
Abstract
Importance of proteins and their proper folding in maintaining cellular homeostasis could never be more appreciated than in the current era. Scrutinizing the etiological factors of various debilitating diseases like Parkinson’s disease, atherosclerosis, Li-Fraumeni syndrome, cystic fibrosis, diabetes mellitus type 2, etc., that are currently affecting a significant fraction of world human population reveals protein misfolding to be the common thread in all. The cytotoxic symptoms observed in these diseases could be easily explained by genetic mutations directly affecting primary structure and hence conformation of the protein, resulting in either their cellular deficiency due to excessive degradation or accumulation of their cytotoxic oligomers and amyloid fibrils. In this chapter, we have emphasized the expanse of protein misfolding disorders and explained their pathogenesis and pathophysiology. In addition, the medications presently being employed to patients and the recent advances in the development of novel treatment strategies both in primeval and clinical trial stage have been elaborately discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
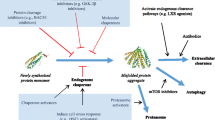
Current medical and technological advances have increased life expectancy. But, this luxury is eclipsed with the emergence of multiple degenerative disorders like Parkinson’s disease, cystic fibrosis, muscular dystrophy, atherosclerosis, diabetes, etc. As prodigious it might sound, these enlarged ensemble of diseases have not stemmed from any pathogenic invasion but rather by imbalanced homeostasis of proteins. It is evidently known that proteins are structural and functional basis of a cell, therefore, their improper folding, overexpression, or excessive loss is found to be detrimental. To avoid such deleterious circumstances, during the course of evolution, cells have evolved an efficient protein quality control system, consisting of chaperones, ubiquitin proteasomal system (UPS), and autophagy. Generation of molecular chaperones is the very first response cells usually show against misfolded protein. It is the responsibility of chaperones to guide and promote correct folding of newly translated polypeptides and prevent their perilous interactions with other cellular proteins and organelles. In spite of that, a large fraction of newly translated protein fails to fold correctly, generating extensible burden of defective polypeptides, which are then directed by the chaperones for degradation either by ubiquitin-proteasome system or autophagy. However, under certain circumstances, this quality control system fails to identify and remove misfolded proteins, leading to formation of soluble or insoluble aggregates, which may accumulate as amyloidogenic plaques (Taylor et al. 2002). These aggregates can then move to different places in the cell and gain toxicity of various natures, leading to different pathological consequences in the cell. Alternatively, overwhelming of this system might also elaborate proteasome mediated enhanced degradation of the protein.
Protein misfolding is now considered the pathological hallmark of a large number of human diseases. Although, this was not the case initially, as earlier abnormal protein conformation was generally linked with just defective enzyme function and related disorders. However, with increasing pace of research in this field, various familial and sporadic diseases are springing out to be the consequence of protein misfolding in almost every tissue and organ system. In the following chapter, we have summarized the protein-related disorders under the following sections, namely – protein misfolding-related neurodegenerative disorders, cardiovascular disorders, metabolic disorders, pulmonary disorders, and ophthalmic disorders. Also, for the ease of classification under these sections, most of the systemic disorders are kept under the category of other disorders. Through this chapter, we have revised a few disorders of each category and tried to provide the recent developments that have been done in those fields. In the later sections, the understanding gained until now regarding the causes and the mechanisms of toxicity has been elaborated. Finally, the most essential aspect of all these research – the therapeutic advances – has been discussed.
2 Proteopathies: The Devil Widespread from Head to Toe
Proteopathies is an umbrella term used to represent the disorders associated with structural abnormality of proteins. Protein misfolding-related disorders are quite prevalent and are extensively investigated in nervous system, but of late they have been found to be associated with diseases localized in almost every organ system. This section provides the detailed overview of representative diseases of various organ systems, describing their clinical manifestations and connection with improper protein folding (See Table 8.1 for list of various proteopathies).
2.1 Neurological Disorders
Neurodegenerative diseases (ND) are some of the most debilitating disorders, affecting thinking, skilled movements, feelings, cognition, and memory. The name for these diseases is derived from a Greek word υρο-, néuro, “nerval” and a latin verb dēgenerāre, “to decline” or “to worsen”. Despite significant dissimilarities in clinical manifestation, this diverse group of diseases including Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), frontotemporal lobar degeneration (FTLD-U), Huntington disease (HD), Prion diseases (PrP), and many more share some common features such as their advent late in life, the widespread neuronal loss and synaptic aberrations, and the existence of cerebral accumulation of misfolded protein aggregates in neuronal inclusions and plaques. These deposits are emblematic signature of most of the neurological disorders and can trigger cascade of events ultimately resulting in synapse loss and neuron death with devastating clinical consequences.
Albeit the differences in the primary sequence of the respective protein in each ND, these proteins have similar morphological, structural, and staining characteristics and are anticipated to have destabilized secondary and/or tertiary structures in vivo and also referred as intrinsically disordered proteins. However in each ND, despite the distinctive localization, distribution, and composition of protein aggregates, cross-β sheets were a common feature in all disease-related amyloids.
2.1.1 Alzheimer’s Disease
Alzheimer’s disease (AD), the most common neurodegenerative disorder, is the late-onset dementing illness, with progressive loss of memory, task performance, speech, and recognition of people and objects. Familial AD has been correlated with mutations of the amyloid precursor protein (APP) gene, presenelin-1 gene, and presenelin-2 gene. These autosomal dominant mutations are all involved in the metabolism of APP, whose cleavage by β-secretase and γ-secretase yield β-amyloid (Aβ) peptides, particularly Aβ1–40 and Aβ1–42 fragments. The Aβ1-40 form is the more common of the two, but Aβ1-42 is more prone to aggregate and is thus associated with disease states. Moreover, presenelin-1 and its homolog presenelin-2 associate with three additional proteins – nicastrin, aph-1, and pen2 – to form a tetrameric complex which functions as γ-secretase (Bi 2010). Sporadic cases of AD seem to be allied with the gene coding for apolipoprotein E (ApoE). Studies have shown the population with ApoE4 allele is at greater risk of developing AD than the general population, while the ApoE2 allele confers relative protection (Farrer et al. 1997). However, the exact nature of ApoE and AD connection is still ambiguous. A few among the other multiple genes whose mutations have been implicated in AD pathogenesis are gene for insulin-degrading enzyme, SORL1gene, and ubiquilin-1 (Minati et al. 2009).
AD has been exemplified by the formation of two types of protein aggregates in the brain, senile plaques and cerebral amyloid angiopathy, which are accretions of Aβ peptide (1), and (2) the intracellular neurofibrillary tangles comprised of hyperphosphorylated filaments of the microtubule-associated protein tau. Using NMR studies, Aβ fibrils were revealed to have cross-β structure, a dimer of β-sheets parallel to the fibril axis formed by interaction of β1- and β2-strands of two Aβ peptides. Contradictory evidences from various experiments suggest the pathogenic agents responsible in amyloid-related diseases to be either the transient, prefibrillar Aβ assemblies or oligomers preceding the formation of mature fibrils (Benilova et al. 2012). In both cases, however, the cellular toxicity is believed to manifest through the progression of events such as synaptic failure, inflammation, and tau hyperphosphorylation (Reitz 2012). Furthermore, hyperphosphorylation of tau protein reduces its proficiency to bind with microtubules, bringing about its self-aggregation into paired helical fragments, which then deposits into neurofibrillary tangles. These aggregates of tau protein are believed to further aggravate the toxic insults of Aβ amyloids by (i) triggering synaptic excitotoxicity caused due to enhanced interactions between NMDA receptors and PSD95 proteins (Ittner et al. 2010), and (ii) collapsing the cellular scaffolding to produce widespread brain cell degeneration and dysfunction (Handoko et al. 2013).
2.1.2 Parkinson’s Disease
Parkinson’s disease (PD) is characterized by resting tremor, bradykinesia (slowness of movement), rigidity, and postural instability, and the patients with PD have characteristic hallmarks of loss of dopaminergic neurons and α-synuclein (aSyn) aggregate depositions, also called Lewy bodies, in the cytoplasm of neurons of the substantia nigra (SN) in the brain. aSyn is expressed as multiple isoforms spanning 98-, 112-, 126-, and 140-amino acid residues as a result of alternative pre-mRNA splicing of SNCA gene (also known as PARK1). Variants of SNCA gene, namely A53T, A30P, and E46K, as well as the presence of multiple copies of Wt SNCA gene are related to early-onset familial PD. Genome-wide association studies (GWAS) have linked SNCA with sporadic PD also (Stefanis 2012). Additionally, cohort studies have reported the family with carriers of G51D SNCA mutation to display neuropathological features of both PD and multiple system atrophy, reinforcing the connection of SNCA with PD progression (Kiely et al. 2013). PD is also attributable to mutations in genes other than SNCA, such as parkin, DJ-1, PINK1, and LRRK2.
aSyn is considered to be natively unfolded in its monomeric state as its purified form at neutral pH lacks an ordered structure and function. However, inside cell, they attain alpha-helical conformation after binding with cell membrane. Recent investigations have even reported endogenous aSyn isolated from brain tissue, living human cells, and neuronal and non-neuronal cell lines to exist as a tetramer of about 58 kDa (Bartels et al. 2011). The interesting feature of these tetrameric species was its ability to resist aggregation, suggesting aSyn to disassemble into monomers under diseased conditions prior to its aggregation into pathogenic amyloid fibrils. Structural studies have implicated the central hydrophobic region of aSyn (61–95), so-called NAC (non-Aβ component), to confer it the β-sheet potential upon aggregation. Moreover, investigations have shown aSyn lacking NAC domain has less toxicity and aggregation propensity (El-Agnaf et al. 1998). This aSyn aggregation process prior to the formation of amorphous aggregates, and amyloid fibrils, also involves the formation of various soluble oligomeric intermediates, collectively termed protofibrils, which assume spherical, ring, and string like characteristics when seen under the electron microscope (Rochet et al. 2012). While there are evidences that describe amyloid fibrils to mediate cellular toxicity by disrupting cellular topography and interfering with normal cellular physiology; recent developments have led to the assumption that oligomers/protofibrils are responsible for toxicity in PD. This theory is supported by certain studies, such as the one done on aSyn variants – A53T and A30P – indicated the neurotoxic effects to be a result of enhanced protofibril formation rather than accelerated fibrillation, and another study done on aSyn variants that promoted oligomer formation also revealed most prominent dopaminergic cell death upon lentiviral injection into rat SN (Rochet et al. 2012). Because of such imperative role of aSyn in PD pathogenesis, the utilization of aSyn levels in colonic mucosal biopsy has been suggested as a biomarker for PD (Shannon et al. 2012). But measurement of only aSyn levels does not seem an efficient and specific biomarker for PD, since AD patients are also known to have significantly increased aSyn levels in the cerebrospinal fluid. However, analysing levels of both aSyn and neurosin, aSyn cleaving enzyme might be a better method to distinguish synucleopathies like PD from AD, since PD patients display lower neurosin and aSyn cerebrospinal fluid levels as compared with non-demented controls and AD patients (Wennstrom et al. 2013).
2.1.3 Huntington Disease
Huntington disease (HD) is a fatal disease with symptoms including involuntary movements (chorea), personality, and cognitive changes. HD is a representative of the congregate of polyglutamine repeat (polyQ) disorders, whose other members include Kennedy disease (spinobulbar muscular atrophy or SBMA), dentatorubropallidoluysian atrophy, and six forms of spinocerebellar ataxia (SCA1, 2, 3, 6, 7, and 17). All sharing a common feature of presence of polyQ tract in the respective protein. In case of HD, the CAG trinucleotide repetition lies within exon 1 of the huntingtin gene, encoding the ubiquitously expressed huntingtin (htt) protein. The longer glutamine portion so introduced within htt modifies its native conformation, instigating inclusion formation in the brain. The threshold polyQ length effect observed in HD patients is quite remarkable, in which the polyQ extensions of ≥40 leads to disease, whereas ≤40 do not. This might be because htt with polyQ extensions ≤40 can be degraded by the proteasome, but when the stretches expand >40, proteasome’s proficiency to degrade them reduces, leading to their deposition in the nucleus, backing the notion that aggregation of htt with polyQ stretches is responsible for disease manifestation (Scherzinger et al. 1999). The data available up till now has suggested polyglutamine aggregates to have a unique secondary structure with compact β-sheets with interspersed β-turns every nine glutamines, famously known as “polar zippers.” The presence of htt inclusions is quite prominent in the striatal neurons and affect mostly the GABA-producing cells, ultimately diminishing the cellular levels of an essential neurotransmitter acetylcholine, hence loss of voluntary muscle control. Additionally, polyQ htt aggregates are believed to impair UPS by engaging with it within the nucleus, disrupt cellular protein homeostasis by recruiting proteins with short polyQ stretches into the nascent htt aggregates, and induce oxidative stress, immune dysfunction, and mitochondrial dysfunction (Ross and Poirier 2004). The polyQ repeats may also perturb htt’s physiological function as a transcription factor, altering the configurations of gene transcription. However, evidences from a few experiments had shown that the toxicity of expanded polyglutamine might not be associated with the formation of visible inclusions, while what remained as a common feature of toxicity in all these experimental systems were insoluble molecular aggregates.
2.1.4 Amyotrophic Lateral Sclerosis (Lou Gehrig’s Disease)
Amyotrophic lateral sclerosis (ALS) predominantly affects the upper motor neurons of the motor cortex, and the lower motor neurons in brainstem and spinal cord, causing muscle weakness, spasticity, atrophy, progressive paralysis, and ultimately death within the few years of the disease commencement. Approximately 90 % of the ALS cases are sporadic in nature and has no known origin, while familial ALS has been found to be associated with mutations of SOD1 gene, TDP gene, FUS gene, and ANG gene (Kiernan et al. 2011). Initially, it was assumed that the mutation of superoxide dismutase (SOD1) reduces its antioxidant activity and enhances the build-up of reactive oxygen species (ROS) in the cell, causing toxicity. However, studies done on transgenic mice with null SOD1 expression did not showed any motor neurodegeneration, discarding functional loss to be the reason for disease progression (Reaume et al. 1996). Interestingly, Wt SOD1 overexpression in mice displayed ALS-like pathology and deposition of SOD1 aggregates in brain, suggesting another pathway for ALS development (Graffmo et al. 2012). In fact, the presence of hyaline inclusions immunoreactive for SOD1 in the neurons and astrocytes is the common manifestations of familial and sporadic ALS (Ivanova et al. 2014). It is supposed that either aggregates or oligomeric precursors of mutant SOD1 disturb cellular proteostasis by disrupting protein quality control system and induces stress by interfering with cellular cytoskeletal transport network (Chattopadhyay and Valentine 2009). Current advances have shown that apart from SOD1, the mutations of profilin 1 (protein essential for actin polymerization) may also have an involvement in ALS development by enhancing axonal retraction and denervation in the adult neuromuscular system (Robberecht and Philips 2003). Additionally, in the spinal cord samples of ALS patients, the upregulation of inflammatory cytokines, such as TNFα, COX-2, and interleukins, has been observed (Wijesekera and Leigh 2009). However, the association between inflammatory network and ALS is yet not understood and needs to be explored.
2.1.5 Prion Diseases
Prion protein (PrP) diseases, also termed transmissible spongiform encephalopathies (TSEs), are a heterogeneous group of disorders that are characterized by ataxia, myoclonus, dementia, insomnia, and psychiatric disturbances. There are both sporadic PrP diseases, such as sporadic Creutzfeldt-Jakob disease (CJD), as well as familial forms including familial CJD, Gerstmann-Straussler-Scheinker disease, and fatal familial insomnia. Pathologically these disorders are distinguished from other ND by their resemblance with viruses to propagate the disease from infected cell to healthy cells. However, in this case, the entity showing infectious nature is prion proteins, and its variants and their amyloid plaques cause the disease (Aguzzi and Calella 2009). The mutations of PRNP, the gene encoding prions, have been found to be associates with both familial and sporadic prion diseases. The known-disease causing variants of PRNP are Met129Val and Glu219Lys. Genome-wide association studies have also revealed the presence of an additional candidate loci upstream of the retinoic acid receptor beta encoding gene to be associated with CJD (Mead et al. 2009).
In PrP diseases, prions undergo conformational changes from its normal conformation, PrPC to its insoluble protease resistant pathological form, PrPSc. Findings accumulated in last three decades via various studies such as understanding prion disease manifestation in Wt animals injected with highly purified PrPSc and finding that PrP knockout mice are resistant to prion infection has given rise to the prion hypothesis (Soto and Estrada 2008). This hypothesis explains the mechanism of conversion of PrPC into PrPSc. According to the hypothesis, only PrPSc not PrPC has the ability to replicate in the brain without requiring nucleic acids, and it catalyzes the conversion of Wt PrPC into the misfolded form. It is believed that oligomeric PrPSc acts as a nuclei to bind PrPC, perhaps in combination with certain ancillary proteins, and incorporate it into the growing oligomer. However, at some point, the long PrPSc polymers break into smaller pieces, which then further act as seeds to drive PrPC conversion, producing a self-propagating amyloid. Interestingly, a recent study done with spleen tissue from a PRNP heterozygous genotype individual had demonstrated to propagate the variant CJD agent, and it was realized that the prions can be present in the other tissues without CNS involvement (Bishop et al. 2013). This development furthers that despite multitudes of investigations, our knowledge regarding this disease is still limited and it is possible that prion disorders affect multiple tissues not just nervous system, increasing the challenge to find its cure.
2.1.6 Are All Neurodegenerative Proteopathies Infectious?
PrP diseases have exemplified that how protein’s conformational change can make a protein to behave like a pathogen and cause an infectious disease. This infectious nature of prions is best explained by seeding-nucleation model as the disease progression lie on the competence of preformed stable misfolded oligomeric proteins to act as a seed to catalyze the misfolding and aggregation process. It is quite possible that the proteins involved in other neurodegenerative disorders also have yet undiscovered ability to be transmissible, since their aggregation and amyloidosis also follow the same model as that of prions. With recent advances, various reports have arose that defends this hypothesis. Aggregates of Aβ peptide have been reported to spread to unaffected cells when injected into the brain of an AD mouse and display Aβ accumulation in a pattern dependent upon both the host and the agent (Rosen et al. 2011). The capability of passing between living cells was also illustrated for aggregates of truncated tau, consisting of the microtubule-binding region and a fluorescent protein tag that can leave and enter cells in culture, and promoted the aggregates and fibrillization of normal tau within them (Jellinger 2011). Another study has reported that tau protein aggregates to spread around neighboring areas of the brain by “jumping” within neurons (Brundin et al. 2010). The various studies done on neuronal cell lines, animal models, and humans, where PD host with grafted dopaminergic neurons shows lewy body pathology, has reinforced the concept that neuronal protein aggregates can display prion-like pathogenic behavior (Angot et al. 2012). This is reflected in propagation pattern of aSyn-rich lesions, in accordance with Braak hypothesis of staging of PD, first through the lower brainstem, the anterior olfactory nucleus, and olfactory bulb, and then subsequently in mesencephalic and neocortical regions. Lately, a groundbreaking work describing the first ever research model that shows both cell-to-cell spread of aSyn aggregates and progressive loss of dopaminergic neurons has reinforced the former hypothesis. In this investigation, single injection of preformed fibrillar aSyn into the striatum of Wt mice was sufficient to induce intraneuronal aSyn accumulation and Lewy body pathology (Luk et al. 2012). The propagation of proteinaceous lesions has also been demonstrated in aggregates of SOD1, TDP-43, and polyQ proteins (Jellinger 2011). The presence of prion-like domain in these proteins has been alleged to influence the disease progression. It is necessary to understand that lack of experimental confirmation for infectious nature of protein aggregates in other ND does not mean that it is a disease-specific phenomenon, and the seeding activity displayed by protein aggregates in all neuronal proteopathies clearly indicates cell-to-cell transmission to be general dominion of amyloids.
2.2 Metabolic Disorders
2.2.1 Diabetes Mellitus Type 2
Type 2 diabetes (T2DM) is a loosely defined clinical syndrome that is characterized by insulin resistance, defective insulin secretion, and loss of β-cell mass (number of β-cells) with increased β-cell apoptosis and islet amyloid deposition. The amyloid lesions within the islets are depositions of misfolded islet amyloid polypeptide (IAPP) oligomers with cross-β sheet structure (Hayden et al. 2005; Patel et al. 2014). The initial site of localization of these lesions in the islets is not known. However, the most probable site seems to be insulin secretory granule (ISG), the storage reservoir of pro-insulin and pro-IAPP. It is here prohormone convertases 1, 2, and 3 process pro-IAPP into their cleaved forms, which are then coreleased into the circulation (Marzban et al. 2004). Any abnormality in this processing has been observed to ensue IAPP-derived islet amyloid deposition, demonstrating pro-IAPP also to be amyloidogenic (Hayden et al. 2005). Imbalance among other components of ISG, such as Ca2+, Zn2+, and C-peptide, that maintain mature insulin and IAPP is also believed to promote aggregate formation (Westermark et al. 1996). Additionally, excessive ROS burden and ER stress in conjugation with excessive insulin production increase protein misfolding, thereby overwhelming cell’s PQC and fostering IAPP aggregation (Back and Kaufman 2012). Other than IAPP and insulin, this islet amyloid also contains serum amyloid P component (SAP), ApoE, and the heparan sulfate proteoglycan, perlecan. Perlecan stabilizes islet amyloid by allowing IAPP to bind to the basement membranes surrounding islet capillaries, resulting in ISG’s decreased secretory response due to thickened basement membrane (Kahn et al. 1999). This event is supposed to activate the structural alterations within islet, thus creating a localized milieu with predisposition for skewed insulin to IAPP ratio.
The physiological function of IAPP is yet not fully apprehended; however, studies with rodent models have alleged its involvement in the regulation of various metabolic constraints including satiety, gastric emptying, adipose accumulation, and blood glucose levels via glucose-simulated insulin secretion inhibition (Abedini and Schmidt 2013). This loss of physiological function could be the additive factor in the toxicity seen in T2DM. However, oligomerization of IAPP seems to be the major contributor for apoptosis of β-cells via instigation of localized inflammation in islet, oxidative stress enhancement, and autophagy dysregulation.
2.2.2 Phenylketonuria
Phenylketonuria (PKU), the first treatable inherited disorder, is an inborn error of metabolism of l-phenylalanine (L-Phe) caused by mutations of the gene encoding phenylalanine-4-hydroxylase (PAH). At present, approximately 500 mutations of PAH gene are known, most of whom are found to be associated with PKU. These mutations either leads to degradation or aggregation of misfolded PAH enzyme, ultimately causing its deficiency in the liver. Since PAH with cofactor tetrahydrobiopterin is involved in the catalysis of L-Phe to l-tyrosine, its deficiency reduces tyrosine levels and elevates blood levels of L-Phe and its metabolites, impairing brain development and function (Donlon et al. 2004). Depending upon the L-Phe levels and loss of PAH activity, PKU is differentiated into various categories – classic PKU with slight or no activity (Phe > 1,200 μmol/l), mild PKU, and non-PKU hyperphenylalaninemia with some activity (Phe > 360–1,200 μmol/l and 120–600 μmol/l, respectively) (Williams et al. 2008).
Although unlike other protein misfolding disorders PKU is treatable, the lifelong dietary control often leads to under nutrition and psychosocial problems (Harris 2014). Therefore, in order to create therapeutic interventions disrupting PAH oligomerization, it is necessary to understand its conformational dynamics during catalysis and aggregation. The investigations done so far have not much advanced our knowledge, what we know is that PAH is a homotetramer, which on binding with L-Phe and tetrahydrobiopterin alters its conformation and gets activated. These structural changes involves fine interplay among PAH monomers and various domains via networks of side-chain interactions. This conformational flexibility is believed to make PAH vulnerable to misfolding. This is buttressed by the studies displaying unstable aggregation and enhanced degradation of variant PAH enzymes (Gersting et al. 2008).
2.3 Cardiovascular Disorders
2.3.1 Atherosclerosis
Atherosclerosis is a progressive vascular disease characterized by chronic inflammation, augmented oxidative stress, and arterial vessel thickening due to atherogenesis and amyloid plaque formation. Smoking, diabetes mellitus, imbalanced lipid metabolism, high blood pressure, and enhanced cholesterol levels are some of its known risk factors (Ursini et al. 2002). The prolonged inflammation observed in atherosclerotic arteries is a general immune system response to injury, but in this case it further aggravates the condition by expanding arterial plaque and narrowing the vessels (Herczenik and Gebbink 2008). Arterial vessel narrowing, if untreated, leads to various cardiac complications for instance, stroke, peripheral arterial disease, stenosis, and myocardial infarction (Wilck and Ludwig 2014). Additionally, rupturing and release of arterial plaque components in blood could cause embolism and prove fatal (Herczenik and Gebbink 2008). Atherosclerotic arterial lesions are generally comprised of cholesterol, calcium, lipoproteins, macrophages, and aggregates of various misfolded proteins such as Aβ, α1-antitrypsin, and members of apolipoprotein (Apo) family (Rocken et al. 2006). Apo proteins are essential for lipid metabolism, since their binding is necessary for the activation of various physiological lipid and cholesterol carriers such as high density lipoprotein (HDL), low density lipoprotein (LDL), etc. Oxidation of LDL has been reported to alter its native conformation and convert α-helix to β-sheets, thereby priming LDL fibrillation and amyloidosis (Maor et al. 1997). However, recent investigations done with 17-β-estradiol showed its binding with modified apoB-100 to inhibit the misfolding and hence aggregation of LDL and apoB-100 complex, suggesting apoB-100 misfolding to be the root cause behind amyloid formation and atherogenesis (Brunelli et al. 2014). Unlike most of the other misfolded proteins, structure destabilization or proteolysis does not seem to be the reason for the deposition of mutated apo protein fibrils in atherosclerotic lesions. This hypothesis is conferred from the structural analysis of apoA-1 variants, which displayed very minute changes in protein stability, α-helical content, protein-lipid interactions, and proteolytic pattern (Das et al. 2014).
2.3.2 ATTR Amyloidosis
ATTR amyloidosis is the most common form of familial cardiac amyloidosis, caused by mutation in the gene for the plasma protein transthyretin (TTR), and is characterized by severe heart failure and arrhythmias. Out of the 100 different amyloidogenic missense point mutations designated for TTR gene, V122I is the most frequently observed variant. This mutation was found in 3.9 % of all Afro-American population and 23 % of African Americans with cardiac amyloidosis and may be responsible for the higher prevalence of heart failure in elderly blacks (Buxbaum et al. 2006). The onset and severity of disease manifestation vary with specific mutation. For instance, TTR with Val30Met mutation is rarely corresponded with amyloid development in cardiomyocytes, while another TTR variant with Thr60Ala mutation develops cardiac amyloidosis early in individuals (Banypersad et al. 2012). It is assumed that mutant TTR act as nuclei and prompts aggregation of Wt TTR. This was braced by the study done in Japan, which reports Wt TTR to be present in the hearts of patients with familial amyloid polyneuropathy (Kholova and Niessen 2005).
2.3.3 Aortic Medial Amyloidosis
Aortic medial amyloidosis is a very common protein deposition disease of aortic media in elderly people. It is characterized by the presence of amyloid fibrils composed of medin protein. Medin is a 50-residue fragment derived from internal splicing of lactadherin, a milk fat globule protein (Stubbs et al. 1990). This disease has no well-established clinical significance but is considered to be involved in the age-related loss of elasticity of the vessels. Not much research is yet done on this aspect of this disorder because of which the cause of misfolding and the mechanism of toxicity are not clear.
2.4 Pulmonary Disorders
2.4.1 Cystic Fibrosis
Cystic fibrosis is one of the most widespread life-shortening inherited disorders among the caucasian population and is characterized by airway infection, inflammation, remodeling, and obstruction, resulting in gradual destruction of the lung tissue and ending in an early death. It follows autosomal recessive inheritance as the disease develops only when both copies of cystic fibrosis transmembrane regulator (CFTR) gene are mutated. CFTR is a cAMP-activated ATP-gated anion channel that transports chloride and thiocyanate ions across epithelial cell membrane to the covering mucus. To maintain electrical balance, sodium ions also depart from the cell, which leads to increased osmolarity of the outer mucosal cells, ensuing water to move out of cell by osmosis. All disease-causing mutations in the CFTR gene prevent the protein from attaining proper conformation and, hence, are retained and degraded. The loss of CFTR function impedes the CFTR channel from functioning properly; inhibits the movement of ions and water out of the cells; causes the development of thick, sticky mucus which then cause obstruction of the passageway; and traps bacteria that give rise to chronic infections (Childers et al. 2007).
CFTR protein quality control is mediated at either of the two stages: (a) Molecular chaperone, heat shock protein (Hsp) 70 stops the misfolded protein processing in the cytosol and present it to the proteasome for degradation; (b) Calnexin, a lectin chaperone of ER bind to glycosylated CFTR and retro-translocates the misfolded CFTR to the cytosol for degradation (Gregersen 2006). In addition, the study done in ΔF508 CFTR yeast model (the most common CFTR variant) had reported human small Hsp, αA-crystallin to preferentially interact with ΔF508 CFTR, maintain its solubility and direct them for ER associated degradation (Ahner et al. 2007). All these studies suggest differential effect of various chaperons on variant CFTR. This is backed by the investigation done on mutant human cystathionine beta-synthase, where the ratio of Hsp70 and Hsp26 was demonstrated to determine the fate of misfolded proteins to be either refolded or degraded (Singh and Kruger 2009). This model for cystic fibrosis pathology is supported by the study in which ΔF508 was observed to have increased sensitivity to proteolytic digestion, as compared with the Wt CFTR (Bellotti and Chiti 2008). Interestingly, CFTR overexpression or proteasome inhibition has been demonstrated to augment the accumulation of Wt and ΔF508 CFTR protein along with ubiquitin and certain chaperones to develop aggresomes that are surrounded by collapsed intermediate filament proteins, expounding CFTR as an aggregation prone protein under stress conditions (Kopito 2000). Genome-wide association studies have indicated another protein, interferon-related development regulator-1, to be associated with cystic fibrosis. A recent study done to understand its role in disease pathology found its upregulated levels in neutrophils to be linked with enhanced ROS generation (Hector et al. 2013). Further studies are required to clearly understand its role in disease pathogenesis.
2.4.2 Emphysema
Emphysema is part of a group of conditions termed chronic obstructive pulmonary disease and is characterized by destruction of lung tissue around the alveoli. Smoking is considered the major causative factor, followed by α1-antitrypsin (AAT) deficiency as the rare cause for emphysema. AAT is a monomeric secretory protein synthesized most abundantly by hepatocytes. After secreting from cells, Wt AAT binds to and inhibits trypsin and also the blood protease, elastase. In the lungs of individual with AAT deficiency, elastase and trypsin degrade the lung tissue that participates in the absorption of oxygen, eventually leading to breathing problems. The reason for AAT deficiency in lungs is the mutation in the gene encoding AAT, causing the production of misfolded protein which is then retained intracellularly and degraded in pre-golgi, non-lysosomal compartment (Stoller and Aboussouan 2005). The study done with transport-impaired PiZ variant (most common variant linked with AAT deficiency) in transfected mouse hematoma cell line demonstrated that these variants form stable soluble aggregates, possibly in the form of homotrimers, which then undergoes a discrete size reduction and degradation within the ER (Le et al. 1992). Severe cases of AAT misfolding may also lead to cirrhosis of the liver, where the AAT aggregates develop a fibrosis that produces scarring and dysfunction.
2.4.3 Pulmonary Alveolar Proteinosis
Pulmonary alveolar proteinosis is a well-known conformation disorder characterized by the presence of amyloid fibrils comprising lung surfactant protein C (SP-C) within the alveoli, interfering with gas exchange (Gustafsson et al. 1999). A latest cohort study done in Japan has reported elevation in its incidence and prevalence rate and discarded its linkage with smoking, one of the common causes of lung diseases (Inoue et al. 2008). Variants of gene encoding SP-C have been correlated with chronic lung disease in children and adults. These mutations are believed to induce pro SP-C protein misfolding, which then cause toxicity in epithelial cells either via its deposition in lungs or pro SP-C trapping in the ER and its rapid degradation via UPS (Bridges et al. 2003). Additionally, fatty acid removal from SP-C is known to induce conformational change, such that it will aggregate to form β-sheet-rich amyloid fibrils (Brasch et al. 2004).
2.5 Muscle Disorders
2.5.1 Congenital Myopathies
Congenital myopathies include various inborn skeletal muscle disorders frequented with depositions of protein fibrils also called rod bodies. Approximately 45 varied types of myopathies are known and classified on the basis of histopathologic characteristics (Bodensteiner 2014). Due to rare occurrence, not all of them are investigated in great depth. However, core-rod myopathy, reducing body myopathy, cap disease, cylindrical spirals myopathy, and nemaline myopathy have been prominently studied. They don’t display any significant morphological differences because skeletal muscle could conceive only limited amount of pathological modifications. So, the major differences observed in their pathogenesis are believed to be because of the associated genetic mutations and related modified proteins. Recent studies have reported mutations in various genes including TPM3, FHL1, TPM2, ACTA1, RYR1, KLHL40, and NEB to be involved with protein inclusion formation (Malfatti et al. 2014; Wilding et al. 2014). These rod bodies generally include fibrils of various proteins, such as troponin T, nebulin, α- and β-tropomyosin, and α-actin.
Nemaline myopathy is a representative member of this diverse group of disorder. Investigations done with cardiac myocytes have confirmed accretions of α-actin fibrils in the sarcomere nucleus as its pathological hallmark (Clarkson et al. 2004). It is assumed that missense mutations in α-actin gene cause structural alterations in the protein in such a way that their tendency to self-associate enhances (Vang et al. 2005). Overexpression of variant α-actin proteins in cultured fibroblasts and myoblast cell lines was also observed to trigger nuclear fibrillation (Costa et al. 2004). The cytotoxicity observed is believed to be a cumulative consequence of loss of sarcomeric actin fibers and generation of toxic protein aggregates (Gregersen 2006).
2.5.2 Muscular Dystrophy
Muscular dystrophy (MD) is an ensemble of genetic disorders exhibiting progressive skeletal muscle weakness, hampered locomotion, muscular tissue degeneration, and accretions of defective proteins (Emery 2002). There exist several forms of MD, such as Duchenne, limb-girdle, myotonic, distal, Becker, Emery-Dreifuss, facioscapulohumeral, and oculopharyngeal muscle dystrophies, each affecting different muscle assemblies with manifestations in multiple organ systems. Congenital MD is generally associated with mutations of genes encoding for proteins involved either in connecting muscle tissue with extracellular assembly or in forming dystrophin-associated protein complex (Emery 2002). Duchenne and Becker forms of congenital MD are characterized with the presence of misfolded or truncated dystrophin protein, causing deficiency of active dystrophin and dystrophin glycoprotein complex (Koenig et al. 1989). Dystrophin act as a mechanical stabilizer, and as a part of dystrophin glycoprotein complex, it helps linking cytoskeleton of each muscle cell to the extracellular matrix via sarcolemma (Hoffman et al. 1987). Oculopharyngeal MD is characterized by the presence of polyalanine tract in the nuclear poly (A)-binding protein 1 (PABPN1) and musculoskeletal symptoms like limb weakness, ptosis, and dysphagia. Studies done with cellular and transgenic mouse models of oculopharyngeal MD have revealed the presence of aggregates of protein PABPN1 with polyalanine extensions in the myonuclei of skeletal muscles (Brais et al. 2014). Interestingly, both Wt and mutant PABPN1 are susceptible to aggregation, but only oligomers of mutant PABPN1 are believed to cause cytotoxicity (Raz et al. 2011). Furthermore, reduced availability of functional PABPN1 aggravates the condition by impairing gene expression, since it is responsible for polyadenylating pre-mRNAs (de Klerk et al. 2012).
2.6 Ophthalmic Disorders
2.6.1 Retinitis Pigmentosa
In patients with retinitis pigmentosa, the functionality of the retina is affected which can lead to severe visual impairment and blindness. Studies have revealed its most common etiology to be mutations in rhodopsin gene, which leads to the generation of unstable opsin protein that can’t bind with its cofactor 11-cis retinal to form functional rhodopsin protein (Anukanth and Khorana 1994; Rosenfeld et al. 1992). Rhodopsin is the pigment found in the rod cells that enables our vision during low-light conditions (Stuart and Birge 1996). The variant opsin protein has an inherent instable structure, which triggers its self-association and binds with Wt opsin proteins to form cytotoxic aggregates (Chen et al. 2014b). These aggregates are resistant from UPS mediated degradation and thus block cellular proteasomes, which incites the production of intracellular inclusions or aggresomes comprising Wt and mutated rhodopsin, certain opsin binding proteins, and other misfolded and unfolded proteins that were destined to be degraded (Saliba et al. 2002). Retinal cells require a rapid protein turnover for its proper functioning, and PQC failure tremendously enhances the misfolded protein load, aiming the cell towards programmed cell death.
2.6.2 Cataract
Cataract or opacification of eye lens is one of the most common causes of blindness and has been associated with mutations in the genes encoding for crystallin proteins (Kosinski-Collins and King 2003; Xi et al. 2014). α and βγ Crystallins are the major structural proteins of eye lens responsible for maintaining transparency and enhancing its refractive index (Mahendiran et al. 2014). Additionally, α crystallin functions as molecular chaperone and assist unfolded or misfolded proteins toward degradation, thereby helping lens in tolerating detrimental effects of protein aggregates at old age (Cheng et al. 2010). Mutations in these proteins lead to unobstructed generation of cytoplasmic inclusions, which lead to enhanced light scattering and hence visual impairment (Moreau and King 2012). However, genetic studies done with families displaying juvenile onset cataract have suggested accumulation of mutant γ-crystallin in nucleus to ensue toxicity through disruption of transcriptional processes and structural disorganization of lens fiber cell nucleus (Stefani 2004).
2.7 Cancer
Millions of people die every year due to cancer, making it the most fatal disorder worldwide (Stewart and Wild 2014). The current treatment strategies, including radiation, chemotherapy, and ultimately surgical removal of affected organs, are not helpful for patients in last stages of cancer. The immortality and metastasizing features of cancerous cells give them an edge over other pathogens, complicating our comprehension of their pathogenesis and pathophysiology. During malignancy, proteins are usually uncontrollably overexpressed or structurally affected because of genetic mutations, resulting in changes in activity and protein-protein interactions in cancer cells (Xu et al. 2011). Despite this, only recently, role of protein aggregation has been implied in cancer and it has been found that many cancers are associated with the abnormal accumulations of aggregates of Wt and mutant p53. p53 is a tumor suppressor nuclear phosphoprotein that triggers cell cycle arrest and apoptosis in response to cell stress. It is comprised of structured, partially folded and natively unfolded segments, which give high conformational flexibility (Stefani 2004). Over 50 % of reported cases of human tumors are found to be associated with p53 mutations. According to IARC TP53 Mutation Database, over 95 % of the malignant mutations occur in the DNA-binding domain of p53, where they cluster in so-called hot spots of mutation (Olivier et al. 2002).
Nuclear and cytoplasmic aggregates of inactive conformational variant of p53 have been described to be associated with tumors such as retinoblastoma, neuroblastoma, colon cancer, and breast cancer (Stefani 2004). Various independent studies done in the last decade have shown that several regions of p53 have the tendency to form β-sheet-rich fibrillar aggregates under physiologic and stress conditions. Moreover, mutant p53 had been demonstrated to colocalize with amyloid-like protein aggregates in cancer biopsies (Levy et al. 2011). Like other misfolded proteins, p53 folding variant also has the ability to drive conformational change in Wt p53 and its paralogs p63 and p73. This has been supported by a recent study, where amyloid fibrils of R248Q, a p53 mutant, were reported to seed the aggregation of Wt p53 in a prion-like fashion (Xu et al. 2011). All these evidences imply that p53 lead to tumor genesis either via loss of the antitumor function or by a gain of toxic function.
2.8 Epithelial Tissue Disorders
2.8.1 Hypotrichosis Simplex of the Scalp
In the last decade, hypotrichosis simplex of the scalp (HSS), an autosomal-dominant form of isolated alopecia, has been recognized as a proteinopathy, which can be characterized by the amyloid-like aggregates of corneodesmosin. Corneodesmosin is a glycoprotein that is expressed in the epidermis and in the inner root sheath of hair follicles (Jonca et al. 2002). It is believed to behave as a keratinocyte adhesion molecule and is encoded by CDSN gene. In vitro and in vivo studies reported mutations in CDSN to induce toxicity in hair follicles and trigger HSS by causing abnormal proteolysis of Wt corneodesmosin, which results in truncated fragments that aggregate to form oligomers in the superficial dermis and the periphery of hair follicles (Levy-Nissenbaum et al. 2003).
2.9 Other Disorders
2.9.1 Immunoglobulin Light Chain Amyloidosis
Immunoglobulin light chain amyloidosis is a clonal but non-proliferative plasma cell disorder, illustrated by the pathologic production of fibrillar proteins containing monoclonal light chains which deposit in various tissues (Rosenzweig and Landau 2011). Clonal population of bone marrow plasma cells that produces a monoclonal light chain of κ or λ type either as an intact molecule or a fragment is the characteristic feature of this disorder (Gertz 2013). Like other proteopathies, in this case also, the light chain Ig protein misfold forms an insoluble β-pleated sheet rich protein deposits which then cause organ dysfunction. In the absence of clonal plasma cells in the bone marrow, light chain amyloid may localized to a single site, most often the skin, larynx, or urinary tract (Rosenzweig and Landau 2011). The clinical features indicative of light chain amyloidosis are nonischemic cardiomyopathy with “hypertrophy” on echocardiography, nondiabetic nephrotic syndrome, chronic inflammatory demyelinating polyneuropathy, weight loss, hepatomegaly, increased alkaline phosphatase value with no imaging abnormalities of the liver, edema, or paresthesias (Gertz 2012). The light chain origin of an amyloid deposit can be confirmed with immunohistochemistry or immunogold assay (Gertz 2011). Current treatment approaches for light chain amyloidosis focus on eradication of the monoclonal plasma cell population and suppression of the pathologic light chains which can result in organ improvement and extend patient survival (Rosenzweig and Landau 2011).
3 Common Origin of Proteopathies
The clinical symptoms realized in almost all proteopathies are either because of cellular protein deficiency, owing to its enhanced degradation, or because of misfolded protein fibrils and oligomer mediated cytotoxicity. However, in certain cases, impaired protein quality control system is the underlying cause for protein enhanced degradation or accumulation of unfolded proteins, thereby triggering proteopathies. These common origins have been discussed in detail in the following section.
3.1 Impairment of Protein Quality Control System
Protein quality control (PQC) system is formed by refined alliance between molecular chaperones localized in cytosol (prefoldin, crystallins, heat-shock proteins, Hsc70) and ER (Grp94, Bip, calnexin), and PQC system including UPS and autophagy. Chaperone-targeted proteolysis is aimed at maintaining protein homeostasis by clearing short-lived or mislocated Wt proteins, overexpressed proteins, proteins that are unable to attain their native conformation due to mutations or posttranslational modifications or interaction with other proteins, damaged proteins that could harm cell, and protein aggregates. Such proteins are initially polyubiquitinated via multienzymatic reactions and are then sent to proteasome for degradation. A recent study done with cardiac proteasomes has reported its different subtypes to have varied susceptibility toward proteasome inhibitors, suggesting that proteasomes are differentially composed and modified to degrade different families of substrates (Kloss et al. 2010). Autophagy provides clearance route for many cytosolic misfolded proteins via micro autophagy, macroautophagy, and chaperone-mediated autophagy. Chaperone-mediated autophagy involves binding of misfolded or unfolded proteins with the receptor protein present on the lysosomal membrane, thereby triggering autophagosome formation, which then moves into lysosomal lumen for degradation. Yeast and cell line model studies have suggested preferential deposition of terminally aggregated proteins like mutant htt and PrPSc in perivacuolar inclusions (Kaganovich et al. 2008).
Impairment or overwhelming of PQC due to aging, oxidative stress, inflammation, blockage by misfolded proteins, etc. can lead to accumulation of abnormal proteins in cell, causing eccentric inclusion formation furthering cellular damage. For instance, atypical PQC is a common occurrence in cardiac diseases including hypertrophy, heart failure, cardiomyopathy, and atherosclerosis. This is supported by the consensus of the chaperone studies which gave the impression that chaperone’s loss of activity compromises heart’s ability to handle stress, whereas chaperone’s presence prevents from cell injury (Su and Wang 2010). Another example that explains the role of PQC impairment in development of protein misfolding diseases is of familial ALS, which is also caused due to failure of UPS to degrade polyubiquitinated, misfolded SOD1. This conception is backed by the discovery of familial ALS-linked mutations in genes encoding for ubiquilin 2, phosphatidylinositol 3,5-bisphosphate-5-phosphatase, charged multivesicular body protein 2b, valosin-containing protein, and optineurin, since all these proteins are directly involved in protein homeostasis (Robberecht and Philips 2003).
However, certain terminally aggregated proteins are resistant to proteasomal degradation; as in the case of HD, proteasome’s active site is incapable of cleaving polyQ extensions of mutant htt, thereby preventing its clearance from the cell (Palombella et al. 1989). Furthermore, current developments done towards understanding the mechanism underlying retinal dystrophies such as McKusick-Kaufman syndrome, Leber congenital amaurosis, retinitis pigmentosa, and Bardet-Biedl syndrome have revealed their appearance to be associated with mutations in the gene encoding opsin chaperones RP2, MKKS, and AIPL1 (Chapple et al. 2001). All this data clearly suggests a prominent role of PQC system in preventing the development of various proteopathies. Unambiguous understanding of the PQC modulation pathways would reveal several therapeutic targets.
3.2 Enhanced Degradation of Proteins
The highly efficient protein quality system with the intention to avoid development of antigenic or misfolded proteins degrades significant number of proteins prior to their maturation. This protects the cell from harmful protein inclusions and proves advantageous against pathogenic infections, where fragments of non self-proteins degraded via UPS are presented as antigen to prompt immune system. But, this stringent scrutiny of PQC sometimes could prove detrimental. For instance, the deletion mutants of gene encoding α-subunit of the lysosomal enzyme β-hexoaminidase associated with Tay Sach’s disease expresses truncated protein, which being rejected by PQC becomes deficient in the cell, and triggers associated clinical symptoms (Lau and Neufeld 1989). The functional loss of mutated proteins because of its retention and enhanced degradation in the ER by cell’s PQC is a common etiology for various other disorders including emphysema, cystic fibrosis, congenital sucrase-isomaltase deficiency, etc.
Sometimes, pathogens and misfolded proteins prevent themselves by utilizing host’s PQC against them, for instance E6 genes of both high-risk and low-risk human papilloma viruses (HPV) have been reported to immortalize human mammary epithelial cells by inducing degradation of p53 protein in vivo (Band et al. 1993). Similarly, HPV-16 E7 protein was found to enhance the degradation of retinoblastoma protein as an additional mechanism of oncogenic transformation (Boyer et al. 1996). Furthermore, a recent investigation done to understand the contribution of ApoE in sporadic AD revealed ApoE to promote β-amyloid degradation by modulating microglial cholesterol levels (Lee et al. 2012).
3.3 Aggregate/Amyloid Formation
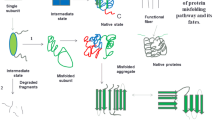
Proteins when not properly folded and prevented from cell’s refolding or degradation mechanisms start intramolecular interactions with other unfolded proteins via their exposed hydrophobic regions, spontaneously leading to their aggregation. It is generally known that mutations, genetic alterations, or hostile solvent (rarely) are the basic cause of protein misfolding. Most of the disease-causing genetic alterations do not allow protein to fold into its lowest free energy native state, thereby accumulating non-functional protein intermediates which are either degraded by cell’s PQC or aggregate to form fibrils. Initially, these intrinsically trapped species aggregate to form soluble oligomers, which then further self-associate to form insoluble amyloid fibrils. Amyloids are ordered aggregates having no universal tertiary or quaternary structure, but are enriched with cross beta-sheet structure with the spasmodic presence of stabilizing rows of hydrophobic interactions that run along the fibril axis (except in case of htt). Under electron microscope, they appear as unbranched fibrils with a consistent diameter of 7.5–12 nm and give apple-green birefringence with histological dyes such as congo red. The process of amyloid fibril formation follows nucleated growth mechanism, which includes a lag phase followed by a rapid exponential growth phase of fibril formation. Lag phase or the time required by peptides to form “nuclei” is the rate-limiting step, while growth phase is the time required by monomers and oligomers to irreversibly associate with the core. However, certain mutations and addition of preformed fibrillar species are known to decrease or eliminate the lag phase, indicating that in these cases amyloid formation is no longer dependent on the formation of a nucleus. Since this process follows second order kinetics, the initial concentration of misfolded proteins is an essential factor to determine its fate.
The eccentric attribute of aggregation prone proteins is their structural flexibility, which enhances their tendency to self-associate in the presence of external stimulants such as mutation, local environment, protein cross-linking with transglutaminase, redox changes, and posttranslational modifications. Presence of phosphorylated, nitrated, and oxidized forms of aSyn in the Lewy bodies and hyper-phosphorylated tau protein in neurofibrillary tangles has suggested an essential role of posttranslational modifications in modulating fibrillation. This is supported by the studies done on fly and mouse models of SCA, where blockage of Ataxin-1 phosphorylation was observed to reduce the extent of inclusion formation and degeneration of purkinje neurons (Ross and Poirier 2004). Prion proteins have been also reported to adopt many of the physical characteristics of PrPSc after posttranslational modifications (Dear et al. 2007). Furthermore, sumoylation of htt protein is believed to decrease its ability to aggregate and increase neurodegeneration by competing with ubiquitin for binding at identical target lysines, preventing its degradation by proteasome (Steffan et al. 2004).
Role of hydrophobicity of amino acid sequence as an important determining factor came to attention with the finding that mutations done in the regions responsible to form nuclei decrease the aggregation propensities, when the hydrophobicity is increased (or decreased) at the site of mutation (Chiti and Dobson 2006). Charge of a protein is another essential factor, as we all know that extreme high or low charge of a protein would impede its self-association. This is augmented by our understanding that interaction of a polypeptide with macromolecules having high compensatory charge increases their aggregation potential. Moreover, the tendency of a protein to attain beta-sheet conformation rather than alpha-helical structure is known to increase their chance of getting aggregated. It seems during the course of evolution, certain proteins have developed in such a way that their propensities to form beta sheet structure are avoided. It is assumed that because of these reasons most of the proteins acquire higher net charge and lower hydrophobicity than natively folded proteins to attenuate their aggregation tendencies in normal physiological conditions (Chiti and Dobson 2006; Uversky 2002).
4 Cellular Mechanisms of Toxicity
It is interesting that how the conduit from protein misfolding to proteopathy development follows almost similar chain of cytotoxic events, irrespective of the organ system affected. This section elaborates such toxicity causing mechanisms or events, such as functional disruption of cell organelles, protein quality control system and immune system, enhanced oxidative stress, generation of amyloid pores, etc.
4.1 Oxidative Stress and Mitochondrial Dysfunction
Mitochondria are ubiquitous intracellular organelles that perform various essential functions inside the cell such as ATP production, cellular calcium buffering, provision of free radicals, and apoptosis regulation. Its dysfunctioing due to the presence of protein aggregates can lead to number of deleterious consequences, such as impaired calcium balance, oxidative stress, and activation of the mitochondrial permeability transition pore (Shigenaga et al. 1994). Under oxidative stress, cellular ROS level elevates, attacking macromolecules like proteins, lipids, nucleic acids, etc. ROS-mediated lipid peroxidation is a known complication of pulmonary proteopathies, which imbalances phospholipid turnover and could rupture cell membranes. Moreover, abrupt oxidation of proteins via ROS could alter their structure so that either they become target for UPS mediated degradation or accumulate to form toxic aggregate species, thus perturbing physiological pathways and furthering cellular damage (Wilck and Ludwig 2014). Its abnormal functioning can also attenuate ATP production, causing reduced proteasome activity and enhanced retention of misfolded proteins in the cell, thereby strengthening the vicious circle of protein aggregates→mitochondrial dysfunction→oxidative stress→UPS failure→protein aggregates.
However, the mechanism of protein aggregation mediated PQC dysfunctioning is yet not clear and is a key area of interest, as it can provide with novel potential therapeutic targets. aSyn aggregates, most probably their protofibrils are believe to do so, by down regulation of complex I activity; mitochondrial fragmentation and excessive mitophagy. However, in the beginning, it was assumed that aSyn disrupts mitochondrial respiratory chain system, located between the outer and inner membranes, by binding with membrane phospholipids. But, a contemporary study done on single SN neurons has indicated that there is no such direct association as they had observed increased levels of respiratory chain complex subunits within neurons containing aSyn pathology (Reeve et al. 2012). On top of that, a recent study done with mice expressing A53T aSyn variant revealed age-dependent changes in both mitochondrial morphology and proteins regulating mitochondrial fission and fusion, indicating aSyn to impair normal mitochondrial dynamics. Additionally, the existence of histological and morphological changes in mitochondria in normal myoblasts with transgenic APP gene had shown involvement of mitochondrial dysfunctioning in AD pathogenesis. Studies done with ALS patients, mice and cellular models have also reported enhanced calcium levels and reduced respiratory chain complexes I and IV activity in mitochondria (Wijesekera and Leigh 2009). Similar observations were reported in CFTR deficient cells, where elevated ROS levels altered ATP utilization rate, impaired calcium metabolism, and perturbed electron transport system (Velsor et al. 2006).
4.2 Cell Membrane Disruption/Amyloid Pores
Membrane permeabilization is a common element of misfolded protein toxicity and is known to be caused via formation of annular pores in cell membranes by oligomeric species of various proteins such as Aβ, IAPP, and aSyn. This can further cause various deleterious effects in cell, for example, Ca2+ immobilization, ER induction, ROS generation and ultimately, cell death (Herczenik and Gebbink 2008). Apart from dopamine/aSyn adduct formation in the cell, another reason for dopaminergic cell death in case of PD is believed to be disruption of ion gradients maintaining neuronal homeostasis because of membrane permeabilization caused by oligomeric aSyn.
4.3 Immune Dysfunction
In addition to their direct toxic effects, misfolded proteins can overwhelm immune system and instigate autoimmune responses. In the brain of AD patients, neuroinflammatory responses are manifested as local stimulation of the complement system, acute-phase responses, enhanced C-reactive protein expression, and activation of inducible nitric oxide synthase and prostaglandin generating cyclooxygenase-2. It has been found that Aβ aggregates trigger microglial cell activation leading to release of a variety of proinflammatory mediators including, complement factors, cytokines, secretory products ROS, and nitric oxide, ultimately causing neuronal cell degeneration (Hickman and El Khoury 2013). Similar kind of chronic inflammation is also visible in most of the other protein misfolding diseases, including ALS, PD, prion diseases, atherosclerosis, cystic fibrosis, etc. However, in the case of atherosclerosis and diabetes mellitus, inflammatory processes are mediated by macrophages not microglial cells. Interestingly, mutant htt seems to have some yet unknown arsenal via which it could compromise immune responses, cause motility and migration deficits in immune cells, thereby making host system susceptible to immune deficiency disorders (Kwan et al. 2012).
4.4 Damaged Protein Quality Control System
Impairment of various components of PQC system due to the presence of protein aggregates is a common toxic insult reported in proteopathies. While healthy PQC system could eliminate toxic aggregates, perturbing its function is sufficient enough to compromise normal cellular homeostasis, thus making them an important target for therapeutic intervention. In cardiomyocytes, reduction of protein aggregation or chaperone overexpression has been reported to prevent proteasomal functional insufficiency, indicating impairment of PQC system to be mediated by aggregates. Few studies have suggested aggregates of polyglutamine expanded proteins like ataxin 1 to derange UPS function (Duenas et al. 2006). aSyn linked toxicity is also seen to be mediated by modulation of PQC mechanisms. aSyn overexpression studies has shown its involvement in the retention of damaged mitochondria in the cytosol by enhancing insolubility of Parkin, an E3 ligase, which leads to aberrant accumulation of toxic proteins like aminoacyl-tRNA synthetase, the far upstream element binding protein 1 and Paris, deregulating mitobiogenesis and ROS metabolism (Rochet et al. 2012). Brain tissues in AD show decreased content of Beclin 1, a protein involved in the initiation of autophagy, thereby suggesting autophagic lysosomal system impairment. . This was supported by the finding that transgenic mice overexpressing APP due to a deletion of one Beclin-1 allele increases the rates of the formation of amyloid plaques and accumulation of morphologically abnormal lysosomes.
4.5 Co-aggregation
Most of the misfolded protein aggregates acquire a peculiar ability to interact with the Wt proteins and coaggregate them as well, leading to fatal insufficiency of that protein along with the enhancement of cellular aggregate burden. Similar mechanism is expended by mutated p53 protein aggregates to coaggregate with Wt protein in healthy cells and spread cancer (Forget et al. 2013). Studies done with polyQ htt protein aggregates suggest primary composition of aggregated peptide to be an essential factor for determining its coaggregating propensities (Bak and Milewski 2010). However, in some of the cases, aggregates have been observed to sequester not only Wt proteins but also other cellular proteins, as in the case of Li-fraumeni syndrome, where p53 aggregates have been observed to nonspecifically interact and coaggregate with other tumor suppressor proteins like p63 and p73 thereby elevating cytotoxicity (Xu et al. 2011). Similar observations were made in a recent finding, where artificial protein aggregates displayed higher tendencies to coaggregate with crucial proteins such as those involved in transcription and cytoskeleton stabilization, explaining the intensified toxic effect on several cellular mechanisms (Olzscha et al. 2011). It has been alluded that proteins usually coaggregate with oppositely charged proteins due to nonspecific hetero-interchain interactions (Trivedi et al. 1997). In physiological conditions, chaperones prevent such conspicuous interactions among proteins, and in diseased cells they bind with aggregates to shield other proteins (Doyle et al. 2013).
5 Therapeutic Advances
This part of the chapter overviews the current treatment strategies employed against proteopathies. The current advances, advantages, and disadvantages of those strategies have also been mentioned.
5.1 Immunotherapy
Active and passive immunizations are aimed at inhibiting generation of protein aggregation and removal of aggregates that are already formed. The concept of passive immunotherapy includes raising monoclonal antibodies against toxic proteins which can then be injected into patients to decrease their amyloid or aggregate burden. Formerly, the so-called “first-in-man” and “first-in-kind” clinical trial for the development of a PD vaccine, PD01A has been initiated. This vaccination aims to educate the immune system to generate antibodies directed against aSyn, and hence neutralize its toxic impact (PRNewswire 2012). However, for AD, several types of Aβ peptide immunotherapy are under investigation, including direct immunization with synthetic intact Aβ1-42, active immunization involving the administration of synthetic fragments of Aβ peptide conjugated to a carrier protein and passive administration with monoclonal antibodies directed against Aβ peptide (Schenk 2002). The first clinical trial was done with anti-Aβ vaccine AN1792, but due to the development of meningoencephalitis in a small percentage of patients all study dosing was halted. Moreover, immunization with Aβ1-42 was efficient in clearing amyloid plaques in patients with AD, but prevented progressive neurodegeneration (Delrieu et al. 2012). Several monoclonal antibodies for AD had also been tested: solanezumab (LY-2062430), PF-04360365, bapineuzumab (AAB-001), R-1450 (RO-4909832), GSK-933776, and MABT-5102A (Mangialasche et al. 2010). Out of these, solanezumab seems to be very promising as it presents less CNS adverse events and is the first therapeutic drug to be evaluated in the antiamyloid treatment in asymptomatic AD prevention clinical trial. Investigators are now planning to test an Aβ-clearing drug in older people thought to be in the presymptomatic stage of AD (Alzforum 2013). Anti-inflammatory therapy is also seen as a potential therapeutic intervention for cardiac amyloidosis. Patients with AL amyloidosis have been reported to survive longer after treatment with melphalan and prednisolone (Kyle et al. 1999). Prednisolone acts by reducing the transcription of inflammatory mediators, cycloxygenase 2, and several cytokines including TNFα, IL-1, and IFNγ, while melphalan is a protein synthesis inhibitor. Additionally, anti-TNFα agents have been reported to substantially suppress the production of serum amyloid A, the protein associated with AA amyloidosis (Banypersad et al. 2012).
Recently, a therapeutic monoclonal antibody that is reactive with all types of amyloid has lately been developed by targeting serum amyloid A protein (SAP) because it is a universal constituent of all amyloid deposits and an excellent immunogen (Bodin et al. 2010). To ensure that anti-SAP antibodies reach residual SAP in the amyloid deposits, circulating human SAP can be depleted by the bis-d-proline compound CPHPC. This novel combined therapy has shown encouraging results in mice models and is expected to be effective for all forms of human systemic and local amyloidosis.
5.2 Antioxidative Therapy
ROS are generally harmful, which makes antioxidant defenses essential and then promising therapeutic targets. These agents can be classified on the basis of their modus operandi – (a) compounds preventing free radical formation; (b) compounds neutralizing free radicals by chemically interacting with them; and (c) compounds enhancing the ROS secondary metabolites thereby limit the extent of cellular damage. Glutathione (GSH), a potent antioxidant belonging to the second category of antioxidant agents, has been shown to relieve oxidative stress in neuronal cells. Recently developed l-dopa-GSH co-drugs with blood-brain barrier-crossing properties have been reported to effectively prevent MAO-mediated metabolism of dopamine (Cacciatore et al. 2012). Furthermore, methionine sulfoxide reductase, an antioxidant repair enzyme, had also been shown to assist the inhibition of aSyn fibrillation, and neurotoxicity by repairing oxidatively damaged protein and by depleting ROS (Liu et al. 2008). Antioxidant therapy has also been tried to treat AD either by increasing the pool of endogenous antioxidants (e.g. vitamins, coenzyme Q10, or melatonin) or by the intake of dietary antioxidants, such as phenolic compounds of flavonoid or non-flavonoid type. However, in clinical trials, these agents had shown limited success may be because of poor distribution or in case of ND, agent’s inherent difficulty to cross the brain–blood barrier. Shockingly, clinical trials to test the efficacy of antioxidants in treating diabetes patients reported contradictory results, describing elevated antioxidative stress agents to be negatively correlated with oxidative stress culmination (Golbidi et al. 2011). But, such results could be because of inadequate study design or selected targets. Further, clinical trial studies are required to clearly understand the importance of antioxidative therapy.
5.3 Modulating PQC Mechanisms
5.3.1 Ubiquitin Proteasome System
Most of the therapeutic strategies targeting UPS aim at enhancing their efficiency to remove misfolded proteins and their deposits from the cell. Following this stratagem, various modulators of UPS have been found and are being clinically tested. Genome-wide association studies have identified negative regulator of ubiquitin-like protein 1 (NUB1) as a modifier of mutant htt abundance. NUB1 overexpression has been reported to rescue neuronal cells from cell death by increasing proteasomal degradation of mutant htt, suggesting interferon-β (known inducer of NUB1) as a potential therapeutic agent in HD treatment (Lu et al. 2013). Similarly, upregulation of the 11S proteasome in mice models with desmin-related cardiomyopathy had been demonstrated to reduce aberrant protein aggregation and prevent cardiac dysfunction (Li et al. 2011). Contrastingly, bortezomib, a selective 26S proteasome inhibitor, has emerged as an effective drug against cancer and cardiac amyloidosis, and its use for treating multiple myeloma has been recently approved by US food and drug administration (Adams and Kauffman 2004). It seems that impairing proteasomal function enhances ER stress and encourages cell to undergo apoptotic cell death without affecting overall functioning of the organ system affected (Hedhli et al. 2008). Currently, for treatment of patients suffering from cardiac amyloidosis with bortezomib in conjugation with other agents is undergoing phase II and phase III clinical trials (Banypersad et al. 2012).
5.3.2 Molecular Chaperones
Upregulation of molecular chaperones is considered to inhibit aggregation by facilitating the refolding of misfolded proteins or by directing the aggregated proteins to cellular clearance pathways. Using various cellular models and in vitro studies, upregulation of heat shock proteins like Hsp70, Hsp40 and Hsp27 have been shown to attenuate aSyn fibril formation (Rochet et al. 2012). Also, Hsp70 overexpression is found to be an effective treatment in mouse model of SCA1 disease and fly model of HD (Chaudhuri and Paul 2006). Interestingly, intermediate concentrations of molecular chaperone Hsp104 in yeast models have been reported to enhance the propagation of yeast non-Mendelian factor [psi+], analogue of mammalian prions, while at higher concentrations, it cleared [psi+] from the cell. These results suggest modulating cellular levels of molecular chaperone as another potential therapeutic target (Chernoff et al. 1995).
5.3.3 Autophagy
Rapamycin is a potent autophagy inducer that works via inhibiting mTOR’s (molecular target of rapamycin) kinase activity. Studies done with cellular models have shown rapamycin treatment to enhance degradation of toxic fibrillated proteins such as mutant ataxin, aSyn, htt, and tau (Hochfeld et al. 2013). Similar results were obtained with fly and mouse models of HD, where rapamycin effectively cleared mutant htt from the cell, reducing aggregate load and toxicity in the cell. This beneficiary was exclusively brought about by autophagy induction as this drug did not prove effective in the fly models of various proteopathies with reduced activity of autophagy genes (Wang et al. 2009). Rapamycin analogue CCI-779 had also been shown to reduce levels of toxin proteins in drosophila models of SCA3 and HD (Menzies et al. 2010; Ravikumar et al. 2004). Since mTOR is also involved in ribosome biogenesis and protein translation, inhibiting its activity for a long duration of time leads to deleterious side effects. Alternatively, various other autophagy inducers lacking these side effects have been introduced such as lithium, sodium valproate, carbamazepine, xestospongin B, and rilmenidine have also been introduced (Hochfeld et al. 2013). They have been shown to reduce toxicity and clear aggregates in fly models of HD, and rilmenidine being the drug with minimal side effects is currently under clinical trials with HD patients (Rose et al. 2010).
5.4 Gene Therapy
The main aim of gene therapy is to replace the mutant gene with the Wt copy wherever it is needed in the body. However, it is the delivery of gene to the target organ that makes it difficult for treating every genetic disease. The major breakthrough came in this regard with the development of a modified virus carrying Wt CFTR gene to be delivered into the lungs of CF patients in the form of aerosols (Laube 2014). Other than viruses, liposomes have also emerged as an alternative vector to deliver Wt CFTR gene (Prickett and Jain 2013). The largest clinical trial of gene therapy for cystic fibrosis is currently being conducted by the UK Cystic Fibrosis Gene Therapy Consortium. It involves analyzing cystic fibrosis patients using an inhaler to breathe in a working copy of the cystic fibrosis gene once a month for a year. Other than CF, discoveries in this regard have been made to treat HD and PKU (Chowhan et al. 2013). Adeno-associated virus-mediated delivery of PAH gene for long-term improvement of PAH deficiency in PKU has been studied via mouse models, though the investigations have not advanced to human trials stage (Ding et al. 2005). However, the clinical trials for gene-therapy mediated treatment of HD have been started in 2014 by Roche and Isis Pharmaceuticals, Inc.
5.5 Reduction/Inhibition of Amyloid Formation
5.5.1 RNA Silencing or Antisense Therapy
The rationale for using antisense therapy against proteopathies is that the RNA interference (RNAi) mediated allele-specific silencing of the disease associated gene will repress gene expression of mutant protein and maintain heterozygous levels of Wt protein in the cell. This will eradicate the possibility of accumulation of misfolded proteins in the cell. Novel drugs aimed at utilizing RNA silencing and antisense oligonucleotide therapies for reducing production of TTR, protein associated with ATTR cardiac amyloidosis has been developed. ALN-TTR01, a systemically delivered RNAi therapeutic, is already in phase I clinical trial (Benson et al. 2006). In addition, the first clinical trial of intrathecal delivery of an antisense oligonucleotide, ISIS 333611, against mutant SOD1 messenger RNA has revealed this antisense therapy to be safe in ALS patients (Miller et al. 2013). It is believed that the gene silencing strategy cannot be utilized in diseases with defective PQC or where its efficiency has declined with age, such as in most cases of PD and AD (Gregersen 2006). But, recently a new specific SOFA-hepatitis delta virus ribozyme has been engineered that was exploited as RNA silencing tool against APP gene in AD cell models and demonstrated to be effective in decreasing variant APP mRNA and protein levels, as well as misfolded Aβ levels (Ben Aissa et al. 2012).
5.5.2 Stabilization of Fibril Precursor Protein
It is known that amyloid formation requires conversion of majority of alpha helices of respective precursor protein into β sheets to form fibril precursor protein which has an alternate aggregate-prone conformation that triggers aggregate accretion and propagation seeding reaction. It has been hypothesized that if we stabilize this soluble precursor protein by binding it to some pharmaceutical, then this process of amyloidosis will be inhibited. This has been lately explored in ATTR cardiac amyloidosis, where effects of drugs like diflunisal and tafamidis on TTR structure stability are going on (Banypersad et al. 2012). Higher affinity superstabilizers, palindromic ligands have also been developed that inhibit amyloid formation in ATTR cardiac amyloidosis through their protein stabilization effect (Kolstoe et al. 2010). Additionally, small peptides are being synthesized that can bind specifically with the fibril precursor protein to block and/or reverse its abnormal conformational change (Estrada and Soto 2006).
Alternatively, extensive investigations have led to the finding of small compounds, such as indomethacin, Congo red, 2,4-dinitrophenol, cyclodextrin derivatives, curcumin, meclocycline, di- and tri-substituted aromatic molecules, and hematin, which can inhibit protein aggregation by stabilizing native conformation of the aggregation prone protein to inhibit their oligomerization (Miroy et al. 1996; Necula et al. 2007).
5.5.3 Modulation of Cellular Enzymes
A recent finding of a specific mutation in the gene encoding for APP that can significantly decrease its cleavage by beta-secretase and confer resistance to the development of AD in patients has reinforced the hypothesis that the inhibition of Aβ cleaving secretases is an important disease modifying approach (Jonsson et al. 2012). Following this trend, Merck USA has started largest phase II/II clinical trial of MK8931, a BACE-1 (β-secretase APP cleaving enzyme) inhibitor in AD (Merck&Co. 2013), instead of the failure of phase 3 trial in 2010 of a gamma-secretase inhibitor drug, semagacestat in prodromal AD because of its adverse effects seen on worsening of cognition (Mckee 2010). Additionally, anticholinesterase inhibitors are currently approved by FDA for AD treatment. They are believed to work either by rescuing AD brains from acetylcholine deficiency or by preventing abrupt neuronal stimulation by antagonizing NMDA-type glutamate receptors (Chen et al. 2014a). Since hyperphosphorylated tau are present in the neurofibrillary tangles in brains of AD patients, inhibitors of tau kinases are also considered an important therapeutic targets, but it is still a relatively unexplored avenue (Huang and Mucke 2012). Recently, enhanced GCase levels have been reported to revert aSyn related pathophysiological attributes and advance cognition in mouse models of gaucher’s disease. It has been hypothesized that correcting GCase deficiency could affect SNCA gene expression and prevent PD development (K Chowhan et al. 2014; Schapira and Gegg 2013). Utilizing cellular enzymes as therapeutic targets have also been experimented for treating PKU, where the introduction of L-Phe metabolizing enzyme, phenylalanine ammonia lyase, in intestinal lumen was observed to efficiently metabolize and digest L-Phe (Harding 2008).
5.5.4 Tetracyclines
The tetracyclines are a family of broad spectrum antibiotics that include tetracycline, doxycyclin, and minocycline. Studies done with transgenic mice models have revealed tetracycline to inhibit IAPP amyloid formation by hampering misfolding of soluble oligomers, and hence delay development of type-2 diabetes (Aitken et al. 2010). Since they can cross blood–brain barrier, they have been utilized in neurodegenerative diseases also. Minocycline has been shown to prevent Aβ and tau protein accumulation in AD models (Noble et al. 2009), while tetracycline was observed to inhibit the conversion of prion protein, PrPC into PrPSc form and hinder prion aggregation (Tagliavini et al. 2000). Furthermore, doxycycline displayed prolonged survival and delayed onset of disease in animal models of prions (Forloni et al. 2002).
5.5.5 Osmolytes
Osmolytes or chemical chaperones are small organic compounds known to have the capability to correct the altered structure of misfolded proteins by influencing the rate or fidelity of their folding reaction, thereby preventing UPS degradation mediated cellular deficiency of that protein. They have been reported to converse the intracellular retention of several different misfolded proteins such as prion protein, α-galactosidase A, aquaporin-2, CFTR, vasopressin V2 receptor, p53, AAT, and P-glycoprotein (Chaudhuri and Paul 2006). Osmolytes such as glycerol and 4-phenylbutyric acid have been reported to increase the secretion efficiency of Wt as well as variant AAT in cell lines and transgenic animals. In CF, detrimental effects of ΔF508 mutation can be reverted by treating cells with osmolytes like glycerol, dimethyl sulfoxide (DMSO), trimethylamine-N-oxide (TMAO), and deuterated water (Brown et al. 1996). However, effects of some osmolytes like TMAO on protein aggregation are concentration dependent. TMAO at higher concentration (>3 M) leads to tight folding of aSyn eventually forming stable oligomers and decreased fibrillation rate, but at low concentrations it forms partially folded intermediate which accelerates fibrillation (Fink 2006). Because of these complex effects of osmolytes on aggregation kinetics, utilizing it for treatment will require very stringent investigations. Additionally, target-based virtual screening has shown pharmacological chaperones as potential candidates to treat PKU via stabilizing PAH and preventing its stability (Harris 2014).
5.5.6 Polyamines
Effects of various naturally occurring cosolutes like polyamines on protein aggregation are extensively investigated in order to find a therapeutically important molecule. A recent gene expression analysis done on differentially affected brainstem regions of PD patients had implicated higher polyamine levels to be one of the causes for induction of aSyn aggregation (Lewandowski et al. 2010). Furthermore, it has been found that the pathological length polyQ proteins like mutated htt induce polyamine synthetic pathway, which further enhance their aggregation and trigger apoptotic cell death (Chowhan and Singh 2012). Thus, indicating reduction of polyamine levels as a potential therapeutic target for PD and HD.
6 Conclusions and Future Perspectives
Protein misfolding is indeed a major etiology for several localized and systemic diseases. Genetic mutations, cellular stress, failed unfolded protein response, and overwhelmed protein folding and trafficking system all together or independently instigate accumulation of misfolded protein aggregates and cause cellular deficiency of its native counterpart. This directly influences the proper functioning of cellular mechanisms involving the misfolded protein in addition to the cytotoxicity caused by its oligomers and amyloid fibrils. Plenty of resources have been expended up till now to find cure for these diseases but what we have achieved so far are more or less symptomatic treatments, nothing providing permanent relief. The novel therapeutic strategies like gene therapy, peptidomimetics, protein stabilizing agents like osmolytes and polyamines, etc. though seem promising have yet to prove their competence in clinical trials. We believe that in addition to counteracting the toxic effects of protein oligomers and amyloid fibrils, developing novel strategies focused on targeting the primary cause of proteopathies, i.e. protein misfolding, will prove to be a universal remedy for all protein misfolding-related disorders. This could be done either by developing agents that could augment correct folding of mutated proteins or by employing tactics to decrease the load of toxic protein oligomers and fibrils. Furthermore, it has to be understood that to discover new treatment approaches, we have to unravel the entire physiological phenomenon involved with a disease, and to gain all this information we require strenuous mechanistic investigations.
References
Abedini A, Schmidt AM (2013) Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett 587(8):1119–1127
Adams J, Kauffman M (2004) Development of the proteasome inhibitor velcade (bortezomib). Cancer Invest 22(2):304–311
Aguzzi A, Calella AM (2009) Prions: protein aggregation and infectious diseases. Physiol Rev 89(4):1105–1152
Ahner A, Nakatsukasa K, Zhang H, Frizzell RA, Brodsky JL (2007) Small heat-shock proteins select deltaf508-cftr for endoplasmic reticulum-associated degradation. Mol Biol Cell 18(3):806–814
Aitken JF, Loomes KM, Scott DW, Reddy S, Phillips AR, Prijic G, Fernando C, Zhang S, Broadhurst R, L’Huillier P, Cooper GJ (2010) Tetracycline treatment retards the onset and slows the progression of diabetes in human amylin/islet amyloid polypeptide transgenic mice. Diabetes 59(1):161–171
Alzforum (2013) Solanezumab selected for alzheimer’s a4 prevention trial. Conference coverage series: Human Amyloid Imaging (HAI) http://www.alzforum.org/news/conference-coverage/solanezumab-selected-alzheimers-a4-preventiontrial
Angot E, Steiner JA, Lema Tome CM, Ekstrom P, Mattsson B, Bjorklund A, Brundin P (2012) Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS One 7(6), e39465
Anukanth A, Khorana HG (1994) Structure and function in rhodopsin. Requirements of a specific structure for the intradiscal domain. J Biol Chem 269(31):19738–19744
Back SH, Kaufman RJ (2012) Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem 81:767–793
Bak D, Milewski M (2010) The composition of the polyglutamine-containing proteins influences their co-aggregation properties. Cell Biol Int 34(9):933–942
Band V, Dalal S, Delmolino L, Androphy EJ (1993) Enhanced degradation of p53 protein in hpv-6 and bpv-1 e6-immortalized human mammary epithelial cells. EMBO J 12(5):1847–1852
Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD (2012) Updates in cardiac amyloidosis: a review. J Am Heart Assoc 1(2), e000364
Bartels T, Choi JG, Selkoe DJ (2011) Alpha-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477(7362):107–110
Bellotti V, Chiti F (2008) Amyloidogenesis in its biological environment: challenging a fundamental issue in protein misfolding diseases. Curr Opin Struct Biol 18(6):771–779
Ben Aissa M, April MC, Bergeron LJ, Perreault JP, Levesque G (2012) Silencing of amyloid precursor protein expression using a new engineered delta ribozyme. Int J Alzheimers Dis 2012:947147
Benilova I, Karran E, De Strooper B (2012) The toxic aβ oligomer and Alzheimer’s disease: an emperor in needs of clothes. Nat Neurosci 15:349–357
Benson MD, Kluve-Beckerman B, Zeldenrust SR, Siesky AM, Bodenmiller DM, Showalter AD, Sloop KW (2006) Targeted suppression of an amyloidogenic transthyretin with antisense oligonucleotides. Muscle Nerve 33(5):609–618
Bi X (2010) Alzheimer disease: update on basic mechanisms. J Am Osteopath Assoc 110(9 Suppl 8):S3–9
Bishop MT, Diack AB, Ritchie DL, Ironside JW, Will RG, Manson JC (2013) Prion infectivity in the spleen of a prnp heterozygous individual with subclinical variant creutzfeldt-jakob disease. Brain 136(Pt 4):1139–1145
Bodensteiner JB (2014) Congenital myopathies. In: Neuromuscular disorders in clinical practice. Springer, New York, Heidelberg, Dordrecht, London, Edition 2nd (2013); pp 1295–1310
Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, Hutchinson WL, Mangione PP, Gallimore JR, Millar DJ, Minogue S, Dhillon AP, Taylor GW, Bradwell AR, Petrie A, Gillmore JD, Bellotti V, Botto M, Hawkins PN, Pepys MB (2010) Antibodies to human serum amyloid p component eliminate visceral amyloid deposits. Nature 468(7320):93–97
Boyer SN, Wazer DE, Band V (1996) E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res 56(20):4620–4624
Brais B, Chrestian N, Dupré N, Bouchard J-P, Rouleau G (2014) Oculopharyngeal muscular dystrophy. In: Neuromuscular disorders in clinical practice. Springer, New York, pp 1277–1283
Brasch F, Birzele J, Ochs M, Guttentag SH, Schoch OD, Boehler A, Beers MF, Muller KM, Hawgood S, Johnen G (2004) Surfactant proteins in pulmonary alveolar proteinosis in adults. Eur Respir J 24(3):426–435
Bridges JP, Wert SE, Nogee LM, Weaver TE (2003) Expression of a human surfactant protein c mutation associated with interstitial lung disease disrupts lung development in transgenic mice. J Biol Chem 278(52):52739–52746
Brown CR, Hong-Brown LQ, Biwersi J, Verkman AS, Welch WJ (1996) Chemical chaperones correct the mutant phenotype of the delta f508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones 1(2):117–125
Brundin P, Melki R, Kopito R (2010) Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol 11(4):301–307
Brunelli R, De Spirito M, Mei G, Papi M, Perrone G, Stefanutti C, Parasassi T (2014) Misfolding of apoprotein b-100, ldl aggregation and 17-β-estradiol in atherogenesis. Curr Med Chem 21(20):2276–2283
Buxbaum J, Jacobson DR, Tagoe C, Alexander A, Kitzman DW, Greenberg B, Thaneemit-Chen S, Lavori P (2006) Transthyretin V122I in African americans with congestive heart failure. J Am Coll Cardiol 47(8):1724–1725
Cacciatore I, Baldassarre L, Fornasari E, Mollica A, Pinnen F (2012) Recent advances in the treatment of neurodegenerative diseases based on GSH delivery systems. Oxid Med Cell Longev 2012, e240146
Chapple JP, Grayson C, Hardcastle AJ, Saliba RS, van der Spuy J, Cheetham ME (2001) Unfolding retinal dystrophies: a role for molecular chaperones? Trends Mol Med 7(9):414–421
Chattopadhyay M, Valentine JS (2009) Aggregation of copper-zinc superoxide dismutase in familial and sporadic als. Antioxid Redox Signal 11(7):1603–1614
Chaudhuri TK, Paul S (2006) Protein-misfolding diseases and chaperone-based therapeutic approaches. FEBS J 273(7):1331–1349
Chen H, Wan Y, Jiang S, Cheng Y (2014a) Alzheimer’s disease research in the future: bibliometric analysis of cholinesterase inhibitors from 1993 to 2012. Scientometrics 98(3):1865–1877
Chen Y, Jastrzebska B, Cao P, Zhang J, Wang B, Sun W, Yuan Y, Feng Z, Palczewski K (2014b) Inherent instability of the retinitis pigmentosa p23h mutant opsin. J Biol Chem 289(13):9288–9303
Cheng C, Xia CH, Huang Q, Ding L, Horwitz J, Gong X (2010) Altered chaperone-like activity of alpha-crystallins promotes cataractogenesis. J Biol Chem 285(52):41187–41193
Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW (1995) Role of the chaperone protein hsp104 in propagation of the yeast prion-like factor [psi+]. Science 268(5212):880–884
Childers M, Eckel G, Himmel A, Caldwell J (2007) A new model of cystic fibrosis pathology: lack of transport of glutathione and its thiocyanate conjugates. Med Hypotheses 68(1):101–112
Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75:333–366
Chowhan RK, Singh LR (2012) Polyamines in modulating protein aggregation. J Proteins Proteomics 3(2):139–148
Chowhan RK, Warepam M, Dar TA, Singh LR (2013) Recent trends in treating neuronal proteinopathies. J Proteins Proteomics 4(2)
Chowhan RK, Mittal S, Dar TA, Kamal MA, Singh LR (2014) Ignored avenues in alpha-synuclein associated proteopathy. CNS Neurol Disord Drug Targets 13(7):1246–1257
Clarkson E, Costa CF, Machesky LM (2004) Congenital myopathies: diseases of the actin cytoskeleton. J Pathol 204(4):407–417
Costa CF, Rommelaere H, Waterschoot D, Sethi KK, Nowak KJ, Laing NG, Ampe C, Machesky LM (2004) Myopathy mutations in alpha-skeletal-muscle actin cause a range of molecular defects. J Cell Sci 117(Pt 15):3367–3377
Das M, Mei X, Jayaraman S, Atkinson D, Gursky O (2014) Amyloidogenic mutations in human apolipoprotein A-I are not necessarily destabilizing-a common mechanism of apolipoprotein A-I misfolding in familial amyloidosis and atherosclerosis. FEBS J 281(11):2525–42
de Klerk E, Venema A, Anvar SY, Goeman JJ, Hu O, Trollet C, Dickson G, den Dunnen JT, van der Maarel SM, Raz V, t Hoen PA (2012) Poly(a) binding protein nuclear 1 levels affect alternative polyadenylation. Nucleic Acids Res 40(18):9089–9101
Dear DV, Young DS, Kazlauskaite J, Meersman F, Oxley D, Webster J, Pinheiro TJ, Gill AC, Bronstein I, Lowe CR (2007) Effects of post-translational modifications on prion protein aggregation and the propagation of scrapie-like characteristics in vitro. Biochim Biophys Acta 1774(7):792–802
Delrieu J, Ousset PJ, Caillaud C, Vellas B (2012) ‘Clinical trials in Alzheimer’s disease’: immunotherapy approaches. J Neurochem 120(Suppl 1):186–193
Ding Z, Georgiev P, Thony B (2005) Administration-route and gender-independent long-term therapeutic correction of phenylketonuria (pku) in a mouse model by recombinant adeno-associated virus 8 pseudotyped vector-mediated gene transfer. Gene Ther 13(7):587–593
Donlon J, Levy H, Scriver CR (2004) Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. Online metabolic and molecular basis of inherited disease. http://ommbid.mhmedical.com/content.aspx?bookid=474§ionid=45374059
Doyle SM, Genest O, Wickner S (2013) Protein rescue from aggregates by powerful molecular chaperone machines. Nat Rev Mol Cell Biol 14(10):617–629
Duenas AM, Goold R, Giunti P (2006) Molecular pathogenesis of spinocerebellar ataxias. Brain 129(Pt 6):1357–1370
El-Agnaf OM, Bodles AM, Guthrie DJ, Harriott P, Irvine GB (1998) The n-terminal region of non-a beta component of Alzheimer’s disease amyloid is responsible for its tendency to assume beta-sheet and aggregate to form fibrils. Eur J Biochem 258(1):157–163
Emery AEH (2002) The muscular dystrophies. Lancet 359(9307):687–695
Estrada LD, Soto C (2006) Inhibition of protein misfolding and aggregation by small rationally-designed peptides. Curr Pharm Des 12(20):2557–2567
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein e genotype and Alzheimer disease. A meta-analysis. Apoe and Alzheimer disease meta analysis consortium. JAMA 278(16):1349–1356
Fink AL (2006) The aggregation and fibrillation of alpha-synuclein. Acc Chem Res 39(9):628–634
Forget KJ, Tremblay G, Roucou X (2013) P53 aggregates penetrate cells and induce the co-aggregation of intracellular p53. PLoS One 8(7), e69242
Forloni G, Iussich S, Awan T, Colombo L, Angeretti N, Girola L, Bertani I, Poli G, Caramelli M, Grazia Bruzzone M, Farina L, Limido L, Rossi G, Giaccone G, Ironside JW, Bugiani O, Salmona M, Tagliavini F (2002) Tetracyclines affect prion infectivity. Proc Natl Acad Sci U S A 99(16):10849–10854
Gersting SW, Kemter KF, Staudigl M, Messing DD, Danecka MK, Lagler FB, Sommerhoff CP, Roscher AA, Muntau AC (2008) Loss of function in phenylketonuria is caused by impaired molecular motions and conformational instability. Am J Hum Genet 83(1):5–17
Gertz MA (2011) Immunoglobulin light chain amyloidosis: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol 86(2):180–186
Gertz MA (2012) Immunoglobulin light chain amyloidosis: 2012 update on diagnosis, prognosis, and treatment. Am J Hematol 87:184–189
Gertz MA (2013) Immunoglobulin light chain amyloidosis: 2013 update on diagnosis, prognosis, and treatment. Am J Hematol 88(5):416–425
Golbidi S, Ebadi SA, Laher I (2011) Antioxidants in the treatment of diabetes. Curr Diabetes Rev 7(2):106–125
Graffmo KS, Forsberg K, Bergh J, Birve A, Zetterström P, Andersen PM, Marklund SL, Brännström T (2012) Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis. Hum Mol Genet dds399
Gregersen N (2006) Protein misfolding disorders: pathogenesis and intervention. J Inherit Metab Dis 29(2–3):456–470
Gustafsson M, Thyberg J, Naslund J, Eliasson E, Johansson J (1999) Amyloid fibril formation by pulmonary surfactant protein c. FEBS Lett 464(3):138–142
Handoko M, Grant M, Kuskowski M, Zahs KR, Wallin A, Blennow K, Ashe KH (2013) Correlation of specific amyloid-beta oligomers with tau in cerebrospinal fluid from cognitively normal older adults. JAMA Neurol 70(5):594–599
Harding C (2008) Progress toward cell-directed therapy for phenylketonuria. Clin Genet 74(2):97–104
Harris GME (2014) Identification of pharmacological chaperones for phenylalanine hydroxylase. A virtual screening approach to discover novel drug candidates for treatment of phenylketonuria. Master thesis, The University of Bergen
Hayden MR, Tyagi SC, Kerklo MM, Nicolls MR (2005) Type 2 diabetes mellitus as a conformational disease. JOP 6(4):287–302
Hector A, Kormann M, Kammermeier J, Burdi S, Marcos V, Rieber N, Mays L, Illig T, Klopp N, Falkenstein F, Kappler M, Riethmueller J, Graepler-Mainka U, Stern M, Eickmeier O, Serve F, Zielen S, Doring G, Griese M, Hartl D (2013) Expression and regulation of interferon-related development regulator-1 in cystic fibrosis neutrophils. Am J Respir Cell Mol Biol 48(1):71–77
Hedhli N, Lizano P, Hong C, Fritzky LF, Dhar SK, Liu H, Tian Y, Gao S, Madura K, Vatner SF, Depre C (2008) Proteasome inhibition decreases cardiac remodeling after initiation of pressure overload. Am J Physiol 295(4):H1385–1393
Herczenik E, Gebbink MF (2008) Molecular and cellular aspects of protein misfolding and disease. Faseb J 22(7):2115–2133
Hickman SE, El Khoury J (2013) The neuroimmune system in Alzheimer’s disease: The glass is half full. J Alzheimers Dis 33(Suppl 1):S295–302
Hochfeld WE, Lee S, Rubinsztein DC (2013) Therapeutic induction of autophagy to modulate neurodegenerative disease progression. Acta Pharmacol Sin 34(5):600–604
Hoffman EP, Brown RH Jr, Kunkel LM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51(6):919–928
Huang Y, Mucke L (2012) Alzheimer mechanisms and therapeutic strategies. Cell 148(6):1204–1222
Inoue Y, Trapnell BC, Tazawa R, Arai T, Takada T, Hizawa N, Kasahara Y, Tatsumi K, Hojo M, Ichiwata T, Tanaka N, Yamaguchi E, Eda R, Oishi K, Tsuchihashi Y, Kaneko C, Nukiwa T, Sakatani M, Krischer JP, Nakata K (2008) Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med 177(7):752–762
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Gotz J (2010) Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142(3):387–397
Ivanova MI, Sievers SA, Guenther EL, Johnson LM, Winkler DD, Galaleldeen A, Sawaya MR, Hart PJ, Eisenberg DS (2014) Aggregation-triggering segments of sod1 fibril formation support a common pathway for familial and sporadic als. Proc Natl Acad Sci 111(1):197–201
Jellinger KA (2011) Interaction between alpha-synuclein and other proteins in neurodegenerative disorders. ScientificWorldJournal 11:1893–1907
Jonca N, Guerrin M, Hadjiolova K, Caubet C, Gallinaro H, Simon M, Serre G (2002) Corneodesmosin, a component of epidermal corneocyte desmosomes, displays homophilic adhesive properties. J Biol Chem 277(7):5024–5029
Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jonsson EG, Palotie A, Behrens TW, Magnusson OT, Kong A, Thorsteinsdottir U, Watts RJ, Stefansson K (2012) A mutation in app protects against Alzheimer’s disease and age-related cognitive decline. Nature 488(7409):96–99
Kaganovich D, Kopito R, Frydman J (2008) Misfolded proteins partition between two distinct quality control compartments. Nature 454(7208):1088–1095
Kahn SE, Andrikopoulos S, Verchere CB (1999) Islet amyloid: a long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes 48(2):241–253
Kholova I, Niessen HW (2005) Amyloid in the cardiovascular system: a review. J Clin Pathol 58(2):125–133
Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, Proukakis C, Quinn N, Lees AJ, Hardy J, Revesz T, Houlden H, Holton JL (2013) Alpha-synucleinopathy associated with g51d snca mutation: a link between parkinson’s disease and multiple system atrophy? Acta Neuropathol 125(5):753–769
Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC (2011) Amyotrophic lateral sclerosis. Lancet 377(9769):942–955
Kloss A, Meiners S, Ludwig A, Dahlmann B (2010) Multiple cardiac proteasome subtypes differ in their susceptibility to proteasome inhibitors. Cardiovasc Res 85(2):367–375
Koenig M, Beggs AH, Moyer M, Scherpf S, Heindrich K, Bettecken T, Meng G, Müller CR, Lindlöf M, Kaariainen H (1989) The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet 45(4):498
Kolstoe SE, Mangione PP, Bellotti V, Taylor GW, Tennent GA, Deroo S, Morrison AJ, Cobb AJ, Coyne A, McCammon MG, Warner TD, Mitchell J, Gill R, Smith MD, Ley SV, Robinson CV, Wood SP, Pepys MB (2010) Trapping of palindromic ligands within native transthyretin prevents amyloid formation. Proc Natl Acad Sci U S A 107(47):20483–20488
Kopito RR (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10(12):524–530
Kosinski-Collins MS, King J (2003) In vitro unfolding, refolding, and polymerization of human gammaD crystallin, a protein involved in cataract formation. Protein Sci 12(3):480–490
Kwan W, Trager U, Davalos D, Chou A, Bouchard J, Andre R, Miller A, Weiss A, Giorgini F, Cheah C, Moller T, Stella N, Akassoglou K, Tabrizi SJ, Muchowski PJ (2012) Mutant huntingtin impairs immune cell migration in Huntington disease. J Clin Invest 122(12):4737–4747
Kyle RA, Gertz MA, Greipp PR, Witzig TE, Lust JA, Lacy MQ, Therneau TM (1999) Long-term survival (10 years or more) in 30 patients with primary amyloidosis. Blood 93(3):1062–1066
Lau MM, Neufeld EF (1989) A frameshift mutation in a patient with Tay-Sachs disease causes premature termination and defective intracellular transport of the alpha-subunit of beta-hexosaminidase. J Biol Chem 264(35):21376–21380
Laube BL (2014) The expanding role of aerosols in systemic drug delivery, gene therapy and vaccination: an update. Transl Respir Med 2(1):3
Le A, Ferrell GA, Dishon DS, Le QQ, Sifers RN (1992) Soluble aggregates of the human piz alpha 1-antitrypsin variant are degraded within the endoplasmic reticulum by a mechanism sensitive to inhibitors of protein synthesis. J Biol Chem 267(2):1072–1080
Lee CY, Tse W, Smith JD, Landreth GE (2012) Apolipoprotein e promotes beta-amyloid trafficking and degradation by modulating microglial cholesterol levels. J Biol Chem 287(3):2032–2044
Levy CB, Stumbo AC, Ano Bom AP, Portari EA, Cordeiro Y, Silva JL, De Moura-Gallo CV (2011) Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int J Biochem Cell Biol 43(1):60–64
Levy-Nissenbaum E, Betz RC, Frydman M, Simon M, Lahat H, Bakhan T, Goldman B, Bygum A, Pierick M, Hillmer AM, Jonca N, Toribio J, Kruse R, Dewald G, Cichon S, Kubisch C, Guerrin M, Serre G, Nothen MM, Pras E (2003) Hypotrichosis simplex of the scalp is associated with nonsense mutations in CDSN encoding corneodesmosin. Nat Genet 34(2):151–153
Lewandowski NM, Ju S, Verbitsky M, Ross B, Geddie ML, Rockenstein E, Adame A, Muhammad A, Vonsattel JP, Ringe D, Cote L, Lindquist S, Masliah E, Petsko GA, Marder K, Clark LN, Small SA (2010) Polyamine pathway contributes to the pathogenesis of Parkinson disease. Proc Natl Acad Sci U S A 107(39):16970–16975
Li J, Horak KM, Su H, Sanbe A, Robbins J, Wang X (2011) Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest 121(9):3689–3700
Liu F, Hindupur J, Nguyen JL, Ruf KJ, Zhu J, Schieler JL, Bonham CC, Wood KV, Davisson VJ, Rochet JC (2008) Methionine sulfoxide reductase a protects dopaminergic cells from Parkinson’s disease-related insults. Free Radic Biol Med 45(3):242–255
Lu B, Al-Ramahi I, Valencia A, Wang Q, Berenshteyn F, Yang H, Gallego-Flores T, Ichcho S, Lacoste A, Hild M, Difiglia M, Botas J, Palacino J (2013) Identification of NUB1 as a suppressor of mutant huntingtin toxicity via enhanced protein clearance. Nat Neurosci 16(5):562–570
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338(6109):949–953
Mahendiran K, Elie C, Nebel JC, Ryan A, Pierscionek BK (2014) Primary sequence contribution to the optical function of the eye lens. Sci Rep 4
Malfatti E, Monges S, Bohm J, Quijano-Roy S, Brochier G, Lubieniecki F, Behin A, Laforêt P, Stojkovic T, Viou MT (2014) Gp 264: congenital myopathies with protein aggregates and inclusions: importance of extensive muscle biopsy analysis in the diagnostic workup. Neuromuscul Disord 24(9):896
Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M (2010) Alzheimer’s disease: clinical trials and drug development. Lancet Neurol 9(7):702–716
Maor I, Hayek T, Coleman R, Aviram M (1997) Plasma LDL oxidation leads to its aggregation in the atherosclerotic apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol 17(11):2995–3005
Marzban L, Trigo-Gonzalez G, Zhu X, Rhodes CJ, Halban PA, Steiner DF, Verchere CB (2004) Role of beta-cell prohormone convertase (pc)1/3 in processing of pro-islet amyloid polypeptide. Diabetes 53(1):141–148
Mckee S (2010) Lilly hit by spectacular failure of phase III Alzheimer’s candidate. Pharma Times, 18 Aug 2010
Mead S, Poulter M, Uphill J, Beck J, Whitfield J, Webb TE, Campbell T, Adamson G, Deriziotis P, Tabrizi SJ, Hummerich H, Verzilli C, Alpers MP, Whittaker JC, Collinge J (2009) Genetic risk factors for variant Creutzfeldt-Jakob disease: a genome-wide association study. Lancet Neurol 8(1):57–66
Menzies FM, Huebener J, Renna M, Bonin M, Riess O, Rubinsztein DC (2010) Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 133(Pt 1):93–104
Merck&Co. 2013. Merck advances development program for investigational Alzheimer’s disease therapy, mk-8931. Merck Making News
Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, Andres PL, Mahoney K, Allred P, Alexander K, Ostrow LW, Schoenfeld D, Macklin EA, Norris DA, Manousakis G, Crisp M, Smith R, Bennett CF, Bishop KM, Cudkowicz ME (2013) An antisense oligonucleotide against sod1 delivered intrathecally for patients with sod1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol 12(5):435–442
Minati L, Edginton T, Bruzzone MG, Giaccone G (2009) Reviews: current concepts in Alzheimer’s disease: a multidisciplinary review. Am J Alzheimers Dis Other Demen 24(9):95–121
Miroy GJ, Lai Z, Lashuel HA, Peterson SA, Strang C, Kelly JW (1996) Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc Natl Acad Sci U S A 93(26):15051–15056
Moreau KL, King JA (2012) Protein misfolding and aggregation in cataract disease and prospects for prevention. Trends Mol Med 18(5):273–282
Necula M, Kayed R, Milton S, Glabe CG (2007) Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J Biol Chem 282(14):10311–10324
Noble W, Garwood CJ, Hanger DP (2009) Minocycline as a potential therapeutic agent in neurodegenerative disorders characterised by protein misfolding. Prion 3(2):78–83
Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P (2002) The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat 19(6):607–614
Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, Vendruscolo M, Hayer-Hartl M, Hartl FU, Vabulas RM (2011) Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144(1):67–78
Palombella V, Rando O, Goldberg A, Maniatis T (1989) The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 78:773–785
Patel HR, Pithadia AS, Brender JR, Fierke CA, Ramamoorthy A (2014) In search of aggregation pathways of IAPP and other amyloidogenic proteins: finding answers through NMR spectroscopy. J Phys Chem Lett 5(11):1864–1870
Prickett M, Jain M (2013) Gene therapy in cystic fibrosis. Transl Res 161(4):255–264
PRNewswire (2012) Parkinson’s vaccine- worldwide first clinical trial in Vienna-press release. http://www.prnewswire.com/news-releases/affiris-ag-parkinsons-vaccine---worldwide-first-clinical-study-invienna-157198595.html
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36(6):585–595
Raz V, Abraham T, van Zwet EW, Dirks RW, Tanke HJ, van der Maarel SM (2011) Reversible aggregation of PABPN1 pre-inclusion structures. Nucleus 2(3):208–218
Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH Jr, Scott RW, Snider WD (1996) Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 13(1):43–47
Reeve A, Park T, Jaros E (2012) Relationship between mitochondria and α-synuclein: a study of single substantia nigra neurons. Arch Neurol 69(3):385–393
Reitz C (2012) Alzheimer’s disease and the amyloid cascade hypothesis: a critical review. Int J Alzheimers Dis 2012, e369808
Robberecht W, Philips T (2003) The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 14(4):248–264
Rochet JC, Hay BA, Guo M (2012) Molecular insights into Parkinson’s disease. Prog Mol Biol Transl Sci 107:125–188
Rocken C, Tautenhahn J, Buhling F, Sachwitz D, Vockler S, Goette A, Burger T (2006) Prevalence and pathology of amyloid in atherosclerotic arteries. Arterioscler Thromb Vasc Biol 26(3):676–677
Rose C, Menzies FM, Renna M, Acevedo-Arozena A, Corrochano S, Sadiq O, Brown SD, Rubinsztein DC (2010) Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum Mol Genet 19(11):2144–2153
Rosen RF, Fritz JJ, Dooyema J, Cintron AF, Hamaguchi T, Lah JJ, LeVine H 3rd, Jucker M, Walker LC (2011) Exogenous seeding of cerebral beta-amyloid deposition in betaAPP-transgenic rats. J Neurochem 120(5):660–666
Rosenfeld PJ, Cowley GS, McGee TL, Sandberg MA, Berson EL, Dryja TP (1992) A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat Genet 1(3):209–213
Rosenzweig M, Landau H (2011) Light chain (al) amyloidosis: update on diagnosis and management. J Hematol Oncol 4(1):1–8
Ross CA, Poirier MA (2004) Protein aggregation and neurodegenerative disease. Nat Med 10(Suppl):S10–17
Saliba RS, Munro PM, Luthert PJ, Cheetham ME (2002) The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J Cell Sci 115(Pt 14):2907–2918
Schapira AH, Gegg ME (2013) Glucocerebrosidase in the pathogenesis and treatment of Parkinson disease. Proc Natl Acad Sci U S A 110(9):3214–3215
Schenk D (2002) Amyloid-β immunotherapy for Alzheimer’s disease: the end of the beginning. Nat Rev Neurosci 3(10):824–828
Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE (1999) Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc Natl Acad Sci U S A 96(8):4604–4609
Shannon KM, Keshavarzian A, Dodiya HB, Jakate S, Kordower JH (2012) Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov Disord 27(6):716–719
Shigenaga MK, Hagen TM, Ames BN (1994) Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A 91(23):10771–10778
Singh LR, Kruger WD (2009) Functional rescue of mutant human cystathionine beta-synthase by manipulation of hsp26 and hsp70 levels in saccharomyces cerevisiae. J Biol Chem 284(7):4238–4245
Soto C, Estrada LD (2008) Protein misfolding and neurodegeneration. Arch Neurol 65(2):184–189
Stefani M (2004) Protein misfolding and aggregation: new examples in medicine and biology of the dark side of the protein world. Biochim Biophys Acta 1739(1):5–25
Stefanis L (2012) Alpha-synuclein in Parkinson’s disease. Cold Spring Harbor Perspect Med 2(2):a009399
Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL (2004) Sumo modification of huntingtin and Huntington’s disease pathology. Science 304(5667):100–104
Stewart BW, Wild CP (2014) World cancer report 2014. IARC Press, International Agency for Research on Cancer, Lyon
Stoller JK, Aboussouan LS (2005) Alpha1-antitrypsin deficiency. Lancet 365(9478):2225–2236
Stuart JA, Birge RR (1996) Characterization of the primary photochemical events in bacteriorhodopsin and rhodopsin. In: Biomembranes: a multi-volume treatise, vol 2. pp 33–139
Stubbs JD, Lekutis C, Singer KL, Bui A, Yuzuki D, Srinivasan U, Parry G (1990) cDNA cloning of a mouse mammary epithelial cell surface protein reveals the existence of epidermal growth factor-like domains linked to factor VIII-like sequences. Proc Natl Acad Sci U S A 87(21):8417–8421
Su H, Wang X (2010) The ubiquitin-proteasome system in cardiac proteinopathy: a quality control perspective. Cardiovasc Res 85(2):253–262
Tagliavini F, Forloni G, Colombo L, Rossi G, Girola L, Canciani B, Angeretti N, Giampaolo L, Peressini E, Awan T, De Gioia L, Ragg E, Bugiani O, Salmona M (2000) Tetracycline affects abnormal properties of synthetic PrP peptides and PrP(Sc) in vitro. J Mol Biol 300(5):1309–1322
Taylor JP, Hardy J, Fischbeck KH (2002) Toxic proteins in neurodegenerative disease. Science 296(5575):1991–1995
Trivedi VD, Raman B, Rao CM, Ramakrishna T (1997) Co-refolding denatured-reduced hen egg white lysozyme with acidic and basic proteins. FEBS Lett 418(3):363–366
Ursini F, Davies KJ, Maiorino M, Parasassi T, Sevanian A (2002) Atherosclerosis: another protein misfolding disease? Trends Mol Med 8(8):370–374
Uversky V (2002) Natively unfolded proteins: a point where biology meets physics. Protein Sci 11:739–756
Vang S, Corydon TJ, Borglum AD, Scott MD, Frydman J, Mogensen J, Gregersen N, Bross P (2005) Actin mutations in hypertrophic and dilated cardiomyopathy cause inefficient protein folding and perturbed filament formation. FEBS J 272(8):2037–2049
Velsor LW, Kariya C, Kachadourian R, Day BJ (2006) Mitochondrial oxidative stress in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am J Respir Cell Mol Biol 35(5):579–586
Wang T, Lao U, Edgar BA (2009) Tor-mediated autophagy regulates cell death in drosophila neurodegenerative disease. J Cell Biol 186(5):703–711
Wennstrom M, Surova Y, Hall S, Nilsson C, Minthon L, Bostrom F, Hansson O, Nielsen HM (2013) Low CSF levels of both alpha-synuclein and the alpha-synuclein cleaving enzyme neurosin in patients with synucleinopathy. PLoS One 8(1), e53250
Westermark P, Li ZC, Westermark GT, Leckstrom A, Steiner DF (1996) Effects of beta cell granule components on human islet amyloid polypeptide fibril formation. FEBS Lett 379(3):203–206
Wijesekera LC, Leigh PN (2009) Amyotrophic lateral sclerosis. Orphanet J Rare Dis 4:3
Wilck N, Ludwig A (2014) Targeting the ubiquitin-proteasome system in atherosclerosis: status quo, challenges, and perspectives. Antioxid Redox Signal
Wilding BR, McGrath MJ, Gl B, Mitchell CA (2014) Fhl1 mutants that cause clinically distinct human myopathies form protein aggregates and impair myoblast differentiation. J Cell Sci 127(10):2269–2281
Williams RA, Mamotte CD, Burnett JR (2008) Phenylketonuria: an inborn error of phenylalanine metabolism. Clin Biochem 29(1):31–41
Xi Y-B, Zhang K, Dai A-B, Ji S-R, Yao K, Yan Y-B (2014) Cataract-linked mutation r188h promotes βb2-crystallin aggregation and fibrillization during acid denaturation. Biochem Biophys Res Commun 447(2):244–249
Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, Cornelis A, Rozenski J, Zwolinska A, Marine JC, Lambrechts D, Suh YA, Rousseau F, Schymkowitz J (2011) Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol 7(5):285–295
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer India
About this chapter
Cite this chapter
Chowhan, R.K., Dar, T.A., Singh, L.R. (2015). Proteopathies: Biological, Molecular and Clinical Perspectives. In: Singh, L.R., Dar, T.A., Ahmad, P. (eds) Proteostasis and Chaperone Surveillance. Springer, New Delhi. https://doi.org/10.1007/978-81-322-2467-9_8
Download citation
DOI: https://doi.org/10.1007/978-81-322-2467-9_8
Publisher Name: Springer, New Delhi
Print ISBN: 978-81-322-2466-2
Online ISBN: 978-81-322-2467-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)