
Abstract
Cancer immunotherapy, which leverages features of the immune system to target neoplastic cells, has revolutionized the treatment of cancer. The use of these therapies has rapidly expanded in the past two decades. Immune checkpoint inhibitors represent one drug class within immunotherapy with its first agent FDA-approved in 2011. Immune checkpoint inhibitors act by disrupting inhibitory signals from neoplastic cells to immune effector cells, allowing activated T-cells to target these neoplastic cells. Unique adverse effects associated with immune checkpoint inhibitors are termed immune-related adverse effects (irAEs) and are usually immunostimulatory in nature. Almost all organ systems may be affected by irAEs including the dermatologic, gastrointestinal, pulmonary, endocrine, and cardiovascular systems. These effects range from mild to life-threatening, and their onset can be delayed several weeks or months. For mild irAEs, symptomatic care is usually sufficient. For higher grade irAEs, discontinuation of therapy and initiation of immunosuppressive therapy may be necessary. The management of patients with irAEs involves multidisciplinary care coordination with respect to the long-term goals the individual patient. Clinicians must be aware of the unique and sometimes fatal toxicologic profiles associated with immunotherapies to ensure prompt diagnosis and appropriate management.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
The pharmacologic treatment of cancer has evolved in the last two decades as agents with novel mechanisms of action have successfully transitioned from the bench to the bedside. Traditional cytotoxic chemotherapy, which nonspecifically targets cell proliferation, is associated with sometimes serious adverse effects to multiple organ systems. The immune system is a commonly affected organ system and cytotoxic chemotherapy frequently results in immunosuppression in addition to anti-tumor effects. Targeted therapies have since been developed which leverage features of the body’s innate immune system to direct immune cells to target neoplastic cells [1]. These therapies are collectively termed immunotherapy.
Since the introduction of rituximab as the first FDA-approved monoclonal antibody for the treatment of cancer in 1997, immunotherapy agents, mechanisms, and indications have expanded [2]. Given the rapid growth of immunotherapy, clinicians must be aware of the unique adverse effects associated with their use to ensure timely diagnosis and appropriate management. The purpose of this two-part narrative review is to mechanistically categorize the immunotherapy agents used in the treatment of cancer, examine their respective toxicities, and describe treatment considerations. This first part of a two-part series will focus on immune checkpoint inhibitors.
Methods
Relevant articles were identified through PubMed using the following query: "Drug-Related Side Effects and Adverse Reactions""[Mesh]) AND ""Antineoplastic Agents""[Mesh]) AND ""Immunotherapy""[Mesh]. The database was queried for all article types, including clinical trials, meta-analyses, reviews, and practice guidelines. We searched through the date August 8, 2020. Additional references were identified through the search of publications’ bibliographies. For the purposes of this two-part review, immunotherapy will be subcategorized as immune checkpoint inhibitors, adoptive cellular therapies, kinase inhibitors, other monoclonal antibodies, and oncolytic viruses [1].
Immune Checkpoint Inhibitors
Ipilimimab became the first FDA-approved immune checkpoint inhibitor (ICI) in 2011 for the treatment of advanced melanoma [3]. Since then, 6 other ICIs have been approved for use in the United States (Table 1), with several others in clinical trials. Initial studies indicated improved mortality in patients with advanced-stage melanoma [3,4,5]. Subsequent studies have highlighted the efficacy of these medications, even in patients who were previously considered untreatable [6,7,8,9,10]. Indications continue to expand and include Hodgkin’s lymphoma and several solid tumors such as non-small cell lung cancer, colorectal cancer, and renal cell cancer [11]. ICIs are used either as monotherapy or in conjunction with other agents, including other ICIs (dual immune checkpoint blockade). They are typically dosed in cycles every 2-3 weeks [12].
Mechanism of Action
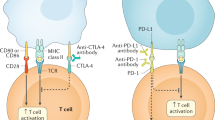
ICIs are monoclonal IgG antibodies, which act by disrupting inhibitory signals which inactivate cellular immune effector cells. The physiologic role of immune checkpoints is to limit the immune response and therefore allow self-tolerance by turning cytotoxic T-cells “off” [13]. Native cells use these checkpoints to avoid tissue damage from activated T-cells. Some cancer cells exploit these checkpoints by interacting with receptors on cytotoxic T-cells in order to evade host immunity. Immune checkpoints are therefore both a mechanism by which cancer cells can escape immuno-surveillance and also a promising therapeutic target [14]. By disrupting the interaction between immune checkpoints and cancer cells, ICIs allow T-cells to remain activated and target these cells (Fig. 1). Current surface receptor and ligand targets for ICIs include cytotoxic T-lymphocyte antigen 4 (CTLA-4), programmed death receptor 1 (PD-1), and its ligand, programmed death-ligand 1 (PD-L1). These receptors and ligands are overexpressed in certain tumor microenvironments, explaining their efficacy in cancer therapy [15].

Immune checkpoint inhibitor mechanism of action (PD-1) (reproduced with permission from Terese Winslow LLC). PD-1, programmed death receptor 1; PD-L1 programmed death-ligand 1.
Overview of Toxicity
In clinical trials for several cancer types, checkpoint inhibitors have shown favorable outcomes in both tumor regression and patient survival. However, unique adverse effects are common due to non-specific immunostimulation which may cause organ-specific inflammation, tissue damage, and autoimmunity [11]. Non-cancerous tissue infiltration by dysregulated T-lymphocytes is a likely mechanism of toxicity, although increased production of antibodies has been described [16,17,18,19,20]. Clinical experience with ICIs and their unique adverse effects, termed immune-related adverse effects (irAEs), has increased since 2011 [21]. To date, there are no well-described reports of ICI overdose in the medical literature.
While ICIs typically produce more mild toxicity than traditional cytotoxic chemotherapy, irAEs can still cause significant morbidity and are sometimes fatal [22, 23]. Up to 85% of patients report irAEs after treatment with ipilimumab, which targets CTLA-4, and up to 70% of patients report irAEs after treatment with inhibitors of the PD-1 axis [16, 24]. High grade organ-specific toxicities for agents which act on the PD-1 axis are similar between agents but some organ systems appear to be more sensitive to certain agents [25]. These specific agents are discussed in more detail in the sections corresponding to specific organs. Toxicity is dose-related for certain agents, and patients receiving combination ICI therapy appear to have an increased incidence of toxicity [22, 26]. Severe irAEs were nearly 40% higher with dual checkpoint blockade compared to monotherapy in one study [7]. Typically, irAEs are organ-specific with certain organ systems displaying increased vulnerability, such as the skin and gastrointestinal tract. Additionally, there is variability in the likelihood of individual organ irAEs based on the specific ICI. In general, colitis and hypophysitis are more common with anti-CTLA-4 antibodies, and thyroid dysfunction and pneumonitis are more common with antibodies impairing the PD-1 axis [27, 28].
Toxicities due to ICIs are due to immunostimulation and mimic autoimmune diseases [11]. In order to standardize reporting of irAEs, they are graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE, Table 2). Although these criteria were primarily developed to standardize reporting of AEs for clinical trials, they are used clinically to direct management [13]. In general, patients with grade 1 or 2 toxicity can either continue treatment with an ICI or have a brief interruption for treatment and monitoring. Patients with severe toxicity (grades 3 or 4) should have their ICI stopped and are generally not re-challenged with ICIs in the future due to the risk of toxicity recurrence [30, 31]. Patients with grade 3 and 4 irAEs are typically managed in a hospital setting. Any decision to restart an ICI should be made following a discussion between the patient and their oncologist with consideration of their organ-specific toxicity, likelihood of recurrence, prognosis, and potential alternative therapies.
The development of immunostimulation, as evidenced by the presence of irAEs, appears to be associated with a beneficial cancer response and prolonged survival. Several irAEs, including vitiligo, hypophysitis, enterocolitis, and pneumonitis, have been associated with favorable tumor response or prolonged survival [32]. These findings, however, should be interpreted with caution due to potential survivorship bias as patients with a positive response to ICI therapy survive longer and therefore have a longer time course in which to develop and report irAEs [13, 33].
Management of irAEs is organ-specific and requires a multidisciplinary approach to ensure the best clinical outcome with respect to the long-term goals of individual patients. General management is with immunosuppression typically using corticosteroids with or without cessation of the ICI depending on the organ involved and severity of toxicity. For grade 2 irAEs, topical or oral corticosteroids may be appropriate initially. For high-grade irAEs, systemic intravenous corticosteroids are recommended. Although corticosteroid response may be immediate, treatment is typically needed for a month or longer for severe irAEs [26]. Although consideration of the adverse effects due to the prolonged use of corticosteroids is necessary for clinicians, they are beyond the scope of this review. There is emerging evidence that as many as one-third of patients with severe irAEs may not respond to systemic corticosteroids [24]. While evidence is limited to case reports and case series, other immunomodulatory agents may be indicated in the management of severe corticosteroid refractory irAEs [34]. Agents such as anti-thymocyte anti-globulin, infliximab (anti-TNF-alpha mAb), tocilizumab, and rituximab have all been used for their immunosuppressive effect [29, 34,35,36]. Given the organ-specific nature of toxicity, this review will use an organ system-based approach to describe potential complications of ICI therapy.
Time-Course
It is important for both patients and clinicians to appreciate the time-course associated with irAEs. Compared to cytotoxic chemotherapy, the development of irAEs can be delayed. The average time to onset for irAEs is 6 to 12 weeks from the initial dose, although rarely some may occur up to one year from last administration [18, 37,38,39]. Dermatologic and gastrointestinal toxicities are among the first to develop, while endocrine toxicity often arises after more than 6 weeks from the last administration [30]. Clinicians must maintain a high index of suspicion for irAEs in patients who have had ICIs within the past year. In patients who have had ICI therapy, any symptom must be considered as a potential irAE [40]. Similarly, patients should be counseled on irAEs and be aware that initial symptoms may be vague or mild [41]. Predictive biomarkers for the development of specific irAEs are not commonly used clinically but represent a promising future area of research, which may potentially allow for earlier recognition of mild irAEs [42,43,44].
Cardiac
ICI-related cardiotoxicity is rare but potentially fatal, requiring a high index of suspicion by treating clinicians. It has been estimated to occur in approximately 0.09% of patients receiving ICI therapy although the true incidence may be higher [17, 35, 45]. Combination ICI therapy is associated with a 5 times higher risk for ICI-related cardiotoxicity when compared to monotherapy [17]. Both PD-1 and PD-L1 are expressed in human cardiomyocytes, lending plausibility to lymphocyte-medicated cardiac toxicity [46]. Myocarditis, dilated cardiomyopathy, pericarditis, pericardial effusion, and arrhythmias have all been described along with fatal cases of heart failure [35, 46, 47]. The median time to onset is 17 to 34 days from treatment initiation but may occur 6 weeks or longer into treatment [48,49,50,51]. Mortality due to ICI-associated cardiotoxicity is high, with estimates as high as 27%-50% [49, 52].
Symptoms of ICI-related cardiotoxicity vary widely and include fatigue, myalgias, chest pain, dyspnea, and syncope [45]. Diagnosis is aided by the presence of cardiac biomarker elevation, which is almost universally present in cases of ICI-related myocarditis [35, 49]. Elevations in serum troponin levels may be predictive of major cardiac sequelae such as cardiac arrest or cardiogenic shock in patients with ICI-related myocarditis [47, 49]. The median admission troponin elevation in patients with ICI-related myocarditis and major adverse cardiac event was almost forty times the upper limit of normal [49]. Additional laboratory testing with brain natriuretic peptide (BNP) or N-terminal pro-brain natriuretic peptide (NT-proBNP) may be helpful in the evaluation of volume overload due to heart failure but is neither diagnostic nor prognostic in the setting of ICI-related myocarditis [45, 52]. Electrocardiogram (ECG) changes are common but not always present. Echocardiography may show left ventricular systolic dysfunction in up to 79% of patients with ICI-associated cardiotoxicity and should be performed in all patients with suspected ICI-associated cardiotoxicity given the risk for pericardial effusion and cardiac tamponade [52]. Cardiac MRI may not have adequate sensitivity for the evaluation of ICI-related myocarditis [53]. Endomyocardial biopsy is the gold standard for diagnosis of ICI-associated myocarditis but the decision to perform a biopsy should be made after weighing potential benefits in diagnosis against potential risks of the procedure including myocardial perforation, arrhythmia, and heart block [35]. In general, biopsy should only be performed if the diagnosis is in question after other diagnostic studies and will impact treatment decisions. A T-cell predominant lymphocytic infiltration of the myocardium is the expected finding if biopsy is performed [49]. However, due to the patchy involvement of ICI-associated myocarditis, negative results should be interpreted with caution [45].
Patients should be closely monitored for life-threatening complications, including arrhythmias and heart failure, and managed in consultation with a cardiologist. Pericardial effusion with tamponade physiology warrants prompt pericardial drainage. Given the potential for sudden cardiac death, ICI cessation and immediate initiation of systemic high-dose corticosteroid treatment are critical [54]. An initial dose of 1-2 mg/kg/day of intravenous methylprednisolone is likely sufficient in the acute setting followed by a slow taper of oral prednisone or prednisolone in the outpatient setting [35, 53, 54]. Corticosteroid therapy may increase the likelihood of left ventricular function recovery by up to 50% [52]. For patients without an immediate response to corticosteroids, dose escalation to methylprednisolone 1 gram/day should be considered [54]. Other immunosuppressive agents such as mycophenolate mofetil, anti-thymocyte globulin (ATG), and tacrolimus, as well as plasmapheresis have been proposed for the treatment of steroid-refractory myocarditis but there is currently a dearth of evidence supporting their use [35]. Infliximab, a monoclonal antibody against the inflammatory cytokine TNF-alpha, has also been proposed but carries the potential to precipitate new-onset congestive heart failure, possibly worsening the course of ICI-associated cardiotoxicity [55]. In contrast to most other irAEs, it is recommended that ICI therapy be temporarily discontinued for grade 1 toxicity and permanently discontinued for cardiac adverse events of grade 2 or higher [29, 56].
Cytokine Release Syndrome
Cytokine release syndrome is a systemic inflammatory response characterized by the systemic release of cytokines. Patients will typically develop fever, chills, hypotension, and tachycardia [11]. It is a rare complication of ICI therapy. Case reports have described the spectrum of symptoms and complications seen with cytokine release syndrome associated with ICI therapy [57,58,59,60,61,62]. An international registry study found that cytokine release syndrome represented only 0.14% of all ICI-related adverse drug reactions [63]. This contrasts with adoptive cellular therapies, another type of cancer immunotherapy, where cytokine release syndrome is far more common. The timing of cytokine release syndrome following ICI therapy ranges from 1 to 18 weeks with a median of 4 weeks [63]. Severe cases may be life-threatening. The diagnosis of ICI-related cytokine release syndrome is difficult due to its rarity, similarity with other disease processes such as sepsis, and potentially delayed onset. It is therefore vital for clinicians to elicit a history of ICI therapy and maintain a broad differential diagnosis. Simultaneous treatment for cytokine release syndrome and sepsis may therefore be prudent until a definitive diagnosis can be established. Further discussion of the diagnostic considerations and management of cytokine release syndrome is provided in part II of this review in the section on adoptive cellular therapies.
Dermatologic
Dermatologic toxicities are the most common irAEs, occurring in 40-50% of patients treated with ipilimumab and 30-40% of patients treated with ICIs which target the PD-1 axis [64]. Of agents acting on the PD-1 axis, nivolumab carries the highest risk [25]. While rarely life-threatening, they may greatly diminish the quality of life for patients [65]. Common skin irAEs include erythema, pruritus, vitiligo-like hypopigmentation, lichenoid reactions, eczema, and morbilliform eruptions. The presence of vitiligo appears to only occur in patients with melanoma and is associated with a favorable cancer prognosis [66, 67]. Rarely, eruptions such as papulopustular eruptions and ulcerations mimicking pyoderma gangrenosum may occur. Low-grade dermatologic irAEs include any of the above eruptions which occupy less than 10% of total body surface area (grade 1) or between 10% and 30% of total body surface area (grade 2). Grade 3 reactions affect over 30% of the total body surface area or significantly affect activities of daily living. Grade 4 reactions are life-threatening [68]. Grade 3 and 4 reactions are rare (1-4% of dermatologic irAEs) but include exfoliative diseases such as Stevens-Johnson syndrome/toxic epidermal necrolysis and drug reaction with eosinophilia and systemic symptoms (DRESS) [64, 68, 69]. Hair re-pigmentation and alopecia have also been reported with ICI therapy but are of unclear clinical significance [70, 71]. Dermatologic complications will typically appear 3 to 8 weeks after the initiation of therapy and are dose-dependent [3, 65].
Skin toxicity is typically mild and managed with symptomatic therapies and topical agents. Patients should be counseled on photoprotection with clothing, hats, and sunscreens to avoid sunburn, especially if vitiligo is present. If the diagnosis of a dermatologic irAE is in question, consultation with a dermatologist should be sought. Skin biopsy may assist in the diagnosis, particularly for a prolonged course of rash or for a persistent rash unresponsive to treatment [66]. Grade 1 and grade 2 skin toxicity can be managed with topical corticosteroids, skin emollients, and antihistamines without interruption of the ICI treatment schedule [31, 72]. For grade 1 and 2 skin toxicity that does not improve with this approach, ICI treatment should be temporarily interrupted and a systemic corticosteroid considered [66]. For rare grade 3 or 4 toxicity, systemic corticosteroids should be initiated and the ICI therapy permanently discontinued.
Endocrine
Endocrine dysfunction following ICI therapy is common and potentially severe. While the thyroid gland is most commonly affected, dysfunction of the pituitary gland is often life-threatening. Other endocrine irAEs include diabetes mellitus and adrenal insufficiency, but these are less common [73]. Thyroid dysfunction can manifest as hypothyroidism, hyperthyroidism, or thyroiditis. Hypothyroidism may be primary, due to thyroid dysfunction, or secondary, due to pituitary gland dysfunction. ICI-related thyroid dysfunction is reported in up to 20% of patients on ICI therapy and is more common in patients treated with anti-PD-1 therapy [74]. This is possibly due to the expression of PD-1 on all B cell surfaces including those of IgM-secreting memory B cells [43]. As such, antibody-mediated thyroid dysfunction is common in patients undergoing therapy with PD-1-based ICIs. Anti-thyroid antibodies have been detected in up to 80% of patients with ICI-related hypothyroidism, highlighting the similarities with autoimmune thyroid disease [75]. Hyperthyroidism is often transient, likely underdiagnosed due to lack of symptoms, and typically requires no treatment unless the patient is significantly symptomatic [76, 77]. ICI-related hyperthyroidism will typically lead to a permanent hypothyroid state within 3 to 6 weeks [78,79,80]. Preventative corticosteroids have not been shown to prevent progression to hypothyroidism in patients with ICI-related hyperthyroidism. Diagnosis of thyroid dysfunction can be confirmed with laboratory testing for serum thyroid-stimulating hormone (TSH), triiodothyronine (T3), and thyroxine (T4). Lifelong thyroid hormone replacement is necessary in patients with symptomatic immune-related hypothyroidism as thyroid gland dysfunction is typically permanent [79].
Pituitary gland inflammation, termed hypophysitis, was previously a rare disease characterized by immune-cell infiltration of the pituitary gland. Since the approval of ipilimumab, hypophysitis has become more common. The incidence of hypophysitis ranges from 1-4% with low-dose ipilimumab therapy to up to 17% with high-dose therapy [81, 82]. The incidence with other ICIs is less than 0.5% [28]. The exact pathophysiologic mechanism for this discrepancy remains unclear, but is possibly related to CTLA-4 expression in human pituitary cells as well as an increased production of antibodies against pituitary cells [83]. It is more common in males, but this may be partially attributable to the greater incidence of melanoma in males [76, 81]. The typical time from ICI-initiation to the diagnosis of hypophysitis is 6-12 weeks but it has been reported up to 16 weeks after ICI treatment [76]. In patients receiving low-dose ipilimumab, the median time of onset is more delayed (11 weeks) than with high-dose therapy, indicating a potential cumulative effect following repeated doses [84]. Hypophysitis presents in most patients with vague and non-specific symptoms such as headache, fatigue, and malaise, making early diagnosis difficult especially in older adults [11, 85]. More severe symptoms include confusion, lethargy, and altered mental status [73, 79]. Visual complaints due to optic pathway disruption are rare with ICI-related hypophysitis compared with other causes.
The diagnosis should be confirmed with laboratory findings of hypopituitarism and MRI abnormalities, given the nonspecific symptoms and potentially insidious onset [86]. MRI evaluation of will typically reveal pituitary enlargement, heterogeneous enhancement, and stalk thickening, although imaging be normal in some cases [86,87,88]. One or multiple pituitary hormone axes may be affected. Deficiencies in TSH and adrenocorticotropic hormone (ACTH) are the most common and lead to central hypothyroidism and central adrenal insufficiency, respectively [73, 79]. Central adrenal insufficiency may be life-threatening if untreated. Less commonly, gonadotrophin and growth hormone deficiencies may be seen [73]. If endocrine dysfunction is suspected, patients should undergo laboratory evaluation including serum TSH, T3, T4, and ACTH.
Physiologic hormone replacement in consultation with an endocrinologist is the mainstay of treatment for ICI-associated hypophysitis following a thorough evaluation of endocrine hormone dysfunction. Systemic high-dose steroids should be commenced prior to thyroid hormone replacement to prevent an adrenal crisis. While adrenal recovery is rare, thyroid and gonadal function does recover in some patients. ICI therapy should be permanently discontinued in patients with hypophysitis.
Gastrointestinal and Hepatic
Immune checkpoint inhibitors are frequently associated with both luminal gastrointestinal (GI) adverse effects as well as hepatic effects. Immune-related pancreatitis is rare but has also been described in patients with melanoma as well as those with solid tumors [89, 90]. The GI effects mimic idiopathic inflammatory bowel disease and differentiation between these conditions is difficult [91]. Colitis is a common irAE and may be severe. In patients treated with ipilimumab, diarrhea occurs in approximately one-third of patients with 8-23% experiencing colitis [92]. GI irAEs are less common with ICIs which act on the PD-1 axis, with colitis occurring in fewer than 4% of patients [92]. An exception to this is nivolumab, which carries an incidence for diarrhea of 10-13% [25]. The onset of symptoms is highly variable and ranges from 11 days to 4 months with a median of 34 days following ICI-therapy [93, 94]. Diarrhea is the most common symptom of colitis, and additional symptoms can include abdominal pain, emesis, fever, weight loss, and hematochezia [95]. Electrolyte abnormalities including hypokalemia and hyponatremia may occur [96]. In addition to an immunostimulatory mechanism of colitis, there is evidence that alterations in the gastrointestinal microbiome may predispose patients to colitis [43, 97, 98]. Patients and providers should therefore be cautious in initiating antibiotic therapy unless clearly indicated. Colonic perforation is a rare complication and possibly dose-dependent [93, 99]. The differential diagnosis for patients on ICI therapy with diarrhea is broad, and patients should be evaluated for other causes including Clostridium difficile toxin and other diarrhea-causing pathogens.
Definitive diagnosis is with biopsy, although frequently unnecessary as other diagnostic modalities such as computed tomography (CT) may provide evidence of colitis [22]. CT findings share a similar appearance with inflammatory bowel disease [100]. Specific findings include diffuse colitis involving more than one segment of the colon, mural bowel wall thickening, and submucosal edema [88]. Treatment of grade 1 toxicity is largely supportive, with fluid and electrolyte repletion. The addition of corticosteroids should be considered with grade 2 colitis and commenced in all cases of grade 3 or 4 colitis [95]. Gastroenterologist consultation and endoscopic evaluation are recommended in cases of grade 2 toxicity that do not respond to supportive measures and in most cases of grade 3 and 4 toxicity if the diagnosis is uncertain [93]. Endoscopic examination typically shows a continuous pattern of inflammatory changes with exudates, granularity, and ulcerations [91]. Biopsy can be performed during endoscopy if the diagnosis is in doubt but must be weighed against the risk for perforation [101]. For grade 3 and 4 colitis, ICI therapy should be permanently discontinued and systemic corticosteroids commenced (1-2 mg/kg/day of methylprednisolone or its equivalent) [93]. A single dose of infliximab (5 mg/kg) has been used successfully in cases where there is a failure to respond to corticosteroids after 3-5 days [102,103,104]. Relapses of colitis are common. As such, corticosteroids should be tapered over a period of 6 to 8 weeks. Prophylactic corticosteroids have not been shown to prevent the development of diarrhea or colitis [105, 106].
Immune-related hepatitis resembles an autoimmune-like drug-induced liver injury and consists of asymptomatic elevations of hepatic transaminases [107]. Rarely, serious hepatitis and liver failure can occur [108, 109]. ICI-associated hepatitis is most often panlobular with a hepatocellular injury pattern [110, 111]. Hepatic irAEs are less common than luminal irAEs, occurring in around 4% of patients [112]. The risk is higher in patients receiving ICI therapy for the treatment of hepatocellular carcinoma [113, 114]. CTCAE grading for hepatitis is based on serum transaminase and total bilirubin measurements [85]. Transaminase levels of 3-5, 5-20, and 20x the upper limit of normal are categorized as grades 2, 3, and 4, respectively. Similarly, total bilirubin concentrations of 1.5-3, 3-10, and 10x the upper limit of normal are categorized as grades 2, 3, and 4, respectively. The risk for severe (grade 3 or 4) hepatitis is dose-dependent and more frequent with dual immune checkpoint inhibition [115]. Immune-related hepatitis typically presents 8 to 12 weeks after initiation of ICI therapy [91]. The diagnosis of ICI-related hepatitis may be suggested by histologic examination of the liver [116,117,118]. Alternative causes of hepatitis, including viral hepatitides and other drug-induced hepatitides, should be excluded [118]. For grade 2 hepatitis, ICI therapy should be temporarily interrupted until serum transaminase levels are declining [36]. For grade 3 or 4 hepatitis, ICI should be stopped and systemic corticosteroids (methylprednisolone 1-2 mg/kg/day) commenced. Corticosteroids should be tapered over a time period of at least one month or until toxicity is downgraded to grade 1 [113]. Based on published case reports, mycophenolate mofetil is recommended in cases unresponsive to corticosteroids [29, 36]. Other agents, such as anti-thymocyte globulin, have been used but available evidence is currently limited to case reports. Infliximab should be avoided due to the potential provocation of fulminant hepatitis [119].
Neurologic
Neurologic irAEs occur in 6 to 12% of patients on ICI therapy and are generally of low grade. Mild neurologic irAEs include nonspecific symptoms such as headache, dizziness, and sensory impairment [120]. High-grade neurologic AEs are rare, occurring in fewer than 1% of patients, and should be managed in consultation with a neurologist [121]. These include meningitis, encephalitis, Bell’s palsy, Guillain-Barre syndrome, central nervous system demyelination, and myasthenia gravis [69, 122]. Seizure activity is a common presenting symptom of encephalitis [120]. Antibodies to the N-methyl-D-aspartate and Hu receptors have been detected in some patients with encephalitis associated with ICI therapy [123, 124]. Diagnosis may be difficult as progression of cancer should also be on the differential diagnosis in any patient with malignancy and a neurologic abnormality. Central nervous system imaging studies and cerebrospinal fluid analysis (CSF) should be performed as needed to evaluate for alternative causes for the clinical presentation. In cases of ICI-related aseptic meningitis, a lymphocyte predominance will be noted on CSF analysis [125, 126]. If a neurologic irAE is suspected and severity is grade 2 or higher, ICI therapy should be interrupted and systemic corticosteroids (methylprednisolone 0.5-1 mg/kg/day for grade 2, greater than 1 mg/kg/day for higher grades) commenced, even for conditions for which corticosteroids are not typically administered (e.g., Guillain-Barre syndrome) [127]. For the treatment of myasthenia gravis and Guillain-Barre syndrome, intravenous immunoglobulin (400 mg/kg for 5 days) or plasmapheresis should be commenced along with pyridostigmine (myasthenia gravis) in consultation with a neurologist [34, 120, 122, 128]. Rituximab, an anti-CD20 monoclonal antibody, has been used in cases of ICI-associated encephalitis unresponsive to corticosteroids and intravenous immunoglobulin but available literature is limited to case reports [124, 129].
Ocular
Ocular irAEs in patients receiving ICI therapy are rare (<1% of patients) [130]. The most commonly reported irAE is dry eyes but several others have been described including uveitis, ulcerative keratitis, choroidal neovascularization, and orbital inflammation [131,132,133]. Mild irAEs can be treated with ocular lubricants and topical corticosteroids, while systemic corticosteroids are reserved for severe inflammation or for those who do not respond to topical therapy. Ophthalmologic evaluation should be arranged for all visual complaints.
Pulmonary
Pneumonitis is an uncommon irAE but rapidly progressive and potentially fatal when present [22, 86]. With the exception of atezolizumab, it is more common following therapy with PD-1 and PD-L1 therapies compared to ipilimumab [25, 31]. The median time to onset is 10-12 weeks following ICI therapy and initial symptoms may be non-specific [134]. Clinicians must maintain a high index of suspicion for immune-related pneumonitis given the late onset and potentially catastrophic outcome. The most common presenting symptoms are dyspnea and cough. These symptoms in a patient with underlying cancer should prompt a broad diagnostic workup including laboratory analyses and chest imaging for diagnoses such as pulmonary embolism, pneumonia, viral infection, and cancer progression. Radiographic findings of ICI-related pneumonitis include interstitial pneumonia, organizing pneumonia, and hypersensitivity pneumonitis [135]. These findings often mimic those from infectious etiologies, making definitive diagnosis difficult. Bronchoscopy with bronchoalveolar lavage may assist in excluding infectious etiologies but lung biopsy is seldom required. If there is suspicion for pneumonitis, ICI therapy should be withheld and corticosteroids commenced [136]. As infection cannot always be promptly ruled out prior to the initiation of immunosuppressive therapy, it is reasonable to initiate broad-spectrum antibiotics in conjunction with corticosteroid therapy. Patients should have periodic monitoring of pulmonary function and continuous pulse oximetry. Supplemental oxygen may be necessary [22]. Patients with grade 3 or 4 toxicity should be admitted to the hospital, receive expert consultation, and receive high-dose systemic corticosteroid therapy [31]. Patients with grade 2 toxicity should also receive systemic corticosteroids and hospital admission if their symptoms progress during an initial observation period of 3 to 6 hours. If there is steroid-resistance after 48 hours, as evidenced by lack of clinical or radiologic improvement, the initiation of infliximab, mycophenolate mofetil, or cyclophosphamide therapy should be considered [137]. Pneumonitis can recur after reinstitution of ICI therapy in patients with grade 1 or 2 pneumonitis. Patients and providers should be aware of this risk when considering restarting ICI therapy.
Renal
Renal irAEs have been reported in 2.2% of patients following ICI therapy but may be underreported due to the high prevalence of kidney disease in patients with cancer [138, 139]. It is estimated that up to 29% of patients may have a low-grade irAE following ICI therapy [139, 140]. High-grade acute kidney injury necessitating hemodialysis, however, is rare [138, 141, 142]. Combination ICI therapy is a risk factor for immune-related renal toxicity when compared to monotherapy. The median onset of acute kidney injury ranges from 2 months with anti-CTLA-4 treatment to 3 to 10 months following ICIs which act on the PD-1 axis [140, 143]. Agents which act on the PD-1 axis have similar incidents of renal toxicity [25]. Symptoms include oliguria, hematuria, or peripheral edema [144]. Acute interstitial nephritis is the most commonly reported cause of immune-related AKI, although lupus nephritis and thrombotic microangiopathy have also been described. Case reports have noted both proteinuria and the presence of anti-double-stranded DNA antibodies [142]. Renal biopsy with lymphocyte infiltration can confirm the diagnosis of immune-related kidney injury in patients taking ICI therapy [139, 145]. If suspected, ICI therapy should be interrupted and systemic corticosteroids initiated after consultation with a nephrologist [16].
Rheumatologic
Several rheumatologic complications have been described during ICI therapy including arthralgia, inflammatory arthritis, rheumatoid arthritis-like disease, inflammatory myopathy, scleroderma, and vasculitis [146, 147]. These complications are more common with ICIs which act on the PD-1 axis and tend to occur later than most other irAEs [148]. Clinicians should have a high suspicion for inflammatory arthritis and arrange prompt evaluation by a rheumatologist as permanent erosive joint damage can occur within weeks [143]. While typically managed with prednisone, steroid-resistant cases have been described and treated with disease-modifying anti-rheumatic drugs or TNF-alpha inhibitors such as infliximab. Duration of therapy can be prolonged and patients sometimes require chronic therapy [143, 149]. Suspected immune-related vasculitis should also be evaluated promptly given the concern for organ-specific complications which may lead to significant morbidity and mortality.
Conclusion
Immunotherapy has revolutionized the pharmacologic care of patients with cancer over the past two decades. Clinicians must be aware of these rapidly expanding classes of xenobiotics and their unique toxicologic profiles. Immune checkpoint inhibitors, first FDA-approved in 2011, block inhibitory signals from cancer cells to T-cells to allow the body’s innate immune system to target cancer cells. Adverse effects unique to ICIs, termed irAEs, represent a heterogeneous classification of immunostimulatory effects to multiple organ systems. While most irAEs are mild, severe cases do occur and may be rapidly fatal. Management of moderate to severe irAEs often includes cessation of ICI therapy and initiation of immunosuppressive agents such as corticosteroids. Appropriate management of patients with irAEs involves multidisciplinary care coordination with respect to patients’ individual long-term goals. Clinician recognition of irAEs and their sometimes delayed onset is critical to ensure appropriate and timely treatment.
References
Yousefi H, Yuan J, Keshavarz-Fathi M, Murphy JF, Rezaei N. Immunotherapy of cancers comes of age. Expert Rev Clin Immunol. 2017;13:1001–15. https://doi.org/10.1080/1744666X.2017.1366315.
Maloney DG, Grillo-López AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, et al. IDEC-C2B8 (rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood. 1997;90:2188–95.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. https://doi.org/10.1056/NEJMoa1003466.
Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26. https://doi.org/10.1056/NEJMoa1104621.
Eggermont AMM, Chiarion-Sileni V, Grob J-J, Dummer R, Wolchok JD, Schmidt H, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. 2016;375:1845–55. https://doi.org/10.1056/NEJMoa1611299.
Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–17. https://doi.org/10.1056/NEJMoa1414428.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. https://doi.org/10.1056/NEJMoa1504030.
Hodi FS, Lee S, McDermott DF, Rao UN, Butterfield LH, Tarhini AA, et al. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA. 2014;312:1744–53. https://doi.org/10.1001/jama.2014.13943.
McDermott D, Lebbé C, Hodi FS, Maio M, Weber JS, Wolchok JD, et al. Durable benefit and the potential for long-term survival with immunotherapy in advanced melanoma. Cancer Treat Rev. 2014;40:1056–64. https://doi.org/10.1016/j.ctrv.2014.06.012.
Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–32. https://doi.org/10.1056/NEJMoa1503093.
Kroschinsky F, Stölzel F, von Bonin S, Beutel G, Kochanek M, Kiehl M, et al. New drugs, new toxicities: severe side effects of modern targeted and immunotherapy of cancer and their management. Crit Care Lond Engl. 2017;21:89. https://doi.org/10.1186/s13054-017-1678-1.
Peterson JJ, Steele-Moses SK. Update on new therapies with immune checkpoint inhibitors. Clin J Oncol Nurs. 2016;20:405–10. https://doi.org/10.1188/16.CJON.405-410.
Palmieri DJ, Carlino MS. Immune checkpoint inhibitor toxicity. Curr Oncol Rep. 2018;20:72. https://doi.org/10.1007/s11912-018-0718-6.
Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59–73. https://doi.org/10.1038/nri2216.
Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–9. https://doi.org/10.1038/nature10673.
Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer Oxf Engl 1990. 2016;54:139–48. https://doi.org/10.1016/j.ejca.2015.11.016.
Johnson DB, Pollack MH, Sosman JA. Emerging targeted therapies for melanoma. Expert Opin Emerg Drugs. 2016;21:195–207. https://doi.org/10.1080/14728214.2016.1184644.
Socinski MA. Incorporating immunotherapy into the treatment of non-small cell lung cancer: practical guidance for the clinic. Semin Oncol. 2015;42(Suppl 2):S19–28. https://doi.org/10.1053/j.seminoncol.2015.09.017.
Ibraheim H, Perucha E, Powell N. Pathology of immune-mediated tissue lesions following treatment with immune checkpoint inhibitors. Rheumatol Oxf Engl. 2019;58:vii17–28. https://doi.org/10.1093/rheumatology/kez465.
Weinmann SC, Pisetsky DS. Mechanisms of immune-related adverse events during the treatment of cancer with immune checkpoint inhibitors. Rheumatol Oxf Engl. 2019;58:vii59–67. https://doi.org/10.1093/rheumatology/kez308.
Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16:563–80. https://doi.org/10.1038/s41571-019-0218-0.
Hryniewicki AT, Wang C, Shatsky RA, Coyne CJ. Management of immune checkpoint inhibitor toxicities: a review and clinical guideline for emergency physicians. J Emerg Med. 2018;55:489–502. https://doi.org/10.1016/j.jemermed.2018.07.005.
Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378:158–68. https://doi.org/10.1056/NEJMra1703481.
Horvat TZ, Adel NG, Dang T-O, Momtaz P, Postow MA, Callahan MK, et al. Immune-related adverse events, need for systemic immunosuppression, and effects on survival and time to treatment failure in patients with melanoma treated with ipilimumab at Memorial Sloan Kettering Cancer Center. J Clin Oncol Off J Am Soc Clin Oncol. 2015;33:3193–8. https://doi.org/10.1200/JCO.2015.60.8448.
Wang P-F, Chen Y, Song S-Y, Wang T-J, Ji W-J, Li S-W, et al. Immune-related adverse events associated with anti-PD-1/PD-L1 treatment for malignancies: a meta-analysis. Front Pharmacol. 2017;8:730. https://doi.org/10.3389/fphar.2017.00730.
Fecher LA, Agarwala SS, Hodi FS, Weber JS. Ipilimumab and its toxicities: a multidisciplinary approach. Oncologist. 2013;18:733–43. https://doi.org/10.1634/theoncologist.2012-0483.
Hwang WL, Pike LRG, Royce TJ, Mahal BA, Loeffler JS. Safety of combining radiotherapy with immune-checkpoint inhibition. Nat Rev Clin Oncol. 2018;15:477–94. https://doi.org/10.1038/s41571-018-0046-7.
Barroso-Sousa R, Barry WT, Garrido-Castro AC, Hodi FS, Min L, Krop IE, et al. Incidence of endocrine dysfunction following the use of different immune checkpoint inhibitor regimens: a systematic review and meta-analysis. JAMA Oncol. 2018;4:173–82. https://doi.org/10.1001/jamaoncol.2017.3064.
Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol Off J Am Soc Clin Oncol. 2018;36:1714–68. https://doi.org/10.1200/JCO.2017.77.6385.
Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30:2691–7. https://doi.org/10.1200/JCO.2012.41.6750.
Yang L, Yu H, Dong S, Zhong Y, Hu S. Recognizing and managing on toxicities in cancer immunotherapy. Tumour Biol J Int Soc Oncodevelopmental Biol Med. 2017;39:1010428317694542. https://doi.org/10.1177/1010428317694542.
Saleh K, Khalife-Saleh N, Kourie HR. Do immune-related adverse events correlate with response to immune checkpoint inhibitors? Immunotherapy. 2019;11:257–9. https://doi.org/10.2217/imt-2018-0201.
Conner SC, Trinquart L. Survivorship bias in analyses of immune checkpoint inhibitor trials. JAMA Oncol. 2019;5:1226. https://doi.org/10.1001/jamaoncol.2019.1187.
Martins F, Sykiotis GP, Maillard M, Fraga M, Ribi C, Kuntzer T, et al. New therapeutic perspectives to manage refractory immune checkpoint-related toxicities. Lancet Oncol. 2019;20:e54–64. https://doi.org/10.1016/S1470-2045(18)30828-3.
Yang S, Asnani A. Cardiotoxicities associated with immune checkpoint inhibitors. Curr Probl Cancer. 2018;42:422–32. https://doi.org/10.1016/j.currproblcancer.2018.07.002.
Baraibar I, Melero I, Ponz-Sarvise M, Castanon E. Safety and tolerability of immune checkpoint inhibitors (PD-1 and PD-L1) in cancer. Drug Saf. 2019;42:281–94. https://doi.org/10.1007/s40264-018-0774-8.
Nishino M, Sholl LM, Hodi FS, Hatabu H, Ramaiya NH. Anti-PD-1-related pneumonitis during cancer immunotherapy. N Engl J Med. 2015;373:288–90. https://doi.org/10.1056/NEJMc1505197.
Champiat S, Lambotte O, Barreau E, Belkhir R, Berdelou A, Carbonnel F, et al. Management of immune checkpoint blockade dysimmune toxicities: a collaborative position paper. Ann Oncol Off J Eur Soc Med Oncol. 2016;27:559–74. https://doi.org/10.1093/annonc/mdv623.
Parakh S, Cebon J, Klein O. Delayed autoimmune toxicity occurring several months after cessation of anti-PD-1 therapy. Oncologist. 2018;23:849–51. https://doi.org/10.1634/theoncologist.2017-0531.
Thallinger C, Füreder T, Preusser M, Heller G, Müllauer L, Höller C, et al. Review of cancer treatment with immune checkpoint inhibitors : current concepts, expectations, limitations and pitfalls. Wien Klin Wochenschr. 2018;130:85–91. https://doi.org/10.1007/s00508-017-1285-9.
Wood LS, Moldawer NP, Lewis C. Immune checkpoint inhibitor therapy: key principles when educating patients. Clin J Oncol Nurs. 2019;23:271–80. https://doi.org/10.1188/19.CJON.271-280.
Kostine M, Chiche L, Lazaro E, Halfon P, Charpin C, Arniaud D, et al. Opportunistic autoimmunity secondary to cancer immunotherapy (OASI): an emerging challenge. Rev Med Interne. 2017;38:513–25. https://doi.org/10.1016/j.revmed.2017.01.004.
Sanchez K, Page DB, Urba W. Immunotherapy toxicities. Surg Oncol Clin N Am. 2019;28:387–401. https://doi.org/10.1016/j.soc.2019.02.009.
Kumar V, Chaudhary N, Garg M, Floudas CS, Soni P, Chandra AB. Current diagnosis and management of immune related adverse events (irAEs) induced by immune checkpoint inhibitor therapy. Front Pharmacol. 2017;8:49. https://doi.org/10.3389/fphar.2017.00049.
Ganatra S, Parikh R, Neilan TG. Cardiotoxicity of immune therapy. Cardiol Clin. 2019;37:385–97. https://doi.org/10.1016/j.ccl.2019.07.008.
Varricchi G, Marone G, Mercurio V, Galdiero MR, Bonaduce D, Tocchetti CG. Immune checkpoint inhibitors and cardiac toxicity: an emerging issue. Curr Med Chem. 2018;25:1327–39. https://doi.org/10.2174/0929867324666170407125017.
Brumbaugh AD, Narurkar R, Parikh K, Fanucchi M, Frishman WH. Cardiac immune-related adverse events in immune checkpoint inhibition therapy. Cardiol Rev. 2019;27:97–107. https://doi.org/10.1097/CRD.0000000000000217.
Moslehi JJ, Salem J-E, Sosman JA, Lebrun-Vignes B, Johnson DB. Increased reporting of fatal immune checkpoint inhibitor-associated myocarditis. Lancet Lond Engl. 2018;391:933. https://doi.org/10.1016/S0140-6736(18)30533-6.
Mahmood SS, Fradley MG, Cohen JV, Nohria A, Reynolds KL, Heinzerling LM, et al. Myocarditis in patients treated with immune checkpoint inhibitors. J Am Coll Cardiol. 2018;71:1755–64. https://doi.org/10.1016/j.jacc.2018.02.037.
Semper H, Muehlberg F, Schulz-Menger J, Allewelt M, Grohé C. Drug-induced myocarditis after nivolumab treatment in a patient with PDL1- negative squamous cell carcinoma of the lung. Lung Cancer Amst Neth. 2016;99:117–9. https://doi.org/10.1016/j.lungcan.2016.06.025.
Johnson DB, Chandra S, Sosman JA. Immune checkpoint inhibitor toxicity in 2018. JAMA. 2018;320:1702–3. https://doi.org/10.1001/jama.2018.13995.
Escudier M, Cautela J, Malissen N, Ancedy Y, Orabona M, Pinto J, et al. Clinical features, management, and outcomes of immune checkpoint inhibitor-related cardiotoxicity. Circulation. 2017;136:2085–7. https://doi.org/10.1161/CIRCULATIONAHA.117.030571.
Zhang J-C, Chen W-D, Alvarez JB, Jia K, Shi L, Wang Q, et al. Cancer immune checkpoint blockade therapy and its associated autoimmune cardiotoxicity. Acta Pharmacol Sin. 2018;39:1693–8. https://doi.org/10.1038/s41401-018-0062-2.
Zarifa A, Lopez-Mattei J, Palaskas N, Iliescu C, Durand J-B, Kim PY. Immune checkpoint inhibitors (ICIs)-related cardiotoxicity. Adv Exp Med Biol. 2020;1244:277–85. https://doi.org/10.1007/978-3-030-41008-7_15.
Kwon HJ, Coté TR, Cuffe MS, Kramer JM, Braun MM. Case reports of heart failure after therapy with a tumor necrosis factor antagonist. Ann Intern Med. 2003;138:807–11. https://doi.org/10.7326/0003-4819-138-10-200305200-00008.
Zarifa A, Albittar A, Kim PY, Hassan S, Palaskas N, Iliescu C, et al. Cardiac toxicities of anticancer treatments: chemotherapy, targeted therapy and immunotherapy. Curr Opin Cardiol. 2019;34:441–50. https://doi.org/10.1097/HCO.0000000000000641.
Rassy EE, Assi T, Rizkallah J, Kattan J. Diffuse edema suggestive of cytokine release syndrome in a metastatic lung carcinoma patient treated with pembrolizumab. Immunotherapy. 2017;9:309–11. https://doi.org/10.2217/imt-2016-0134.
Rotz SJ, Leino D, Szabo S, Mangino JL, Turpin BK, Pressey JG. Severe cytokine release syndrome in a patient receiving PD-1-directed therapy. Pediatr Blood Cancer. 2017;64. https://doi.org/10.1002/pbc.26642.
Zhao L, Yang Y, Li W, Li T, Gao Q. Nivolumab-induced cytokine-release syndrome in relapsed/refractory Hodgkin’s lymphoma: a case report and literature review. Immunotherapy. 2018;10:913–7. https://doi.org/10.2217/imt-2018-0025.
Dimitriou F, Matter AV, Mangana J, Urosevic-Maiwald M, Micaletto S, Braun RP, et al. Cytokine release syndrome during sequential treatment with immune checkpoint inhibitors and kinase inhibitors for metastatic melanoma. J Immunother Hagerstown Md 1997. 2019;42:29–32. https://doi.org/10.1097/CJI.0000000000000236.
Honjo O, Kubo T, Sugaya F, Nishizaka T, Kato K, Hirohashi Y, et al. Severe cytokine release syndrome resulting in purpura fulminans despite successful response to nivolumab therapy in a patient with pleomorphic carcinoma of the lung: a case report. J Immunother Cancer. 2019;7:97. https://doi.org/10.1186/s40425-019-0582-4.
Ohira J, Kawamoto M, Sugino Y, Kohara N. A case report of fulminant cytokine release syndrome complicated by dermatomyositis after the combination therapy with immune checkpoint inhibitors. Medicine (Baltimore). 2020;99:e19741. https://doi.org/10.1097/MD.0000000000019741.
Ceschi A, Noseda R, Palin K, Verhamme K. Immune checkpoint inhibitor-related cytokine release syndrome: analysis of WHO Global Pharmacovigilance Database. Front Pharmacol. 2020;11:557. https://doi.org/10.3389/fphar.2020.00557.
Macdonald JB, Macdonald B, Golitz LE, LoRusso P, Sekulic A. Cutaneous adverse effects of targeted therapies: Part II: inhibitors of intracellular molecular signaling pathways. J Am Acad Dermatol. 2015;72:221–36; quiz 237–8. https://doi.org/10.1016/j.jaad.2014.07.033.
Lacouture M, Sibaud V. Toxic side effects of targeted therapies and immunotherapies affecting the skin, oral mucosa, hair, and nails. Am J Clin Dermatol. 2018;19:31–9. https://doi.org/10.1007/s40257-018-0384-3.
Sibaud V. Dermatologic reactions to immune checkpoint inhibitors : skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19:345–61. https://doi.org/10.1007/s40257-017-0336-3.
Teulings H-E, Limpens J, Jansen SN, Zwinderman AH, Reitsma JB, Spuls PI, et al. Vitiligo-like depigmentation in patients with stage III-IV melanoma receiving immunotherapy and its association with survival: a systematic review and meta-analysis. J Clin Oncol Off J Am Soc Clin Oncol. 2015;33:773–81. https://doi.org/10.1200/JCO.2014.57.4756.
Ng CY, Chen C-B, Wu M-Y, Wu J, Yang C-H, Hui RC-Y, et al. Anticancer drugs induced severe adverse cutaneous drug reactions: an updated review on the risks associated with anticancer targeted therapy or immunotherapies. J Immunol Res. 2018;2018:5376476–9. https://doi.org/10.1155/2018/5376476.
Voskens CJ, Goldinger SM, Loquai C, Robert C, Kaehler KC, Berking C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 2013;8:e53745. https://doi.org/10.1371/journal.pone.0053745.
Zarbo A, Belum VR, Sibaud V, Oudard S, Postow MA, Hsieh JJ, et al. Immune-related alopecia (areata and universalis) in cancer patients receiving immune checkpoint inhibitors. Br J Dermatol. 2017;176:1649–52. https://doi.org/10.1111/bjd.15237.
Rivera N, Boada A, Bielsa MI, Fernández-Figueras MT, Carcereny E, Moran MT, et al. Hair repigmentation during immunotherapy treatment with an anti-programmed cell death 1 and anti-programmed cell death ligand 1 agent for lung cancer. JAMA Dermatol. 2017;153:1162–5. https://doi.org/10.1001/jamadermatol.2017.2106.
Habre M, Habre SB, Kourie HR. Dermatologic adverse events of checkpoint inhibitors: what an oncologist should know. Immunotherapy. 2016;8:1437–46. https://doi.org/10.2217/imt-2016-0074.
Parthymos I, Liamis G, Dounousi E, Pentheroudakis G, Mauri D, Zarkavelis G, et al. Metabolic consequences of immune checkpoint inhibitors: a new challenge in clinical practice. Crit Rev Oncol Hematol. 2020;151:102979. https://doi.org/10.1016/j.critrevonc.2020.102979.
Morganstein DL, Lai Z, Spain L, Diem S, Levine D, Mace C, et al. Thyroid abnormalities following the use of cytotoxic T-lymphocyte antigen-4 and programmed death receptor protein-1 inhibitors in the treatment of melanoma. Clin Endocrinol. 2017;86:614–20. https://doi.org/10.1111/cen.13297.
Osorio JC, Ni A, Chaft JE, Pollina R, Kasler MK, Stephens D, et al. Antibody-mediated thyroid dysfunction during T-cell checkpoint blockade in patients with non-small-cell lung cancer. Ann Oncol Off J Eur Soc Med Oncol. 2017;28:583–9. https://doi.org/10.1093/annonc/mdw640.
Corsello SM, Barnabei A, Marchetti P, De Vecchis L, Salvatori R, Torino F. Endocrine side effects induced by immune checkpoint inhibitors. J Clin Endocrinol Metab. 2013;98:1361–75. https://doi.org/10.1210/jc.2012-4075.
Lee H, Hodi FS, Giobbie-Hurder A, Ott PA, Buchbinder EI, Haq R, et al. Characterization of thyroid disorders in patients receiving immune checkpoint inhibition therapy. Cancer Immunol Res. 2017;5:1133–40. https://doi.org/10.1158/2326-6066.CIR-17-0208.
de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A systematic review and meta-analysis of endocrine-related adverse events associated with immune checkpoint inhibitors. Horm Metab Res Horm Stoffwechselforschung Horm Metab. 2019;51:145–56. https://doi.org/10.1055/a-0843-3366.
Chang L-S, Barroso-Sousa R, Tolaney SM, Hodi FS, Kaiser UB, Min L. Endocrine toxicity of cancer immunotherapy targeting immune checkpoints. Endocr Rev. 2019;40:17–65. https://doi.org/10.1210/er.2018-00006.
Iyer PC, Cabanillas ME, Waguespack SG, Hu MI, Thosani S, Lavis VR, et al. Immune-related thyroiditis with immune checkpoint inhibitors. Thyroid Off J Am Thyroid Assoc. 2018;28:1243–51. https://doi.org/10.1089/thy.2018.0116.
Faje AT, Sullivan R, Lawrence D, Tritos NA, Fadden R, Klibanski A, et al. Ipilimumab-induced hypophysitis: a detailed longitudinal analysis in a large cohort of patients with metastatic melanoma. J Clin Endocrinol Metab. 2014;99:4078–85. https://doi.org/10.1210/jc.2014-2306.
Albarel F, Gaudy C, Castinetti F, Carré T, Morange I, Conte-Devolx B, et al. Long-term follow-up of ipilimumab-induced hypophysitis, a common adverse event of the anti-CTLA-4 antibody in melanoma. Eur J Endocrinol. 2015;172:195–204. https://doi.org/10.1530/EJE-14-0845.
Iwama S, De Remigis A, Callahan MK, Slovin SF, Wolchok JD, Caturegli P. Pituitary expression of CTLA-4 mediates hypophysitis secondary to administration of CTLA-4 blocking antibody. Sci Transl Med. 2014;6:230ra45. https://doi.org/10.1126/scitranslmed.3008002.
Juszczak A, Gupta A, Karavitaki N, Middleton MR, Grossman AB. Ipilimumab: a novel immunomodulating therapy causing autoimmune hypophysitis: a case report and review. Eur J Endocrinol. 2012;167:1–5. https://doi.org/10.1530/EJE-12-0167.
Bhandari S, Gill AS, Perez CA, Jain D. Management of immunotherapy toxicities in older adults. Semin Oncol. 2018;45:226–31. https://doi.org/10.1053/j.seminoncol.2018.09.001.
Puzanov I, Diab A, Abdallah K, Bingham CO, Brogdon C, Dadu R, et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer. 2017;5:95. https://doi.org/10.1186/s40425-017-0300-z.
Carpenter KJ, Murtagh RD, Lilienfeld H, Weber J, Murtagh FR. Ipilimumab-induced hypophysitis: MR imaging findings. AJNR Am J Neuroradiol. 2009;30:1751–3. https://doi.org/10.3174/ajnr.A1623.
Sidhu P, Menzies AM, Long G, Carlino M, Lorens S, Kapoor R. Radiological manifestations of immune-related adverse effects observed in patients with melanoma undergoing immunotherapy. J Med Imaging Radiat Oncol. 2017;61:759–66. https://doi.org/10.1111/1754-9485.12653.
Kottschade L, Brys A, Peikert T, Ryder M, Raffals L, Brewer J, et al. A multidisciplinary approach to toxicity management of modern immune checkpoint inhibitors in cancer therapy. Melanoma Res. 2016;26:469–80. https://doi.org/10.1097/CMR.0000000000000273.
George J, Bajaj D, Sankaramangalam K, Yoo JW, Joshi NS, Gettinger S, et al. Incidence of pancreatitis with the use of immune checkpoint inhibitors (ICI) in advanced cancers: a systematic review and meta-analysis. Pancreatol Off J Int Assoc Pancreatol IAP Al. 2019;19:587–94. https://doi.org/10.1016/j.pan.2019.04.015.
Cramer P, Bresalier RS. Gastrointestinal and hepatic complications of immune checkpoint inhibitors. Curr Gastroenterol Rep. 2017;19:3. https://doi.org/10.1007/s11894-017-0540-6.
Dougan M. Checkpoint blockade toxicity and immune homeostasis in the gastrointestinal tract. Front Immunol. 2017;8:1547. https://doi.org/10.3389/fimmu.2017.01547.
Gupta A, De Felice KM, Loftus EV, Khanna S. Systematic review: colitis associated with anti-CTLA-4 therapy. Aliment Pharmacol Ther. 2015;42:406–17. https://doi.org/10.1111/apt.13281.
Marthey L, Mateus C, Mussini C, Nachury M, Nancey S, Grange F, et al. Cancer immunotherapy with anti-CTLA-4 monoclonal antibodies induces an inflammatory bowel disease. J Crohns Colitis. 2016;10:395–401. https://doi.org/10.1093/ecco-jcc/jjv227.
Reddy HG, Schneider BJ, Tai AW. Immune checkpoint inhibitor-associated colitis and hepatitis. Clin Transl Gastroenterol. 2018;9:180. https://doi.org/10.1038/s41424-018-0049-9.
Messmer M, Upreti S, Tarabishy Y, Mazumder N, Chowdhury R, Yarchoan M, et al. Ipilimumab-induced enteritis without colitis: a new challenge. Case Rep Oncol. 2016;9:705–13. https://doi.org/10.1159/000452403.
Fessler J, Matson V, Gajewski TF. Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer. 2019;7:108. https://doi.org/10.1186/s40425-019-0574-4.
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–7. https://doi.org/10.1126/science.aan3706.
Gordon R, Kasler MK, Stasi K, Shames Y, Errante M, Ciccolini K, et al. Checkpoint inhibitors: common immune-related adverse events and their management. Clin J Oncol Nurs. 2017;21:45–52. https://doi.org/10.1188/17.CJON.S2.45-52.
O’Regan KN, Jagannathan JP, Ramaiya N, Hodi FS. Radiologic aspects of immune-related tumor response criteria and patterns of immune-related adverse events in patients undergoing ipilimumab therapy. AJR Am J Roentgenol. 2011;197:W241–6. https://doi.org/10.2214/AJR.10.6032.
Nishida T, Iijima H, Adachi S. Immune checkpoint inhibitor-induced diarrhea/colitis: endoscopic and pathologic findings. World J Gastrointest Pathophysiol. 2019;10:17–28. https://doi.org/10.4291/wjgp.v10.i2.17.
Beck KE, Blansfield JA, Tran KQ, Feldman AL, Hughes MS, Royal RE, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol Off J Am Soc Clin Oncol. 2006;24:2283–9. https://doi.org/10.1200/JCO.2005.04.5716.
Johnston RL, Lutzky J, Chodhry A, Barkin JS. Cytotoxic T-lymphocyte-associated antigen 4 antibody-induced colitis and its management with infliximab. Dig Dis Sci. 2009;54:2538–40. https://doi.org/10.1007/s10620-008-0641-z.
Minor DR, Chin K, Kashani-Sabet M. Infliximab in the treatment of anti-CTLA4 antibody (ipilimumab) induced immune-related colitis. Cancer Biother Radiopharm. 2009;24:321–5. https://doi.org/10.1089/cbr.2008.0607.
Weber J, Thompson JA, Hamid O, Minor D, Amin A, Ron I, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15:5591–8. https://doi.org/10.1158/1078-0432.CCR-09-1024.
Berman D, Parker SM, Siegel J, Chasalow SD, Weber J, Galbraith S, et al. Blockade of cytotoxic T-lymphocyte antigen-4 by ipilimumab results in dysregulation of gastrointestinal immunity in patients with advanced melanoma. Cancer Immun. 2010;10:11.
Lombardi A, Mondelli MU. Review article: immune checkpoint inhibitors and the liver, from therapeutic efficacy to side effects. Aliment Pharmacol Ther. 2019;50:872–84. https://doi.org/10.1111/apt.15449.
Tian Y, Abu-Sbeih H, Wang Y. Immune checkpoint inhibitors-induced hepatitis. Adv Exp Med Biol. 2018;995:159–64. https://doi.org/10.1007/978-3-030-02505-2_8.
Suzman DL, Pelosof L, Rosenberg A, Avigan MI. Hepatotoxicity of immune checkpoint inhibitors: an evolving picture of risk associated with a vital class of immunotherapy agents. Liver Int Off J Int Assoc Study Liver. 2018;38:976–87. https://doi.org/10.1111/liv.13746.
Zen Y, Yeh MM. Checkpoint inhibitor-induced liver injury: a novel form of liver disease emerging in the era of cancer immunotherapy. Semin Diagn Pathol. 2019;36:434–40. https://doi.org/10.1053/j.semdp.2019.07.009.
Zen Y, Yeh MM. Hepatotoxicity of immune checkpoint inhibitors: a histology study of seven cases in comparison with autoimmune hepatitis and idiosyncratic drug-induced liver injury. Mod Pathol Off J U S Can Acad Pathol Inc. 2018;31:965–73. https://doi.org/10.1038/s41379-018-0013-y.
Di Giacomo AM, Biagioli M, Maio M. The emerging toxicity profiles of anti-CTLA-4 antibodies across clinical indications. Semin Oncol. 2010;37:499–507. https://doi.org/10.1053/j.seminoncol.2010.09.007.
Nadeau BA, Fecher LA, Owens SR, Razumilava N. Liver toxicity with cancer checkpoint inhibitor therapy. Semin Liver Dis. 2018;38:366–78. https://doi.org/10.1055/s-0038-1667358.
Brown ZJ, Heinrich B, Steinberg SM, Yu SJ, Greten TF. Safety in treatment of hepatocellular carcinoma with immune checkpoint inhibitors as compared to melanoma and non-small cell lung cancer. J Immunother Cancer. 2017;5:93. https://doi.org/10.1186/s40425-017-0298-2.
Haanen JBAG, Carbonnel F, Robert C, Kerr KM, Peters S, Larkin J, et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol Off J Eur Soc Med Oncol. 2017;28:iv119–42. https://doi.org/10.1093/annonc/mdx225.
Wang W, Lie P, Guo M, He J. Risk of hepatotoxicity in cancer patients treated with immune checkpoint inhibitors: a systematic review and meta-analysis of published data. Int J Cancer. 2017;141:1018–28. https://doi.org/10.1002/ijc.30678.
Jennings JJ, Mandaliya R, Nakshabandi A, Lewis JH. Hepatotoxicity induced by immune checkpoint inhibitors: a comprehensive review including current and alternative management strategies. Expert Opin Drug Metab Toxicol. 2019;15:231–44. https://doi.org/10.1080/17425255.2019.1574744.
Lleo A, Rimassa L, Colombo M. Hepatotoxicity of immune check point inhibitors: approach and management. Dig Liver Dis Off J Ital Soc Gastroenterol Ital Assoc Study Liver. 2019;51:1074–8. https://doi.org/10.1016/j.dld.2019.06.017.
Rossi RE, Parisi I, Despott EJ, Burroughs AK, O’Beirne J, Conte D, et al. Anti-tumour necrosis factor agent and liver injury: literature review, recommendations for management. World J Gastroenterol. 2014;20:17352–9. https://doi.org/10.3748/wjg.v20.i46.17352.
Touat M, Talmasov D, Ricard D, Psimaras D. Neurological toxicities associated with immune-checkpoint inhibitors. Curr Opin Neurol. 2017;30:659–68. https://doi.org/10.1097/WCO.0000000000000503.
Cuzzubbo S, Javeri F, Tissier M, Roumi A, Barlog C, Doridam J, et al. Neurological adverse events associated with immune checkpoint inhibitors: review of the literature. Eur J Cancer Oxf Engl 1990. 2017;73:1–8. https://doi.org/10.1016/j.ejca.2016.12.001.
Chang E, Sabichi AL, Sada YH. Myasthenia gravis after nivolumab therapy for squamous cell carcinoma of the bladder. J Immunother Hagerstown Md 1997. 2017;40:114–6. https://doi.org/10.1097/CJI.0000000000000161.
Papadopoulos KP, Romero RS, Gonzalez G, Dix JE, Lowy I, Fury M. Anti-hu-associated autoimmune limbic encephalitis in a patient with PD-1 inhibitor-responsive myxoid chondrosarcoma. Oncologist. 2018;23:118–20. https://doi.org/10.1634/theoncologist.2017-0344.
Williams TJ, Benavides DR, Patrice K-A, Dalmau JO, de Ávila ALR, Le DT, et al. Association of autoimmune encephalitis with combined immune checkpoint inhibitor treatment for metastatic cancer. JAMA Neurol. 2016;73:928–33. https://doi.org/10.1001/jamaneurol.2016.1399.
Vitt JR, Kreple C, Mahmood N, Dickerson E, Lopez GY, Richie MB. Autoimmune pancerebellitis associated with pembrolizumab therapy. Neurology. 2018;91:91–3. https://doi.org/10.1212/WNL.0000000000005781.
Cordes LM, Davarpanah NN, Reoma LB, Gasmi B, Quezado M, Khan OI, et al. Neurotoxicities associated with checkpoint inhibitors: two case reports and a review of the literature. Clin Case Rep. 2020;8:24–32. https://doi.org/10.1002/ccr3.2534.
Zimmer L, Goldinger SM, Hofmann L, Loquai C, Ugurel S, Thomas I, et al. Neurological, respiratory, musculoskeletal, cardiac and ocular side-effects of anti-PD-1 therapy. Eur J Cancer Oxf Engl 1990. 2016;60:210–25. https://doi.org/10.1016/j.ejca.2016.02.024.
Nakatani Y, Tanaka N, Enami T, Minami S, Okazaki T, Komuta K. Lambert-Eaton Myasthenic syndrome caused by nivolumab in a patient with squamous cell lung cancer. Case Rep Neurol. 2018;10:346–52. https://doi.org/10.1159/000494078.
Ito M, Fujiwara S, Fujimoto D, Mori R, Yoshimura H, Hata A, et al. Rituximab for nivolumab plus ipilimumab-induced encephalitis in a small-cell lung cancer patient. Ann Oncol Off J Eur Soc Med Oncol. 2017;28:2318–9. https://doi.org/10.1093/annonc/mdx252.
Antoun J, Titah C, Cochereau I. Ocular and orbital side-effects of checkpoint inhibitors: a review article. Curr Opin Oncol. 2016;28:288–94. https://doi.org/10.1097/CCO.0000000000000296.
Abdel-Rahman O, Eltobgy M, Oweira H, Giryes A, Tekbas A, Decker M. Immune-related musculoskeletal toxicities among cancer patients treated with immune checkpoint inhibitors: a systematic review. Immunotherapy. 2017;9:1175–83. https://doi.org/10.2217/imt-2017-0108.
Kottschade LA. Incidence and management of immune-related adverse events in patients undergoing treatment with immune checkpoint inhibitors. Curr Oncol Rep. 2018;20:24. https://doi.org/10.1007/s11912-018-0671-4.
Miranda Poma J, Ostios Garcia L, Villamayor Sanchez J, D’errico G. What do we know about cancer immunotherapy? Long-term survival and immune-related adverse events. Allergol Immunopathol (Madr). 2019;47:303–8. https://doi.org/10.1016/j.aller.2018.04.005.
Fessas P, Possamai LA, Clark J, Daniels E, Gudd C, Mullish BH, et al. Immunotoxicity from checkpoint inhibitor therapy: clinical features and underlying mechanisms. Immunology. 2020;159:167–77. https://doi.org/10.1111/imm.13141.
Ciccarese C, Iacovelli R, Bria E, Modena A, Massari F, Brunelli M, et al. The incidence and relative risk of pulmonary toxicity in patients treated with anti-PD1/PD-L1 therapy for solid tumors: a meta-analysis of current studies. Immunotherapy. 2017;9:579–87. https://doi.org/10.2217/imt-2017-0018.
Gutierrez C, McEvoy C, Munshi L, Stephens RS, Detsky ME, Nates JL, et al. Critical care management of toxicities associated with targeted agents and immunotherapies for cancer. Crit Care Med. 2020;48:10–21. https://doi.org/10.1097/CCM.0000000000004087.
Spain L, Diem S, Larkin J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat Rev. 2016;44:51–60. https://doi.org/10.1016/j.ctrv.2016.02.001.
Rassy EE, Kourie HR, Rizkallah J, Karak FE, Hanna C, Chelala DN, et al. Immune checkpoint inhibitors renal side effects and management. Immunotherapy. 2016;8:1417–25. https://doi.org/10.2217/imt-2016-0099.
Cortazar FB, Marrone KA, Troxell ML, Ralto KM, Hoenig MP, Brahmer JR, et al. Clinicopathological features of acute kidney injury associated with immune checkpoint inhibitors. Kidney Int. 2016;90:638–47. https://doi.org/10.1016/j.kint.2016.04.008.
Wanchoo R, Karam S, Uppal NN, Barta VS, Deray G, Devoe C, et al. Adverse renal effects of immune checkpoint inhibitors: a narrative review. Am J Nephrol. 2017;45:160–9. https://doi.org/10.1159/000455014.
Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–44. https://doi.org/10.1056/NEJMoa1305133.
Fadel F, El Karoui K, Knebelmann B. Anti-CTLA4 antibody-induced lupus nephritis. N Engl J Med. 2009;361:211–2. https://doi.org/10.1056/NEJMc0904283.
Spiers L, Coupe N, Payne M. Toxicities associated with checkpoint inhibitors-an overview. Rheumatol Oxf Engl. 2019;58:vii7–16. https://doi.org/10.1093/rheumatology/kez418.
Belliere J, Meyer N, Mazieres J, Ollier S, Boulinguez S, Delas A, et al. Acute interstitial nephritis related to immune checkpoint inhibitors. Br J Cancer. 2016;115:1457–61. https://doi.org/10.1038/bjc.2016.358.
Lefebvre J, Glezerman IG. Kidney toxicities associated with novel cancer therapies. Adv Chronic Kidney Dis. 2017;24:233–40. https://doi.org/10.1053/j.ackd.2017.05.006.
Arnaud L, Lebrun-Vignes B, Salem J-E. Checkpoint inhibitor-associated immune arthritis. Ann Rheum Dis. 2019;78:e68. https://doi.org/10.1136/annrheumdis-2018-213470.
Barbosa NS, Wetter DA, Wieland CN, Shenoy NK, Markovic SN, Thanarajasingam U. Scleroderma induced by pembrolizumab: a case series. Mayo Clin Proc. 2017;92:1158–63. https://doi.org/10.1016/j.mayocp.2017.03.016.
Lidar M, Giat E, Garelick D, Horowitz Y, Amital H, Steinberg-Silman Y, et al. Rheumatic manifestations among cancer patients treated with immune checkpoint inhibitors. Autoimmun Rev. 2018;17:284–9. https://doi.org/10.1016/j.autrev.2018.01.003.
Kottschade LA. The future of immunotherapy in the treatment of cancer. Semin Oncol Nurs. 2019;35:150934. https://doi.org/10.1016/j.soncn.2019.08.013.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All authors are listed above and comply with the ICMJE authorship criteria.
Conflicts of Interest
None.
Additional information
Supervising Editor: Michael Hodgman, MD
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chhabra, N., Kennedy, J. A Review of Cancer Immunotherapy Toxicity: Immune Checkpoint Inhibitors. J. Med. Toxicol. 17, 411–424 (2021). https://doi.org/10.1007/s13181-021-00833-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13181-021-00833-8