):1279–1290
Westermark P (2005) Aspects on human amyloid forms and their fibril polypeptides. FEBS Journal 272:5942–5949
Yamada M, Ono K, Hamaguchi T, Noguchi-Shinohara M (2015) Natural phenolic compounds as therapeutic and preventive agents for cerebral amyloidosis. AdvExp Med Biol 863:79–94
Yang S, Dunstan D (2014) A brief overview of amyloids and Alzheimer’s disease. Protein Sci 23:1315–1331
Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM (2005) Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 280:5892–5901
Yang Y, Petkova A, Huang K, Xu B, Hua QX et al (2010) An Achilles’ heel in an amyloidogenic protein and its repair: insulin fibrillation and therapeutic design. J Biol Chem 285:10806–10821
Zheng W, Monnot DA (2012) Regulation of brain iron and copper hypotheisis. Pharmacol Ther 133(2):177–188
Zuroff L, Daley D, Black K, Hamaoui M (2017) Clearance of cerebral Aβ in Alzheimer’s disease: reassessing the role of microglia and monocytes. Cell Mol Life Sci 74:2167–2201
Acknowledgements
The infrastructure provided by IIT (BHU) is acknowledged. DK and KP acknowledge research fellowship by IIT (BHU) Varanasi.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kundu, D., Prerna, K., Chaurasia, R. et al. Advances in protein misfolding, amyloidosis and its correlation with human diseases. 3 Biotech 10, 193 (2020). https://doi.org/10.1007/s13205-020-2166-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-020-2166-x