
Abstract
Endosymbiotic bacteria are common in many herbivorous insects. Bemisia tabaci is a phloem-sapping pest of various crop plants and is known to harbor at least five endosymbionts. This species is a complex of at least 40 genetically distinct but morphologically indistinguishable cryptic species worldwide. Endosymbiont composition has been studied in invasive cryptic species such as MEAM1 and MED, but little information exists regarding the indigenous genetic groups in Asia. Here, we determined the endosymbiont profiles of four indigenous Asian cryptic species (Asia I, Asia II 1, Asia II 5 and Asia II 10) of B. tabaci identified in Bangladesh. Overall, the infection rates of Arsenophonus, Cardinium, Hamiltonella, Rickettsia, and Wolbachia were 93%, 86%, 0%, 31%, and 88%, respectively. Phylogenetic analysis revealed two subgroups in Arsenophonus (A1, A2) and Rickettsia (R1, R2), but only one subgroup in Cardinium (C2) and Wolbachia (W1). Each endosymbiont showed varying rates of infection in the four cryptic species and most were co-infected with various combinations. The results of this study provide important information on the relationships between the endosymbionts and cryptic species of B. tabaci indigenous to Asia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Endosymbionts are common in plant-sapping insects and they have important relationships with their host species (Baumann 2005). Primary endosymbionts coevolve with their hosts, becoming key to host survival by assisting in obtaining essential nutrients (Baumann 2005; Rosell et al. 2010). Secondary endosymbionts have a facultative relationship with their hosts, providing fitness benefits such as in reproduction, host plant specialization, and increased tolerance to thermal stress and parasites (Chiel et al. 2009; Feldhaar 2011; Kaiser et al. 2010; Montllor et al. 2002; Oliver et al. 2003; Sintupachee et al. 2006; Tsuchida et al. 2004). In particular, secondary endosymbionts provide pivotal roles in virus transmission for vectoring species such as B. tabaci, which is a unique vector species of begomoviruses (Czosnek and Ghanim 2011).
Bemisia tabaci is a polyphagous pest species that feeds on various horticultural crops, ornamental crops and weed species (Cahill et al. 1996; Jones 2003). This species harbors primary endosymbiont Portiera aleyrodidarum along with at least six secondary endosymbionts, viz., Arsenophonus, Cardinium, Fritschea, Hamiltonella, Rickettsia, and Wolbachia (Bing et al. 2013a, b; Zchori-Fein et al. 2014). Recently, a new endosymbiont, Candidatus Hemipteriphilus asiaticus, was found to be a cryptic species of B. tabaci (Bing et al. 2013a, b). Secondary endosymbionts influence various biological characteristics, such as reproduction (Himler et al. 2011; Hunter et al. 2003; Zchori-Fein et al. 2001; Zchori-Fein and Perlman 2004), survival (Gottlieb et al. 2010; Kontsedalov et al. 2008; Liu et al. 2007; Thierry et al. 2011), insecticide resistance (Kontsedalov et al. 2008), and capacity for disease transmission to plants (Gottlieb et al. 2010). Given their various roles, endosymbionts are necessary for the modulation of host development and environmental impacts (Thao and Baumann 2004).
Bemisia tabaci is a species complex comprising at least 40 cryptic species that are morphologically indistinguishable, but genetically distinct in biological characteristics (De Barro et al. 2011). Among them, the Middle East-Asia Minor 1 (MEAM1, formerly B biotype) and Mediterranean (MED, formerly Q biotype) species are highly invasive and have dispersed to many countries in different continents (Dalton 2006; Horowitz et al. 2005; Pascual and Callejas 2004). In addition, many indigenous cryptic species of B. tabaci has been observed in Africa and Asia. To date, at least 23 indigenous cryptic species with different geographic distributions had been identified in Asia (Dinsdale et al. 2010; Ahmed et al. 2011; Hameed et al. 2012; Firdaus et al. 2013; Shah et al. 2013; Prasanna et al. 2015; Ellango et al. 2015; Hu et al. 2015, 2017; Götz and Winter 2016; Kumar et al. 2016; Jiu et al. 2017). Recently, Our previous study identified four indigenous cryptic species (Asia I, Asia II 1, Asia II 5 and Asia II 10) in Bangladesh (Khatun et al. 2018).
Each genetic group of B. tabaci exhibits a distinct composition of secondary endosymbionts. Several studies have investigated invasive cryptic species such as MEAM1 and MED from different geographic regions (Bing et al. 2013a, b; Chiel et al. 2007; Chu et al. 2011; Gueguen et al. 2010; Park et al. 2012; Skaljac et al. 2010, 2013). However, little information exists regarding the endosymbiont profiles of indigenous cryptic species in Asian countries.
The objective of this study was to determine the infection profiles of secondary endosymbionts in the cryptic species of B. tabaci from Bangladesh. Results from this study will improve our understanding of the composition of endosymbionts and their relationships with the Asian genetic groups of B. tabaci.
2 Materials and methods
2.1 Sample collection
Adult B. tabaci were collected from crop fields in different regions of Bangladesh from 2015 to 2017. Samples were preserved in 70% ethanol and kept at −20 °C for further analysis (Table S1). Identification and genetic diversity of four cryptic species of B. tabaci in these samples has been reported in a previous study (Khatun et al. 2018).
2.2 DNA extraction
Genomic DNA was extracted from a single adult B. tabaci using a pure link genomic DNA mini kit (Invitrogen, Carlsbad, CA, USA). The sample was placed in a 1.5 mL centrifuge tube containing 180 μL digestion buffer and 20 μL proteinase K (50 μg/mL) then incubated at 55 °C for 4 h. DNA samples were extracted and purified using genomic spin columns following the manufacturer’s protocol. DNA concentration was determined using a NanoPhotometer™ (Implen GmbH, Schatzbogen, Germany).
2.3 Screening of secondary endosymbionts
Five known endosymbionts (Arsenophonus, Cardinium, Hamiltonella, Rickettsia, and Wolbachia) were detected through PCR using 16S or 23S rDNA primers (Table 1). The reaction (25 μL) contained 13 μL Smart-Taq Pre-Mix (Solgent Co., Daejeon, Korea), 1 μL of each primer (10 pmol/μL), and 5 μLtemplate DNA solution (40 ng). Amplicons were separated using 1% agarose gel electrophoresis, stained with ethidium bromide solution, and visualized under Ultraviolet (UV) light. After excision from the gel, the amplicons were purified using the Wizard® PCR Preps DNA Purification System (Wizard® SV Gel, Promega Co., Madison, WI). They were then either sequenced directly or via cloning into the T-Blunt™easy plasmid vector (Promega Co., Madison, WI).
2.4 DNA sequence analysis
Cloned amplicons were sequenced using the BigDye® Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and analyzed with a 3100 Capillary DNA Sequencer (Applied Biosystems, Foster City, CA) at the Solgent Sequencing Facility (Solgent Co., Daejeon, Korea). Sequences were obtained from one individual per primer set and BLASTed against the NCBI database (Schaffer et al. 2001).
2.5 Phylogenetic analyses of secondary endosymbionts
Endosymbiont 16S and 23S rDNA sequences were aligned in Clustal Omega for the construction of a maximum-likelihood (ML) phylogenetic tree in MEGA 6.0. The model was selected based on GTR + G for Arsenophonus and Wolbachia, HKY + G for Cardinium and HKY + I for Rickettsia. Phylogeny robustness was tested with 1000 bootstraps (Felsenstein 1985).
2.6 Correlation analysis
Genetic differentiation among the endosymbionts was determined in DnaSP version 5.10 (Librado and Rozas 2009; Tajima 1989). Pairwise genetic distance was generated from 1023 permutations with the K2P model in MEGA 6.0 (Kimura 1980). Geographic distances were calculated from GPS coordinates measured during B. tabaci sample collections. The correlation between endosymbiotic genetic variability and geographic distances was analyzed using IBM SPSS software, version 23.
3 Results
3.1 Endosymbiont profiles of B. tabaci in Bangladesh
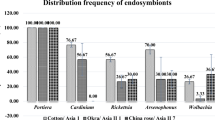
Infection profiles of five endosymbionts were determined from 110 individuals representing four Asian cryptic species of B. tabaci (Asia I, Asia II 1, Asia II 5, and Asia II 10; Table S1). Overall, the infection rates of Arsenophonus, Cardinium, Rickettsia, and Wolbachia were 93.6%, 86.4%, 31.8%, and 88.2%, respectively, but Hamiltonella was not detected in any individuals in this study (Fig. 1, Tables 2 and 3). Phylogenetic analysis of 23S rDNA sequences showed that Arsenophonus belonged to A1 and A2, but not to the A3 and A4 subgroups (Fig. 2). Sequences of both A1 and A2 exhibited a 0.19–3.18% variation and differences between A1 and A2 subgroups were 2.25–3.18% (Table 4, Table S2). The A1 subgroup was detected in Asia I and Asia II 1, while A2 was present in all four cryptic species (Figs. 1 and 2). Cardinium was present in C2 but not in the C1 or C3 subgroups (Figs. 1 and 3). The C2 subgroup was detected in Asia I, Asia II 1, and Asia II 5 with a variation of 0.25–0.74% (Fig. 3, Table 4, Table S3). Rickettsia belonged to the R1 and R2 subgroups;16S rDNA sequence variation ranged from 0.11–13.37% and differences between the R1 and R2 subgroups ranged from 10.15–13.37% (Figs. 1 and 4; Table 4, Table S4). Both R1 and R2 were detected at different rates in Asia I, Asia II 1, and Asia II 5 (Fig. 4). Wolbachia was present in W1 but not in the W2 or W3 subgroups. W1 subgroup was detected in all four cryptic species with a variation of 0.17–0.68% (Figs. 1 and 5; Table 4, Table S5).

Infection frequency (%) of secondary endosymbionts in four cryptic Bemisia tabaci species: Asia I, Asia II 1, Asia II 5, and Asia II 10. Arsenophonus and Rickettsia were divided into subgroups, indicated by different colors: A1 (bright blue), A2 (dark blue), R1 (bright green), and R2 (dark green)

Maximum-likelihood phylogenetic tree of 23S rDNA in Arsenophonus (600 bp) in infected Bemisia tabaci, using a GTR + G substitution model. Evolutionary analyses were conducted in MEGA 6. Sequences from GenBank are indicated in black color and the name of the samples from this study are shown in bright and dark blue color and include the genetic group, collection site, and accession number in Bangladesh

Maximum-likelihood phylogenetic tree of 16S rDNA in Cardinium (400 bp) in infected Bemisia tabaci, using a HKY + G substitution model. Evolutionary analyses were conducted in MEGA 6. Sequences from GenBank are indicated in black color and the name of the samples from this study are shown in pink color and include the genetic group, collection site, and accession number in Bangladesh

Maximum-likelihood phylogenetic tree of 16S rDNA in Rickettsia (900 bp) in infected Bemisia tabaci, using a HKY + I substitution model. Evolutionary analyses were conducted in MEGA 6. Sequences from GenBank are indicated in black color and the name of the samples from this study are shown in bright and dark green color and include the genetic group, collection site, and accession number in Bangladesh

Maximum-likelihood phylogenetic tree of 16S rDNA in Wolbachia (650 bp) in infected Bemisia tabaci, using a GTR + G substitution model. Evolutionary analyses were conducted in MEGA 6. Sequences from GenBank are indicated in black color and the name of the samples from this study are shown in red color and include the genetic group, collection site, and accession number in Bangladesh
The four endosymbionts differed in infection rates depending on B. tabaci genetic groups (Table 2). Arsenophonus was 93.6% infected on average in all four cryptic species, with a range of 90.0–95.5%, except for in Asia II 10. Asia II 10 had 100% infection, but only one individual was examined in this group. The infection rate of A1 was higher than that of A2 in both Asia I and Asia II 1, but only A2 was infected into Asia II 5 and Asia II 10 (Fig. 1, Table 2). The infection rate of Cardinium was 80.6–100%, with the highest rate occurring in Asia II 5. The infection rate of Rickettsia was 13.6–50.0% with R1 being the highest in Asia II 1 (35%) and R2 being the highest in Asia I (25.4%). The infection rate of Wolbachia was 75–100% with the highest rate observed for Asia II 5 (Table 2). Otherwise, we did not find any significant relationships between the infection rates of endosymbionts and crop species infested by B. tabaci (Table S1).
3.2 Multiple infections of B. tabaci endosymbionts
All B. tabaci individuals were multiplied infected in various combinations by the four endosymbionts (Table 3). We identified 12 types of co-infection patterns, with multiple infections of 2, 3, or 4 species of endosymbionts. Single infections were identified only in A1 in 4.5% of Asia I and C2 in 10.0% of Asia II 1 cryptic species. Arsenophonus was the most common endosymbiont found in multiple infections. Infection patterns were more diverse in Asia I (8 patterns) and Asia II 1 (6 patterns) than in Asia II 5 (3 patterns). The most common combination was A1 + C2 + W1, which was the highest in Asia I (41.8%) and Asia II 1 (35.0%), whereas the A2 + C2 + W1 combination was only present in Asia II 5 at a high rate (86.4%). Among Arsenophonus and Rickettsia co-infections A1 was co-infected with R2 but A2 was co-infected with R1 (Table 3).
3.3 Correlation between genetic variation of endosymbionts and geographic distance of cryptic species
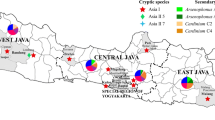
The variations in ribosomal DNA nucleotide sequences of Arsenophonus, Cardinium, and Wolbachia did not correlate with the geographic distribution of B. tabaci in Bangladesh. However, rDNA sequences of Rickettsia differed significantly and had a high distribution frequency (r = 0.428, P = 0.023; Table 5). Namely, the R1 subgroup was mostly present in the north, while R2 was found in the southern regions of Bangladesh (Fig. 6). Thus, Rickettsia subgroups had different distributions in Bangladesh, but the distributions of the other endosymbionts were not related to geographic distribution.

Geographic distribution of Arsenophonus (a) and Rickettsia (b) subgroups of Bemisia tabaci in Bangladesh. Two subgroups of both Arsenophonus and Rickettsia are shown in bright blue (A1), dark blue (A2), bright green (R1), and dark green (R2)
4 Discussion
Our study showed that profiles of the five species of endosymbionts in the indigenous Asian cryptic species of B. tabaci identified in Bangladesh were highly variable. Overall, the infection rate of Arsenophonus was the highest (93.6%), followed by Cardinium and Wolbachia (86.4% and 88.2%, respectively). Rickettsia only showed a moderate level of infection (31.8%). Hamiltonella was not detected in any individuals. These infection rates were complex and varied among the four Asian cryptic species (Asia I, Asia II 1, Asia II 5 and Asia II 10) of B. tabaci.
Arsenophonus was the most prevalent among the four Asian cryptic species. This is consistent with the findings of previous studies (Bing et al. 2013a, b; Ansari et al. 2017; Hashmi et al. 2018). Arsenophonus was detected in all indigenous genetic groups identified from China and India, such as Asia 1, Asia II 1, Asia II 5, and Asia II 7. In India, among all Asian cryptic species, Arsenophonus infection rate was higher in Asia 1 (70.0%) (Ansari et al. 2017; Hashmi et al. 2018). The infection rate of Arsenophonus was higher in Asia II 1 (78.9%) than in Asia II 3 and Asia II 7 from China (Bing et al. 2013a, b). Arsenophonus was also abundant in China I, but had not been previously detected in China (Bing et al. 2013a, b; Tang et al. 2018). Many studies have shown that Arsenophonus infection rates in invasive species are low in MEAM1 (Chiel et al. 2007; Chu et al. 2011; Gottlieb et al. 2008; Gueguen et al. 2010; Marubayashi et al. 2014; Skaljac et al. 2010; Thierry et al. 2011) or absent in MEAM1 and MED (Bing et al. 2013a, b). However, some MED populations from the USA, Montenegro (Q1), Israel (Q2), and Burkina Faso (Q3) were highly infected with Arsenophonus (Bing et al. 2013a, b; Gueguen et al. 2010; Skaljac et al. 2010; Skaljac et al. 2013). Therefore, these findings suggest that Arsenophonus is the most prevalent endosymbiont in Asian cryptic species, which is unlike the profiles of invasive species MEAM1 and MED of B. tabaci.
Cardinium infection rates were high in Asia I, Asia II 1 and Asia II 5 from Bangladesh. In particular, Cardinium infected all investigated individuals (n = 22) of Asia II 5. High infection rates of Cardinium were also reported in Asia II 1 from India (Ansari et al. 2017; Hashmi et al. 2018) and Asia II 3 and Asia II 7 from China (Bing et al. 2013a, b). Otherwise, Cardinium infection is rare in MEAMI and MED (Chu et al. 2011; Gueguen et al. 2010; Thierry et al. 2011). Similar to Arsenophonus, Cardinium had high infection rates in the Asian genetic groups, but low infection rates in the invasive genetic groups.
Hamiltonella was absent in all Asian cryptic species from Bangladesh, and these results are consistent with the reports of previous studies (Bing et al. 2013a, b; Singh et al. 2012). Hamiltonella is known to infect only two cryptic species, MEAM1 and MED and these species are highly invasive worldwide (Bing et al. 2013a, b; Chu et al. 2011; Gottlieb et al. 2008; Gueguen et al. 2010; Marubayashi et al. 2014; Skaljac et al. 2017; Thierry et al. 2011). Hamiltonella infection in MED (previously Q-biotype) is variable in the subgroups of this genetic group. For example, Hamiltonella has high infection rates in Q1 populations in China, but is absent in Q2 and ASL of MED (Bing et al. 2013a, b; Chu et al. 2011; Gottlieb et al. 2008; Gueguen et al. 2010; Marubayashi et al. 2014; Skaljac et al. 2017; Thierry et al. 2011). Hamiltonella is also absent in the sub-Saharan Africa 1–5 cryptic species of B. tabaci (Ghosh et al. 2015).
Rickettsia was identified in MEAM1 of B. tabaci, which is the first recorded occurrence in an insect (Gottlieb et al. 2006). Analysis of its genome sequence revealed that it grouped together with R. bellii (Rao et al. 2012). Our study showed that Rickettsia was detected in three cryptic species in Bangladesh. Its infection rate was moderate, but variable with the highest infection rate in Asia II 1 (50.0%), a moderate infection rate in Asia I (32.8%), and the lowest infection rate in Asia II 5 (13.6%). However, its infection rate was higher in Asia I than in Asia II 1 and Asia II 7 in Indian populations (Hashmi et al. 2018). Rickettsia was also detected in Asia II 3, Asia II 7, China 1, and MEAM1, but was absent from Asia II 1 and MED in China (Bing et al. 2013a, b; Chiel et al. 2007; Chu et al. 2011; Gueguen et al. 2010; Thierry et al. 2011). This suggests that the infection rate of Rickettsia is variable in different genetic groups and geographic regions. For example, our study showed that its infection rate in Asia II 1 in Bangladesh had a significantly different geographic distribution than those in China and India.
Wolbachia is one of the most common insect endosymbionts, with various infection rates for different genetic groups (Correa and Ballard 2016). Our study showed that Wolbachia was highly prevalent in all cryptic species of B. tabaci from Bangladesh, with a 100% infection rate in Asia II 5. High infection rates of Wolbachia were reported in Asia II 1, Asia II 7, and China 1, but Wolbachia was absent in Asia II 3 from China (Bing et al. 2013a, b). However, its infection rate was moderate in Asia 1 and Asia II 7 and low in Asia II 1 from India (Hashmi et al. 2018). This suggests that Wolbachia infection rates vary regardless of the genetic groups of B. tabaci. Otherwise, Wolbachia infection was high in Bangladesh and China, but low in India. However, this difference may not be associated with geographic distribution. In fact, the Wolbachia infection rate is highly variable across regions and populations. For example, the Wolbachia infection rate in both MED and MEAM1 ranges from 0% to 100% throughout China (Bing et al. 2013a, b, 2014; Pan et al. 2012). In Europe and western Africa, Wolbachia infection of MEAM1 and MED was 0–8.3% and 0–33% (Chiel et al. 2007; Chu et al. 2011; Gnankine et al. 2013; Gueguen et al. 2010; Nirgianaki et al. 2003; Skaljac et al. 2010; Thierry et al. 2011).
Phylogenetic analyses showed that both Arsenophonus and Rickettsia were detected in two subgroups, but Cardinium and Wolbachia were detected in a single subgroup in Bangladesh. Arsenophonus was infected into all four cryptic species. Our study showed that A2 infected all four cryptic species, but A1 infected only in Asia I and Asia II 1. This result suggests that Arsenophonus infection depends on the genetic group of B. tabaci. Similarly, two Rickettsia subgroups (R1, R2) were detected in three cryptic species in Bangladesh with different infection rates; R1 was higher in Asia I and R2 was higher in Asia II 1 and Asia II 5. The presence of two Rickettsia subgroups in B. tabaci has been reported in previous studies (Singh et al. 2012; Bing et al. 2013a, b; Ghosh et al. 2015). Singh et al. (2012) observed RI1 and RI2 strains in Indian populations. Bing et al. (2013a, b) reported that one subgroup detected in MEAM1 and Asia II 3 is widely separated from the other group found in Asia II 7 and China 1. Ghosh et al. (2015) described two clusters that had more than 8.5% nucleotide distance in various genetic groups of B. tabaci infesting cassava in Africa. Our results showed that 16S rDNA sequence variation was low within subgroups but subgroups were highly distinct, with differences of 10.15–13.37%. This suggests that there are at least two genetically distinct Rickettsia species in the B. tabaci species complex. Further, the two subgroups were present in the same cryptic species of B. tabaci, although the ratios were different. This suggests that each subgroup of Rickettsia is not confined within certain genetic groups of B. tabaci. This characteristic is also found in Rickettsia infection in the African genetic groups of B. tabaci (Ghosh et al. 2015). For example, sub-Saharan Africa 1- subgroup 3 (SSA 1-SG3) is infected by two groups of Rickettsia at different rates.
Our study also showed that there is a relationship between genetic variation of Rickettsia and geographic distribution, contrary to other endosymbionts. Namely, R1 subgroup was mostly present in the northern region, while R2 subgroup was distributed in the southern region of Bangladesh. Similarly, Singh et al. (2012) identified two subgroups of Rickettsia in B. tabaci collected from various regions of Central and Northern India. R1 was detected in the northern region, but R2 was detected in the central region of India. Our phylogenetic analysis showed that the R2 subgroup in Bangladesh was clustered with R2 strains collected from Akola and Indore, which are in the central region of India. Thus, the geographic distributions of the two Rickettsia subgroups are distinct in these two countries. Results from this study also showed that the distribution of the two Rickettsia subgroups was not associated with genetic groups of B. tabaci, because each Asian cryptic species was infected by both subgroups of Rickettsia at various rates. Further analysis is required to determine the influence of geographic distribution.
Multiple infections of various secondary endosymbionts are common in most host species of insects (Zchori-Fein et al. 2014). We identified 12 types of co-infection patterns among four endosymbionts, particularly among two subgroups of Arsenophonus and Rickettsia. Every individual in this study was infected by at least one endosymbiont. Arsenophonus was the dominant endosymbiont in most co-infections. Interestingly, the combination of Arsenophonus and Rickettsia subgroups was always present as A1 + R2 or A2 + R1. Similarly, data from Israel and Burkina Faso demonstrated A1 + R co-infection in MED (Q2) and A2 + R in MED (Q3), respectively (Gueguen et al. 2010). Arsenophonus in MED (Q1 and Q2) is the R1 subgroup (Skaljac et al. 2017). Together, these data suggest that Asian cryptic B. tabaci may have a unique characteristic that is different from other cryptic species such as MED.
In conclusion, our results revealed the complexity of endosymbiont profiles in four cryptic B. tabaci species from Bangladesh. Specifically, each cryptic species exhibited characteristic patterns of infection rates and combinations of endosymbionts. Notably, Arsenophonuswas a major endosymbiont but Hamiltonella was absent from all Asian cryptic species of B. tabaci. Two genetically distinct subgroups of Rickettsia were associated with geographic distribution. This study provides important insights that can help improve the control techniques of whiteflies.
References
Ahmed MZ, Barro PJ, Greeff JM, Ren SX, Naveed M, Qiu BL (2011) Genetic identity of the Bemisia tabaci species complex and association with high cotton leaf curl disease (CLCuD) incidence in Pakistan. Pest Manag Sci 67:307–317
Ansari PG, Singh RK, Kaushik S, Krishna A, Wada T, Noda H (2017) Detection of symbionts and virus in the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae), a vector of the Mungbean yellow mosaic India virus in Central India. Appl Entomol Zool 52:567–579
Baumann P (2005) Biology of bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu Rev Microb 59:155–189
Bing XL, Ruan YM, Rao Q, Wang XW, Liu SS (2013a) Diversity of secondary endosymbionts among different putative species of the whitefly Bemisia tabaci. Insect Sci 20:194–206
Bing XL, Yang J, Zchori-Fein E, Wang XW, Liu SS (2013b) Characterization of a newly discovered symbiont of the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae). Appl Environ Microbiol 79:569–575
Bing XL, Xia WQ, Gui JD, Yan GH, Wang XW, Liu SS (2014) Diversity and evolution of the Wolbachia endosymbionts of Bemisia tabaci (Hemiptera: Aleyrodidae) whiteflies. Ecol Evol 4:2714–2737
Cahill M, Denholm I, Bryne FJ, Al D (1996) Insecticide resistance in Bemisia tabaci- current status and implications for management. In: Proceedings of Brighton crop protection conference: Pest and diseases, vol 1, pp 75–80
Chiel E, Gottlieb Y, Zchori-Fein E, Mozes-Daube N, Katzir N, Inbar M, Ghanim M (2007) Biotype-dependent secondary symbiont communities in sympatric populations of Bemisia tabaci. Bull Entomol Res 97:407–413
Chiel E, Inbar M, Mozes-Daube N, White JA, Hunter M, Zchori-Fein E (2009) Assessments of fitness effects by the facultative symbiont Rickettsia in the sweetpotato whitefly (Hemiptera: Aleyrodidae). Ann Entomol Soc Am 102:413–418
Chu D, Gao CS, De Barro P, Zhang YJ, Wan FH, Khan IA (2011) Further insights into the strange role of bacterial endosymbionts in whitefly, Bemisia tabaci: comparison of secondary symbionts from biotypes B and Q in China. Bull Entomol Res 101:477–486
Correa CC, Ballard J (2016) Wolbachia associations with insects: winning or losing against a master manipulator. Front Ecol Evol 3:153
Czosnek H, Ghanim M (2011) Bemisia tabaci – tomato yellow leaf curl virus Interaction Causing Worldwide Epidemics. In: Thompson W (ed) The whitefly, Bemisia tabaci (Homoptera: Aleyrodidae) interaction with Geminivirus-infected host plants. Springer, Dordrecht
Dalton R (2006) Whitefly infestations: the Christmas invasion. Nature. 443:898–900
De Barro PJ, Liu S, Boykin LM, Dinsdale AB (2011) Bemisia tabaci: a statement of species status. Annu Rev Entomol 56:1–19
Dinsdale A, Cook L, Riginos C, Buckley YM, Barro PD (2010) Refined global analysis of Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) mitochondrial cytochrome oxidase 1 to identify species level genetic boundaries. Ann Entomol Soc Am 103:196–208
Ellango R, Singh ST, Rana VS, Priya NG, Raina H, Chaubey R, Naveen NC, Mahmood R, Ramamurthy VV, Asokan R (2015) Distribution of Bemisia tabaci genetic groups in India. Environ Entomol 44:1258–1264
Feldhaar H (2011) Bacterial symbionts as mediators of ecologically important traits of insect hosts. Ecol Entomol 36:533–543
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Firdaus S, Vosman B, Hidayati N, Supena ED, Visser RG, Heusden AW (2013) The Bemisia tabaci species complex: additions from different parts of the world. Insect Sci 20:723–733
Ghosh S, Bouvaine S, Maruthi M (2015) Prevalence and genetic diversity of endosymbiotic bacteria infecting cassava whiteflies in Africa. BMC Microbiol 15:93
Gnankine O, Mouton L, Henri H, Terraz G, Houndate T, Martin T, Vavre F, Fleury F (2013) Distribution of Bemisia tabaci (Homoptera: Aleyrodidae) biotypes and their associated symbiotic bacteria on host plants in West Africa. Insect Conserv Divers 6:411–421
Gottlieb Y, Ghanim M, Chiel E, Gerling D, Portnoy V, Steinberg S, Tzuri G, Horowitz AR, Belausov E, Mozes-Daube N (2006) Identification and localization of a Rickettsia sp. in Bemisia tabaci (Homoptera: Aleyrodidae). Appl Environ Microbiol 72:3646–3652
Gottlieb Y, Ghanim M, Gueguen G, Kontsedalov S, Vavre F, Fleury F, Zchori-Fein E (2008) Inherited intracellular ecosystem: symbiotic bacteria share bacteriocytes in whiteflies. FASEB J 22:2591–2599
Gottlieb Y, Zchori-Fein E, Mozes-Daube N, Kontsedalov S, Skaljac M, Brumin M, Sobol I, Czosnek H, Vavre F, Fleury F, Ghanim M (2010) The transmission efficiency of tomato yellow leaf curl virus by the whitefly Bemisia tabaci is correlated with the presence of a specific symbiotic bacterium species. J Virol 84:9310–9317
Götz M, Winter S (2016) Diversity of Bemisia tabaci in Thailand and Vietnam and indications of species replacement. J Asia Pac Entomol 19:537–543
Gueguen G, Vavre F, Gnankine O, Peterschmitt M, Charif D, Chiel E, Gottlieb Y, Ghanim M, Zchori-Fein E, Fleury F (2010) Endosymbiont metacommunities, mtDNA diversity and the evolution of the Bemisia tabaci (Hemiptera: Aleyrodidae) species complex. Mol Ecol 19:4365–4376
Hameed S, Hameed S, Sadia M, Malik SA (2012) Genetic diversity analysis of Bemisia tabaci populations in Pakistan using RAPD markers. Electron J Biotechnol 15
Hashmi TR, Devi SR, Meshram NM, Prasad R (2018) Assessment of bacterial endosymbiont and the host Bemisia tabaci (Hemiptera: Aleyrodidae), using rRNA and mitochondrial cytochrome oxidase I gene sequences. Commun Integr Biol 11:e1433442
Himler AG, Adachi-Hagimori T, Bergen JE, Kozuch A, Kelly SE, Tabashnik BE, Chiel E, Duckworth VE, Dennehy TJ, Zchori-Fein E, Hunter MS (2011) Rapid spread of a bacterial symbiont in an invasive whitefly is driven by fitness benefits and female bias. Science 332:254–256
Horowitz AR, Kontsedalov S, Khasdan V, Ishaaya I (2005) Biotypes B and Q of Bemisia tabaci and their relevance to neonicotinoid and pyriproxyfen resistance. Arch Insect Biochem Physiol 58:216–225
Hu J, Chen YD, Jiang ZL, Nardi F, Yang TY, Jin J, Zhang ZK (2015) Global haplotype analysis of the whitefly Bemisia tabaci cryptic species Asia I in Asia. Mito DNA 26:232–241
Hu J, Zhang X, Jiang Z, Zhang F, Liu Y, Li Z, Zhang Z (2017) New putative cryptic species detection and genetic network analysis of Bemisia tabaci (Hemiptera: Aleyrodidae) in China based on mitochondrial COI sequences. Mito DNA Part A 29:474–484
Hunter MS, Perlman SJ, Kelly SE (2003) A bacterial symbiont in the Bacteroidetes induces cytoplasmic incompatibility in the parasitoid wasp Encarsia pergandiella. Proc Biol Sci 270:2185–2190
Jiu M, Hu J, Wang LJ, Dong JF, Song YQ, Sun HZ (2017) Cryptic species identification and composition of Bemisia tabaci (Hemiptera: Aleyrodidae) complex in Henan Province, China. J Insect Sci 17:1–7
Jones DR (2003) Plant viruses transmitted by whiteflies. Eur J Plant Pathol 109:195–219
Kaiser W, Huguet E, Casas J, Commin C, Giron D (2010) Plant green-island phenotype induced by leaf-miners are mediated by bacterial symbionts. Proc Biol Sci 277:2311–2319
Khatun MF, Jahan SMH, Lee S, Lee KY (2018) Genetic diversity and geographic distribution of the Bemisia tabaci species complex in Bangladesh. Acta Trop 187:28–36
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kontsedalov S, Zchori-Fein E, Chiel E, Gottlieb Y, Inbar M, Ghanim M (2008) The presence of Rickettsia is associated with increased susceptibility of Bemisia tabaci (Homoptera: Aleyrodidae) to insecticides. Pest Manag Sci 64:789–792
Kumar NR, Chang JC, Narayanan MB, Ramasamy S (2016) Phylogeographical structure in mitochondrial DNA of whitefly, Bemisia tabaci Gennadius (Hemiptera: Aleyrodidae) in southern India and Southeast Asia. Mito DNA Part A 28:621–631
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinfo 25:1451–1452
Liu SS, De Barro PJ, Xu J, Luan JB, Zang LS, Ruan YM, Wan FH (2007) Asymmetric mating interactions drive widespread invasion and displacement in a whitefly. Science 318:1769–1772
Marubayashi JM, Kliot A, Yuki VA, Rezende JAM, Krause-Sakate R, Pavan MA, Ghanim M (2014) Diversity and localization of bacterial endosymbionts from whitefly species collected in Brazil. PLoS One 9:e108363
Montllor CB, Maxmen A, Purcell AH (2002) Facultative bacterial endosymbionts benefit pea aphids Acyrthosiphon pisum under heat stress. Ecol Entomol 27:189–195
Nirgianaki A, Banks GK, Frohlich DR, Veneti Z, Braig HR, Miller TA, Bedford ID, Markham PG, Savakis C, Bourtzis K (2003) Wolbachia infections of the whitefly Bemisia tabaci. Curr Microbiol 47:93–101
Oliver KM, Russell JA, Moran NA, Hunter MS (2003) Facultative bacterial symbionts in aphids confer resistance to parasitic wasps. Proc Natl Acad Sci U S A 100:1803–1807
Pan H, Li X, Ge D, Wang S, Wu Q, Xie W, Jiao X, Chu D, Liu B, Xu B, Zhang Y (2012) Factors affecting population dynamics of maternally transmitted endosymbionts in Bemisia tabaci. PLoS One 7:e30760
Park J, Jahan SMH, Song WG, Lee H, Lee YS, Choi HS, Lee KS, Kim CS, Lee S, Lee KY (2012) Identification of biotypes and secondary endosymbionts of Bemisia tabaci in Korea and relationships with the occurrence of TYLCV disease. J Asia Pac Entomol 15:186–191
Pascual S, Callejas C (2004) Intra and interspecific competition between biotypes B and Q of Bemisia tabaci (Hemiptera: Aleyrodidae) from Spain. Bull Entomol Res 94:369–375
Prasanna HC, Kanakala S, Archana K, Jyothsna P, Varma RK, Malathi VG (2015) Cryptic species composition and genetic diversity within Bemisia tabaci complex in soybean in India revealed by mtCOI DNA sequence. J Integr Agr 14:1786–1795
Rao Q, Wang S, Su YL, Bing XL, Liu SS, Wang XW (2012) Draft genome sequence of “candidatus Hamiltonella defensa,” an endosymbiont of the whitefly Bemisia tabaci. J Bacteriol 194:3558
Rosell RC, Blackmer JL, Czosnek H, Inbar M (2010) In: Stansly PA, Naranjos SE (eds) Bemisia: Bionomics and Management of a Global PestMutualistic and dependent relationships with other organisms. Springer, Netherlands, pp 161–183
Schaffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, Koonin EV, Altschul SF (2001) Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res 29:2994–3005
Shah SHJ, Malik AH, Qazi J (2013) Identification of new genetic variant of Bemisia tabaci from Pakistan. Int J Entomol Res 1:16–24
Singh ST, Priya NG, Kumar J, Rana VS, Ellango R, Joshi A, Priyadarshini G, Asokan R, Rajagopal R (2012) Diversity and phylogenetic analysis of endosymbiotic bacteria from field caught Bemisia tabaci from different locations of North India based on 16S r DNA library screening. Infect Genet Evol 12:411–419
Sintupachee S, Milne JR, Poonchaisri S, Baimai V, Kittayapong P (2006) Closely related Wolbachia strains within the pumpkin arthropod community and the potential for horizontal transmission via the plant. Microb Ecol 51:294–301
Skaljac M, Zanic K, Ban SG, Kontsedalov S, Ghanim M (2010) Co-infection and localization of secondary symbionts in two whitefly species. BMC Microbiol 10:142
Skaljac M, Zanic K, Hrncic S, Radonjic S, Perovic T, Ghanim M (2013) Diversity and localization of bacterial symbionts in three whitefly species (Hemiptera: Aleyrodidae) from the east coast of the Adriatic Sea. Bull Entomol Res 103:48–59
Skaljac M, Kanakala S, Zanic K, Puizina J, Pleic IL, Ghanim M (2017) Diversity and phylogenetic analyses of bacterial symbionts in three whitefly species from Southeast Europe. Insects 8:113
Tajima F (1989) A statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tang XT, Cai L, Shen Y, Du YZ (2018) Diversity and evolution of the endosymbionts of Bemisia tabaci in China. PeerJ 6:e5516
Thao ML, Baumann P (2004) Evidence for multiple acquisitions of Arsenophonus by whitefly species (Sternorrhyncha: Aleyrodidae). Curr Microbiol 48:140–144
Thierry M, Becker N, Hajri A, Lett JM, Reynaud B, Delatte H (2011) Symbiont diversity and non-random hybridization among indigenous (Ms) and invasive (B) biotypes of Bemisia tabaci. Mol Ecol 20:2172–2187
Tsuchida T, Koga R, Fukatsu T (2004) Host plant specialization governed by facultative symbiont. Science 303:1989
Weeks AR, Velten R, Stouthamer R (2003) Incidence of a new sex-ratio-distorting endosymbiotic bacterium among arthropods. Proc Biol Sci 270:1857–1865
Zchori-Fein E, Brown JK (2002) Diversity of prokaryotes associated with Bemisia tabaci (Gennadius) (Hemiptera; Aleyrodidae). Ann Entomol Soc Am 95:711–718
Zchori-Fein E, Perlman SJ (2004) Distribution of the bacterial symbiont Cardinium in arthropods. Mol Ecol 13:2009–2016
Zchori-Fein E, Gottlieb Y, Kelly SE, Brown JK, Wilson JM, Karr TL, Hunter MS (2001) A newly discovered bacterium associated with parthenogenesis and a change in host selection behavior in parasitoid wasps. Proc Natl Acad Sci U S A 98:12555–12560
Zchori-Fein E, Lahav T, Freilich S (2014) Variations in the identity and complexity of endosymbiont combinations in whitefly hosts. Front Microbiol 5:1–8
Acknowledgments
We thank Hwal-Su Hwang at Kyungpook National University in the Republic of Korea for his help with sequencing and molecular analysis. This work was supported by the Research Program for Exportation Support of Agricultural Products, Animal and Plant Quarantine Agency, in the Republic of Korea under Grant (#Z-1543086-2017-21-01).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 218 kb)
Rights and permissions
About this article

Cite this article
Khatun, M.F., Shim, JK. & Lee, KY. Genetic diversity and host relationships of endosymbiotic bacteria in the Asian cryptic species of Bemisia tabaci from Bangladesh. Symbiosis 79, 75–87 (2019). https://doi.org/10.1007/s13199-019-00622-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13199-019-00622-6