
Abstract
The objective of the present study was to synthesize ω-3 polyunsaturated fatty acid esters of lutein and to evaluate if esterification can stabilize the both bioactive molecules. Both ω-3 polyunsaturated fatty acid and lutein are prone towards auto-oxidation in their free form. Free lutein extracted from the marigold petals was enzymatically esterified using Candida antarctica NS435 Lipase B, with the ω-3 long-chain polyunsaturated fatty acids. The lutein esters were purified, characterized and finally assessed for their protective role against oxidative degradation in bulk fish oil matrix. The antioxidative effect of these esters was compared with commercial antioxidants of natural origin, i.e., α-tocopherol and a synthetic antioxidant, i.e., tert-butylhydroquinone, at a dosage of 200 mg/L. Both free lutein and lutein–polyunsaturated fatty acid ester had significantly promoted the oxidative stability of bulk fish oil. But based on dose–response relationship, lutein–polyunsaturated fatty acid ester was found to be more efficient than free lutein, in protecting fish oil from secondary oxidation, thereby augmenting their shelf life. Given the high nutraceutical value, potent antioxidative potential and organic origin, it is only relevant to incorporate lutein esters as natural preservative and stabilizers in edible oils.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Antioxidants, both synthetic and naturally occurring, are used industrially to preserve the quality of the food products and extend their shelf-life. Synthetic antioxidants like butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT) and tert-butylhydroquinone (TBHQ) were extensively used in the oil industry. However recent reports suggest that extensive use of such synthetic antioxidants can cause carcinogenesis, mutagenesis, allergic contact dermatitis, etc. (Suja et al. 2004; Eskandani et al. 2014; Li et al. 2015).
Experimental as well as epidemiological reports provide tangible evidence that several dietary phytochemicals can efficiently quench reactive oxygen species both in vitro and in vivo (Wootton-Beard and Ryan 2011; Sen and Chakraborty 2011; Das et al. 2012). Lutein (C40H56O2, β,ε-carotene-3,3′-diol), an oxygenated carotenoid, is an extremely important nutraceutical with therapeutic effects against oxidative stress-related physiological maladies. Lutein, one of the six major carotenoids commonly found in human serum (β-carotene, lycopene, lutein, β-cryptoxanthin, zeaxanthin, and α-carotene) (Fu et al. 2010) serves as an indispensable component of the primate retinal tissue that shields it against the chronic oxidative stress (Olson et al. 2011). Marigold flower petals (Tagetes sp.), serve as the most abundant source where 70–90% of the pigment is lutein, mostly esterified with fatty acids like lauric, myristic and palmitic acids (Sowbhagya et al. 2004). Due to the presence of two hydroxyl groups at both the terminals of the central polyene chain, lutein has shown strong scavenging property against various reactive oxygen species (ROS) and reactive nitrogen species (RNS). Bhattacharyya et al. (2010) and Rodrigues et al. (2012) have revealed the strong protective efficacy of lutein in long-chain polyunsaturated fatty acids (LCPUFAs) like linolenic acid.
But lutein and lutein-enriched foods are sensitive to the degradation and isomerization induced by the thermal treatment beyond 40 °C and extended storage under strong light intensity (Aman et al. 2005; Provesi et al. 2011). Subagio et al. (1999) and Subagio and Morita (2003) have shown that esterification of lutein with fatty acids like myristic acid, lauric acid, and palmitic acid has stabilized lutein and promoted its antioxidant activity in bulk oil. The enhanced thermal stability of these ‘lutein phenolipids’ has been attributed to the esterification of the terminal hydroxyl groups. In similar studies, rutin esterified with LC-PUFAs showed a better antioxidative effect in the bulk phase sunflower oil than rutin esterified with short-chain fatty acids (Viskupicova et al. 2010). In addition to antioxidant activity, LCPUFA conjugated with phenolics have been reported to show significantly improved anti-inflammatory, antiviral and even anticancer activities in comparison to the parent molecules, suggesting that such phenolipids show synergistic activities (Shahidi and Zhong 2010).
The present work endeavored on the hypothesis that esterification of lutein with LC-PUFA concentrate would stabilize both lutein and the LC-PUFAs against oxidation. Lutein, reported in the present study, is extracted from marigold flowers whereas the PUFAs were concentrated from fish oil (FO) by urea complexation. The ester products generated during the conclusion of the experiment were further characterized and evaluated for their antioxidative potential in bulk FO systems. The qualitative, as well as the quantitative impact of the free lutein and the lutein–PUFA esters (LE) in protecting the bulk FO from oxidation, were assessed in comparison to the commercial antioxidants.
Materials and methods
Materials
Fish oil (Maxepa®, Merck Limited, Nani Daman, India) was used as a source for ω-3 LC-PUFAs. Lipase acrylic resin from Candida antarctica (CAS Number: 9001-62-1, Synonym: Novozyme 435, the activity of ≥ 5000 U/g of lipase obtained from Sigma-Aldrich, MO, USA). HPLC grade solvents like acetonitrile and ethyl acetate were procured from Spectrochem (India) and HPLC grade methanol was bought from Sigma-Aldrich (MO, USA). All the other solvents and reagents were of analytical grade and were bought from Sisco Research Laboratories (India).
Extraction of lutein from marigold flowers
Marigold flower petals were collected from local flower market in Kolkata, West Bengal, India and air-dried under shade at 40–45 °C. Air-dried petals were pulverized in a mixer-grinder, in the presence of anhydrous Na2SO4 (4:1, w/w) and passed through a fine mesh before extraction. The extraction was done according to Khalil et al. (2012). Briefly, 500 mg of powdered marigold petals was sonicated with 40 mL solvent (n-hexane/isopropanol, 3:2, v/v) at 20 kHz frequency and 40 W power (Microclean 101, Oscar Ultrasonics; Mumbai, India) for 1 min and incubated for 24 h at room temperature. The sample was then centrifuged at 1500 g for 5 min, and the supernatant was collected. The extraction step was repeated thrice. The extracted supernatants were pooled together in a separating funnel, washed with 20 mL 0.1 M NaCl, and incubating for 30 min until the aqueous, and the organic layer was separated. Hexane was removed at 40 °C under reduced pressure in a rotary evaporator. The remaining solid (marigold oleoresin) was collected and stored at − 20 °C until further use. 300 mg of oleoresin was saponified by refluxing with 5% KOH (50 mL) for 2 h, in the presence of 0.1% BHA to protect the xanthophylls from oxidation. The progress of the saponification reaction was monitored. After completion, 50 mL of distilled water was added to the reaction mix, and the unsaponified matter was extracted with hexane. The lutein-rich stock solution was stored at − 20 °C until further use.
The stock solution of the unsaponified fraction of the oleoresin in hexane was spotted onto silica gel (Silica gel G with CaSO4 binder, Spectrochem Pvt. Ltd., India) coated thin layer chromatographic plates. Hydrophobic mobile phase [Hexane:Diethyl ether (9:1, v/v)] was used for chromatographic separation and subsequent purification of lutein.
Analysis of free lutein by high-pressure liquid chromatography (HPLC)
The extracted, unsaponified samples were analyzed by Reversed phase-HPLC (RP-HPLC) to identify lutein. RP-HPLC analysis was carried out, according to Piccaglia et al. (1998), using an HPLC system (Waters, MA, USA) equipped with an Ultrasphere C18 reversed phase column (4.6 × 250 mm, i.d. 5 µm) (Hichrom, Leicestershire, UK) and a UV–Vis detector (Waters 2487 Dual λ Absorbance Detector, 190–700 nm; Waters, MA, USA). The mobile phase was a gradient solvent system composed of acetonitrile:methanol (9:1, v/v) (A) and ethyl acetate (B). The gradient system was run by linearly increasing solvent B from 0 to 100% over 30 min, at a flow rate of 1 mL/min. The sample injection volume was 10 μL, and the detection wavelength was set at 447 nm.
Preparation of PUFA enriched free fatty acid concentrate from FO
Commercially available FO (Maxepa™, Merck, India) was hydrolyzed to free fatty acids according to the method reported by Mbatia et al. (2011). Briefly, 10 mL of FO was mixed with 25 mL of 90% ethanol containing 1.5 g of NaOH with stirring and refluxed for 2 h. FFAs were recovered by lowering the pH to 2 using 2 N HCl neutralized and dried over anhydrous Na2SO4.
PUFA enrichment of the recovered FFA mixture was done according to the urea-inclusion method described by Chakraborty and Raj (2007). Urea was added to methanol at a ratio of 1:3 (w/v) under constant stirring and heated at 65–70 °C to prepare a transparent homogeneous solution. The fatty acids recovered from saponification of FOwere slowly added to the warm solution at a urea/fatty acid ratio of 4:1 (w/w). After complete dissolution, the suspension was cooled to room temperature and subsequently crystallized at 4 °C and − 20 °C for 24 h each. The urea-fatty acid complex crystals were separated from other fatty acids by vacuum filtration. The filtrate containing the unsaturated fatty acids was evaporated in a rotary vacuum evaporator at a temperature of 40–45 °C to remove the residual methanol, and the concentrated PUFAs were recovered using n-hexane. The PUFA concentrate (1 mL) was converted to fatty acid methyl esters (FAME) and analyzed by gas chromatography (Agilent 6890 N Gas chromatograph equipped with a DB-Wax capillary column; 30 m long × 0.25 mm × i.d. 0.25 mm and a flame ionization detector). The GC inlet temperature and FID detector temperature were maintained at 250 °C, and the oven temperature was maintained at 250 °C for 2 min, then temp was increased at 10 °C/min, up to 280 °C, held at 280 °C for 20 min. The gas flow rate was maintained at 1 mL/min, 300 mL/min and 30 mL/min for N2, H2 and air respectively (Dey et al. 2012).
Lipase-catalyzed esterification of PUFA with Lutein
Lipophilic derivatives of lutein was synthesized by enzymatic esterification method (Mbatia et al. 2011). Briefly, lutein (1 g) and PUFA concentrate (1.9 g) [1:5; Lutein:PUFA molar ratio] were solubilized in moisture-free acetone (100 mL). The enzymatic synthesis was initiated by the addition of immobilized Novozyme 435 lipase (20 g/L) and activated molecular sieves (60 g/L) were used for maintaining the proper water activity (aw < 0.01) to drive the forward reaction toward product formation. Reactions were continued at 40–45 °C with agitation (200 rpm) for up to 8 h and were continually monitored by thin layer chromatography, with a hydrophobic solvent [Hexane:Di-ethyl ether (9:1, v/v)]. To terminate the reaction, enzyme and molecular sieves were filtered off, and acetone was evaporated at 40 °C under reduced pressure.
Isolation of ester products
The ester products were successfully extracted into the organic phase (Mbatia et al. 2011), using 1 part of water and 4 parts of hexane (v/v). The lutein–PUFA ester was separated using a hydrophobic mobile phase containing Hexane:Diethyl ether (9:1) v/v. The solvent was evaporated under reduced pressure, and the yield of the product was estimated gravimetrically.
Characterization of the ester
Quantitative estimation of PUFA incorporation in the ester
A known amount of ester product was again saponified by refluxing for 1–2 h, with 5% methanolic KOH soln. The unsaponifiable part was extracted in n-hexane, washed with 0.1 M NaCl until the pH becomes neutral and finally collected as a moisture-free solution, by passing through a layer of anhydrous Na2SO4. The separated soap was treated with 2 N HCl, and the free fatty acids were extracted in hexane. The fatty acid part was converted to FAMEs and analyzed by GC, to assess the efficiency and specificity of the fatty acids towards the ester formation (Dey et al. 2012).
Analysis of lutein–PUFA esters by HPLC
Purified lutein–PUFA esters were extracted from the thin layer chromatography plate for analysis by RP-HPLC according to Piccaglia et al. (1998), as described earlier.
FT-IR analysis
Lutein esters and the free lutein that used as the substrate for the esterification were assessed for their IR absorption spectra. 100 µL from each of n-hexane solution of Lutein esters and the free lutein were separately absorbed in between two transparent of anhydrous KBr (4 mg for each pellet) and was analyzed across wavenumbers ranging from 4000 to 400 cm−1, in a Jasco FT/IR-6300 Fourier Transform Infrared spectrometer (Easton, MD, USA), using 64 scans to reach a spectral resolution of 4 cm−1. The successful esterification of the terminal hydroxyl groups of lutein were determined by the absorption bands corresponding to the functional groups like ester, carboxyl, and hydroxyl groups (Silverstein et al. 2005).
Calculation of partition coefficient (log P values)
The lipophilicity of the theoretically synthesized derivatives was determined using the Molinspiration online server (http://www.molinspiration.com/cgi-bin/properties; Molinspiration Cheminformatics, Bratislava, Slovak Republic).
Comparison of lutein and lutein–PUFA esters on oxidative stability of FO
Stabilization of FO against oxidation employing synthesized antioxidants
The antioxidative potential of the synthesized lutein–PUFA conjugate to protect commercial FO from oxidative rancidity was investigated. The FO was fortified with lutein and lutein–PUFA esters to a final concentration of 100 to 1000 mg/L. The results were compared to the antioxidative performance of a natural anti-oxidant, i.e., α-tocopherol and a synthetic antioxidant, i.e., TBHQ, at a standard dosage of 200 mg/L. A control FO sample with no added antioxidant was included. The samples were maintained for 15 days in a temperature controlled oven set at 37 °C. The formation of primary and secondary oxidation products was measured by assessing acid value, peroxide value and p-anisidine value according to AOCS Official Methods of Analysis, at different time points. The extent of lipoperoxidation was also determined by the Thiobarbituric Acid Reactive Substances assay (TBARS) (Harlina et al. 2015) at various time points. Briefly, 5 mg of oil sample was mixed with 2 mL of 0.67% TBA and 1 mL of 20% TCA, incubated in boiling water bath for 10 min. Thus, the formation of malondialdehyde (MDA) was measured spectrophotometrically at 532 nm, using reagent blank. The standard curve of malondialdehyde was used to calculate the concentration of MDA and expressed as mg of MDA/g sample.
Statistical analysis
All the experiments were done in triplicate, and the data were expressed as mean ± SD. One-way ANOVA was computed using Origin Pro 8 SR0 (v8.0724) software package (Northampton, MA, USA). The differences between means were accepted significant at P < 0.05.
Results and discussion
Extraction of de-esterified Lutein from marigold
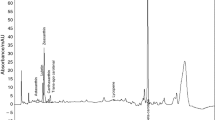
The present report deals with the extraction of lutein from marigold flower (Tagetes patula L.) petals of mixed cultivars. Micellar extraction of dried and ground marigold flower petals had an oleoresin yield of 8.69 ± 0.67% (w/w), rich in lutein esters of fatty acids from C10:0 to C18:3 (Table 1). KOH mediated saponification of this oleoresin had yielded free lutein from these esters. 31.43 ± 0.83% (w/w) of unsaponifiable carotenoid fraction was recovered from the oleoresin, of which, the free lutein concentration was found to be 71.7%, corresponding to the area under the curve (AUC) covered by Peak 1, in Fig. 1. Sowbhagya et al. (2004) had shown that lutein comprised 70% to 73% of the unsaponified carotenoids extracted from petals of Tagetes erecta L. which corroborates our observation. The overall yield of lutein from T. patula L. (Natchigal et al. 2012) was found to be 0.06 to 1.23 wt%. FTIR spectrum of the extracted lutein stock solution in hexane was depicted in Fig. 2. The corresponding –[OH] stretching vibration band between 3200 and 3600 cm−1 was represented by a broad peak. The bands corresponding to the symmetrical stretching vibration of the CH2 groups were represented by the peaks between 2862 and 3000 cm−1. The band assigned to the stretching vibrations of the –[C=C]– groups was at 1650 cm−1. The bands corresponding to bending vibrations in the –CH3 groups were found at 1449 cm−1 and 1386 cm−1 (Boonnoun et al. 2013).

HPLC profile of a carotenoids fraction extracted from marigold petals (red) and the lutein–PUFA ester (black). Peak-1 was identified as lutein (retention time 4.841 min; area % = 71.7%). Peaks 2, 3 and 4 represent the lutein–PUFA esters (retention time 6.102 min for peak 2) (color figure online)

FTIR spectrum of extracted lutein and synthesized lutein–PUFA ester. Hexane soln. of both the analytes was sandwiched between two KBr pellets and the analyzed along a wavenumbers range of 4000–400 cm−1 with pure hexane acting as a background
Preparation of PUFA concentrate
The fatty acid composition FO and the PUFA concentrate were shown in Table 2. The yield ofPUFA concentrate was 17.56 ± 0.45% (w/w), in term of the initial oil content. The concentration of α-linolenic acid (ALA, C18:3, ω-3), eicosapentaenoic acid (EPA, C20:5, ω-3) and docosahexaenoic acid (DHA, C22:6, ω-3) have accounted for 93.29 ± 0.14% of all fatty acids in the PUFA concentrate.
Enzyme-catalyzed synthesis of lutein–PUFA esters
The TLC based separation yielded of 46.4 ± 5.38% of resultant esters. Comparing IR spectrum of free lutein and synthesized lutein esters (Fig. 2), peaks at 1740 cm−1 (represents the –[C=O] vibrations in carbonyl groups) and 1258 cm−1 were identified as unique to the lutein–PUFA esters. It confirmed the presence of acyl groups, a prominent characteristic of esters (Silverstein et al. 2005). The second peak at 1256 cm−1 represented asymmetric –C–O–C– stretch. Besides, noticeable reduction in the intensity of –[OH] stretching vibration band between 3200 and 3600 cm−1, which corresponds to the formation of ester bond with the terminal –[OH] groups of lutein (Silverstein et al. 2005).
Theoretical lipophilicity of free lutein and lutein ester was expressed by the miLog P value, as calculated by the Molinspiration online server. The synthesized lutein esters with PUFAs like ALA (18:3), EPA (20:5), DHA (22:6) had increased hydrophobicity of the esters than free lutein, as described by their miLog P value (9.31 for free lutein, compared to 10.22–10.76 for lutein mono or diesters with ω-3 PUFA, Table 3). It was evident from the fact that the retention time of the PUFA enriched lutein esters in RP-HPLC chromatogram have increased (6.102 min, Fig. 1), as compared to the free lutein (4.841 min), extracted from the marigold petals.
In the present study, natural sources of lutein ester (Table 2) were restructured, by incorporating ω-3 PUFAs (C18:3 to C22:6, ~ 83%) (Table 2). DHA was quite high in the PUFA concentrate (52.37%) that was esterified with the lutein. Interestingly, the formation of lutein di-docosahexaenoate was sterically hindered due to large aliphatic side-chain of DHA that formed esters with lutein at lesser efficiency compared to other PUFAs. Viskupicova et al. (2010) have previously reported that the conversion yield of fatty acid esters gradually diminished with the increase in the chain length of the PUFAs. C. antarctica lipase B catalyzed the esterification of rutin with fatty acids of C4–C12 chain lengths gave a conversion yield of > 50%. Whereas with the same reaction protocol, significantly lower yields were obtained with longer fatty acids (C12–C18) (Ardhaoui et al. 2004; Viskupicova et al. 2010). In addition to the chain lengths, some unsaturations also influence the lipase specificity to a great extent. C. antarctica lipase B, unlike the other lipases, does not show any restraining effect (Mbatia et al. 2011).
Antioxidant activity of lutein esters
Carotenoids like β-carotene, lutein, zeaxanthin, etc. exert their antioxidant potential by quenching the high energy singlet oxygen or other highly reactive radicals and diffuse the energy through the delocalization of π-electrons over the extended conjugated double bonded polyene chain (Sparrow and Kim 2009; Widomska and Subczynski 2014). Nakagawa et al. (2009) illustrated the antioxidative effect of lutein (as food-grade lutein capsule, 9·67 mg lutein per capsule) on erythrocyte membrane phospholipid and subsequent cytotoxicity. In vitro antioxidative activity of lutein, extracted from marigold flowers was shown to safeguard against multiple reactive oxygen species, nitric oxide and lipid peroxidation in a concentration-dependent manner (Bhattacharyya et al. 2010). Interestingly UV-illumination and high temperature can inflict substantial loss of active lutein, whereas esterification of any one or both the terminal hydroxyl groups has shown to stabilize the lutein molecule against light and temperature-mediated degradation (Subagio et al. 1999).
Due to high lipophilicity of the synthesized lutein ester, the antioxidative activity of the PUFA enriched lutein derivative was assessed based on its capacity to prevent the formation of various oxidation products in FO in a concentration range of 100–1000 mg/L and compared to free lutein. Commercial synthetic antioxidant TBHQ and α-tocopherol at 200 mg/L were used as industry standards. Atmospheric moisture mediated hydrolysis rendered rancidity to the FO when incubated at 37 °C. Interestingly, compared to the industry standards, lutein–PUFA esters have shown a significantly higher protective effect against the rise in acid value (Fig. 3a). Evidently, dosage variation from 100 to 1000 mg/L had only incrementally affected the antioxidative efficiency of the ester. Free lutein was found to be efficient against free fatty acid build-up at higher concentration (≥ 400 mg/L).


Oxidation stability of FO during storage at 37 °C for 15 days in presence of the commercial antioxidants, free lutein (Lut) and synthesized lutein–PUFA ester (LE), in terms of a acid value, b peroxide value, cp-anisidine value, d TBARS value. Data presented as mean ± SD, n = 3. The differences between means were accepted significant at P < 0.05. FLO = fish oil in absence of any antioxidants, TBHQ = tert-butylhydroquinone (200 mg/L), standard synthetic antioxidant for commercial use, Toc = α-tocopherol (200 mg/L), standard natural-origin antioxidant for commercial usage, Lut1–Lut6 = free lutein, extracted from marigold petals, at concentrations 100, 200, 300, 400, 500, 1000 mg/L respectively, LE1–LE6 = enzymatically synthesized lutein–PUFA ester, at concentrations 100, 200, 300, 400, 500, 1000 mg/L respectively
Primary oxidation of results in the formation of hydroperoxides that further lead to the creation of carbonyls (in particular, aldehydes) that give the oil a rancid smell (Shahidi 1994). The concentration of primary oxidation products was measured by peroxide value, whereas secondary peroxidation products were assessed by p-anisidine value. Both lutein and lutein–PUFA ester have shown equivalent efficiency in protecting FO from primary oxidation (Fig. 3b). Antioxidative activity of both free lutein and lutein-LC-PUFA ester were found to be comparable with the commercial standards, at concentrations ≥ 400 mg/L. But results depict lutein–PUFA ester was more proficient in protecting FO from secondary oxidation than free lutein (Fig. 3c). At even 200 mg/L concentration, the antioxidative activity of lutein–PUFA ester was found to be comparable to TBHQ and α-tocopherol. The antioxidative activity of these esters was found to be positively correlated with the increase in concentration and significantly improved than the standards at high concentrations. Similar trends were also observed against the lipoperoxidation of FO, as evinced by TBARS assay (Fig. 3d). Lutein–PUFA ester was found to be more efficient than both the commercial standards; at a concentration > 500 mg/L. Free lutein had shown comparable antioxidant activity to α-tocopherol, at the concentration of 1000 mg/L, whereas among the standards, TBHQ was found to be more efficient than its natural alternative, i.e. α-tocopherol. Previously, Rupasinghe and Yasmin (2010) had shown the activity of natural antioxidants like α-tocopherol to be comparatively less efficient than synthetic antioxidants like BHT in protecting FO against lipoperoxidation.
As lutein esters have significantly improved lipophilicity compared to free lutein, at the same concentration, more lutein ester molecules exist in the oil phase, that explained the better antioxidative activity compared to free lutein. Murata et al. (2004) had described the significance of hydrophobicity of the natural flavonoids and their antioxidative potential. Recent reports have evinced that hydrophobicity had a positive impact on the antioxidative potential of a compound in lipophilic food systems. The long-chain fatty acid derivatives of rutin and vanillyl alcohol provide enhanced protection against oxidation of sunflower oil and PUFA enriched oils, both in bulk and oil in water emulsion systems (Viskupicova et al. 2010; Mbatia et al. 2011).
Conclusion
Bioorganic synthesis of novel “phenolipids” from natural components, for utilization in edible matrices, is the modern trend in health and nutrition research. The pharmacophore that resulted from the combination of both the PUFA and the bioactive natural phenolic, allowed for enhanced and efficient health benefits. Its pronounced activity at both in vitro and in vivo speaks of its popularity. In the present work, a novel lutein-ester, enriched in ω-3 PUFAs was synthesized using Candida antarctica NS435 Lipase B. Esterification with PUFAs has increased the lipophilicity of lutein, which in turn amplified its antioxidative potential in a lipophilic edible medium like FO, to an extent comparable to the antioxidants used in current commercial food products.
References
Aman R, Schieber A, Carle R (2005) Effects of heating and illumination on trans-cis isomerization and degradation of β-carotene and lutein in isolated spinach chloroplasts. J Agric Food Chem 53:9512–9518
American Oil Chemists’ Society, AOCS Official method no. Cd 3d-63 for measuring of acid value of Fats and Oils (reapproved 2009), Official method no. Cd 8b-90 for measuring Peroxide value using Isooctane (reapproved 2011), Official method no. Cd 18-90 for measuring anisidine value (reapproved 2011) in Official Methods and Recommended Practices of the AOCS; Firestone D (ed), 6th edn, 2013. Champaign. ISBN 978-1-893997-74-5
Ardhaoui M, Falcimaigne A, Ognier S, Engasser JM, Moussou P, Pauly G, Ghoul M (2004) Effect of acyl donor chain length and substitutions pattern on the enzymatic acylation of flavonoids. J Biotechnol 110:265–271
Bhattacharyya S, Datta S, Mallick B, Dhar P, Ghosh S (2010) Lutein content and in vitro antioxidant activity of different cultivars of Indian marigold flower (Tagetes patula L.) extracts. J Agric Food Chem 58:8259–8264
Boonnoun P, Nerome H, Machmudah S, Goto M, Shotipruk A (2013) Supercritical anti-solvent micronization of chromatography purified marigold lutein using hexane and ethyl acetate solvent mixture. J Supercrit Fluids 80:15–22
Chakraborty K, Raj RP (2007) Eicosapentaenoic acid enrichment from sardine oil by argentation chromatography. J Agric Food Chem 55:7586–7595
Das L, Bhaumik E, Raychaudhuri U, Chakraborty R (2012) Role of nutraceuticals in human health. Int J Food Sci Technol 49(2):173–183
Dey TK, Ghosh S, Ghosh M, Koley H, Dhar P (2012) Comparative study of gastrointestinal absorption of EPA & DHA rich fish oil from nano and conventional emulsion formulation in rats. Food Res Int 49:72–79
Eskandani M, Hamishehkar H, Dolatabadi JEN (2014) Cytotoxicity and DNA damage properties of tert-butylhydroquinone (TBHQ) food additive. Food Chem 153:315–320
Fu HF, Xie BJ, Fan G, Ma SJ, Zhu XR, Pan SY (2010) Effect of esterification with fatty acid of β-cryptoxanthin on its thermal stability and antioxidant activity by chemiluminescence method. Food Chem 122:602–609
Harlina PW, Shahzad R, Ma M, Geng F, Wang Q, He L, Ding S, Qiu N (2015) Effect of garlic oil on lipid oxidation, fatty acid profiles and microstructure of salted duck eggs. J Food Process Preserv 39:2897–2911
Khalil M, Raila J, Ali M, Islam KMS, Schenk R, Krause J, Schweigert FJ, Rawel H (2012) Stability and bioavailability of lutein ester supplements from Tagetes flower prepared under food processing conditions. J Funct Foods 4:602–610
Li J, Bi Y, Liu W, Sun S (2015) Simultaneous analysis of tertiary butylhydroquinone and 2-tert-butyl-1,4-benzoquinone in edible oils by normal-phase high-performance liquid chromatography. J Agric Food Chem 63:8584–8591
Mbatia B, Kaki SS, Mattiasson B, Mulaa F, Adlercreutz P (2011) Enzymatic synthesis of lipophilic rutin and vanillyl esters from fish byproducts. J Agric Food Chem 59:7021–7027
Molinspiration Cheminformatics, Bratislava, Slovak Republic, http://www.molinspiration.com/cgi-bin/properties. Accessed 12 Sept 2015
Murata Y, Osaki KI, Shimoishi Y, Baba N, Tada M (2004) Degradation of ethyl docosahexaenoate by gamma-ray irradiation and suppression of this degradation by antioxidants. Biosci Biotechnol Biochem 68:743–745
Nakagawa K, Kiko T, Hatade K, Sookwong P, Arai H, Miyazawa T (2009) Antioxidant effect of lutein towards phospholipid hydroperoxidation in human erythrocytes. Br J Nutr 102:1280–1284
Natchigal AM, Stringheta AC, Bertoldi MC, Stringheta PC (2012) Quantification and characterization of lutein from tagetes (Tagetes patula L.) and calendula (Calendula officinalis L.) flowers. Acta Hortic 939:309–314
Olson JH, Erie JC, Bakri SJ (2011) Nutritional supplementation and age-related macular degeneration. Semin Ophthalmol 26(3):131–136
Piccaglia R, Marotti M, Grandi S (1998) Lutein and lutein ester content in different types of Tagetes patula and T. erecta. Ind Crops Prod 8:45–51
Provesi JG, Dias CO, Amante ER (2011) Changes in carotenoids during processing and storage of pumpkin puree. Food Chem 128:195–202
Rodrigues E, Mariutt LR, Mercadante AZ (2012) Scavenging capacity of marine carotenoids against reactive oxygen and nitrogen species in a membrane-mimicking system. Mar Drugs 10:1784–1798
Rupasinghe HPV, Yasmin A (2010) Inhibition of oxidation of aqueous emulsions of omega-3 fatty acids and fish oil by phloretin and phloridzin. Molecules 15:251–258
Sen S, Chakraborty R (2011) The role of antioxidants in human health. In: Andreescu S, Hepel M (eds) Oxidative stress: diagnostics, prevention, and therapy, chap 1. American Chemical Society, Washington, pp 1–37. eISBN 9780841226845
Shahidi F (1994) Assessment of lipid oxidation and off-flavour development in meat and meat products. In: Shahidi F (ed) Flavor of meat and meat products. Springer, Boston, pp 247–266
Shahidi F, Zhong Y (2010) Novel antioxidants in food quality preservation and health promotion. Eur J Lipid Sci Technol 112:930–940
Silverstein RM, Webster FX, Kiemle D (2005) Spectrometric identification of organic compounds, 7th edn. Wiley, San Francisco
Sowbhagya HB, Sampathu SR, Krishnamurthy N (2004) Natural colorant from marigold-chemistry and technology. Food Rev Int 20(1):33–50
Sparrow JR, Kim SR (2009) The carotenoids of macular pigment and bisretinoid lipofuscin precursors in photoreceptor outer segments. In: Landrum JT (ed) Carotenoids: physical, chemical and biological functions and properties, chap 16. CRC Press, Boca Raton, pp 355–363
Subagio S, Morita N (2003) Prooxidant activity of lutein and its dimyristate esters in corn triacylglyceride. Food Chem 81:97–102
Subagio S, Wakaki H, Morita N (1999) Stability of lutein and its myristate esters. Biosci Biotechnol Biochem 63(10):1784–1786
Suja KP, Abraham JT, Thamizh SN, Jayalekshmy A, Arumughan C (2004) Antioxidant efficacy of sesame cake extract in vegetable oil protection. Food Chem 84:393–400
Viskupicova J, Martina D, Miroslav O, Tibor L, Ernest S (2010) Lipophilic rutin derivatives for antioxidant protection of oil-based foods. Food Chem 123:45–50
Widomska J, Subczynski W (2014) Why has nature chosen lutein and zeaxanthin to protect the retina? J Clin Exp Ophthalmol 5:326
Wootton-Beard PC, Ryan L (2011) Improving public health: the role of antioxidant-rich fruit and vegetable beverages. Food Res Int 44(10):3135–3148
Acknowledgements
Debjyoti Paul revised the final manuscript in the final stages of revision with valuable inputs.
Funding
Funding was provided by Council of Scientific and Industrial Research MHRD, Govt. of India (Grant No. 09/028 (0926)/2014, EMR-I, date 07/08/2014).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors also declare that there is no conflict of interest between them.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dey, T.K., Maiti, I., Chakraborty, S. et al. Enzymatic synthesis of lipophilic lutein–PUFA esters and assessment of their stabilization potential in EPA–DHA rich fish oil matrix. J Food Sci Technol 56, 2345–2354 (2019). https://doi.org/10.1007/s13197-019-03588-x
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13197-019-03588-x