
Abstract
In recent years, the importance of microbial diversity and function to ecosystem restoration has been recognized. The aim of this work was to investigate the diversity and composition of bacterial communities in response to reclamation of a soil subsidence area affected by mining activities. Soil samples were taken in two seasons (December 2012 and July 2013) from a mining reclamation region at the Liuxin national reclamation demonstration area in China and an adjacent coal-excavated subsidence region. 454 high-throughput sequencing technology was used to compare the composition and diversity of bacterial communities in reclaimed soil to that in subsided soil. Predominant phyla in soils were Proteobacteria, Actinobacteria, Acidobacteria, and Planctomycetes, with Proteobacteria making up the majority of the community. Long-term reclamation was found to have significant influences on bacterial communities, and the bacterial community diversity and composition varied between reclaimed and subsided soil. Seasonal fluctuations also contributed to variation in soil bacterial diversity and community composition, but were minor in comparison to effects of reclamation. Differences observed in bacterial community structure and diversity were related to both fertilizer treatment and vegetation, likely through the effects of soil attributes. Soil organic matter and total nitrogen and available potassium were important factors shaping the microbial communities. The reclaimed soil had higher community diversity of bacteria than subsided soil, which suggests that long-term applications of organic amendments and vegetation mixed sowing had significant impacts on soil remediation and microbial diversity.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ecological restoration and mine reclamation have become important parts of sustainable development strategies in many countries (Ingram et al. 2005). The need to assess the effectiveness of reclamation strategies on reconstructed soil has been promoted (Sheoran et al. 2010; Lewis et al. 2012). Historically, the criteria for evaluating the success of land reclamation have largely involved assessments of soil quality, physicochemical characteristics, and vegetation coverage (Mummey et al. 2002; Hahn and Quideau 2013). In contrast, the microbial ecology of soil with respect to reclamation remains poorly understood. Microorganisms are particularly sensitive to environmental change, and changes in microbial activity, community structure, and function can serve as indicators of anthropogenic impact and even predict the success of the restoration of degraded environments (Kaschuk et al. 2010). However, it remains unclear what influence land reclamation might have on the recovery of the soil bacterial community. Although several recent studies have assessed the effects of soil reclamation on microbial populations, microbial biomass, and activity (Dimitriu et al. 2010; Fan et al. 2011), little is known about the impact of reclamation on the shaping of bacterial microbiota associated with coal-mining disturbed land. There is a considerable dearth of information regarding the responses of soil microbial communities to edaphic factors, vegetation, and seasons, which are important driving factors determining soil microbial diversity and community composition.
Recently, 454 pyrosequencing technology has allowed investigation of microbial diversity at levels previously not feasible. This technique has already been applied to various investigations focusing on species diversity in forests, aquatic environments, grasslands, and farmland (Kuffner et al. 2012; García-Orenes et al. 2013). To date, limited comprehensive data describing bacterial communities in mining-impacted terrestrial ecosystems using pyrosequencing analysis are available (Dimitriu et al. 2010). This is to a large extent because no study has yet discussed the characterization of bacterial diversity in reclaimed soil in depth.
Understanding the response of soil microbial communities to land reclamation and restoration has important implications for the restoration of soil fertility and reestablishment of microbial communities. The present study provides an assessment of long-term land remediation through pyrosequencing-based comparative analyses of the bacterial community in reclaimed soils (RE) and excavated subsided soils near coal mines (SU). Changes in the structure and diversity of the soil bacterial community were evaluated over two sampling periods. The current study explored how bacterial community structure and diversity changed with different seasons and soil systems and identified possible relationships with soil abiotic factors. It is here hypothesized that (1) both the fertilizer treatment and vegetation involved in reclamation would affect, to varying extents, the soil microorganisms, and that (2) the variation in bacterial communities might also be associated with environmental and soil chemical conditions.
Materials and methods
Study area and sample collection
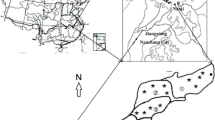
The study was carried out at the Liuxin national reclamation demonstration area (34°23′49″ to 34°24′30″N and 117°06′50″ to 117°08′34″E, Fig. 1), located approximately 60 km north of Xuzhou, Jiangsu Province, in eastern China. Two experimental regions in the Liuxin Mine were chosen for sample collection representing two soil systems: (1) RE, a mining reclamation region with a >13-years reclamation history by means of planting a mixture of legumes with gramineous grass; and (2) SU, an adjacent unreclaimed post-mining region with considerable subsidence that had not previously been treated. Region RE has received fertilizer with organic amendments at a rate of approximately 3–5 Mg ha−1 (once or twice a year). Most of the organic amendments applied have been fresh vegetable residues with high carbon/nitrogen ratios, composted vegetables, and animal waste. The vegetation is dominated by alfalfa (Medicago sativa), white clover (Trifolium repens), and ryegrass (Lolium perenne), which were planted at a sowing ratio of 3:3:2. Region SU (prior to reclamation), the coal-mining subsidence land affected by >20-years underground coal mining activity, was spontaneously colonized by vegetation including Salix babylonica L. and Populus tremula (15–20 years old).

Study area and sampling locations. a Map of two sampling spots located in the Liuxin Mine region; b map of China and the location of Jiangsu province; c location of Liuxin Mine in Jiangsu Province. RE mining reclamation land, SU mining subsidence land
Eleven sites were chosen in both the mining reclamation region and the subsidence region (22 sites in total). Soil sampling was conducted twice, once in December 2013 and again in July 2013. Three soil cores (2.5 cm diameter) from 0 to 20 cm depth were randomly collected and mixed for each sampling site. In this way, a total of 44 soil samples were collected during two seasons, including 11 each from RE-D and RE-J in region RE and 11 each from SU-D and SU-J in region SU. “-D” and “-J” designations for RE and SU indicate that these were samples collected in December and July, respectively. After the soil was sieved through a 2 mm mesh and manually homogenized, all samples were placed in polyethylene bags in triplicate and transported to the laboratory for DNA extraction. One portion was stored at −80 °C prior to molecular analyses, and the other was stored at 4 °C for 24 h, and then air-dried at 40 °C for chemical analysis.
Soil physicochemical analysis
Soil organic matter content (SOM) was determined as reported by Wilson and Sander (1996). Total nitrogen (TN) was determined using Kjeldahl’s method as modified by Bremner and Mulvaney (1978). Total phosphorus (TP) content was measured using the ammonium molybdate spectrophotometric method (Pan et al. 2003).The concentrations of available potassium (AK) were measured using the 1 mol L−1 neutral NH4OAC method (Bao 2008). Soil pH was measured using a Hach pH meter (Hach Company, Loveland, CO, USA) (1:5 soil/water). The 11 soil samples of each group (RE-D, RE-J, SU-D, SU-J) were used for the chemical tests. Analyses were repeated three times, and the data were averaged.
For extraction of soil DNA, the E.Z.N.A.® Soil DNA Kit (Omega Bio-Tek Inc., Norcross, GA, USA) was used with 0.5 g of soil according to the manufacturer’s instructions. To assess DNA quantity and purity, the crude DNA extracts (2 μl) were run on 1 % agarose gel and analyzed by microspectrophotometer. (NanoDrop 2000, NanoDrop Technologies, Wilmington, DE, USA). Extracted DNA was diluted to 2 ng μl−1 for samples from RE-D, 2.2 ng μl−1 for SU-D, 2.1 ng μl−1 for RE-J, and 2.4 ng μl−1 for SU-J before PCR amplification.
Bacterial 16S-rRNA genes were amplified using the following set of primers: 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 533R (5′-TTACCGCGGCTGCTGGCAC-3′) (Cabrera-Rubio et al. 2012). PCR was carried out in triplicate with 20 μl of the reaction mixture comprising 4 μl of five-fold FastPfu buffer, 2 μl of 2.5 mM dNTPs, 5 μM of each primer, 0.4 μl of diluted DNA sample, 0.4 μl of TransStart FastPfu DNA Polymerase, and approximately 10 ng of DNA template using the PCR Gene Amp 9700 (Applied Biosystems, Foster City, CA, USA). The following thermal cycling scheme was used: initial denaturation at 95 °C for 2 min, followed by 25 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and elongation at 72 °C for 30 s followed by a final extension at 72 °C for 5 min. All samples were amplified in triplicate, pooled in equal amounts, and purified using an Axy Prep DNA gel extraction kit as recommended by the manufacturer (Axygen, Biotechnology, Hangzhou, China). Quantification of the PCR products was performed using the PicoGreen® dsDNA Quantitation Reagent (Molecular Probes, Eugene, OR, USA) and a QuantiFluor™-ST Real-time PCR System (Promega, Madison, WI, USA) as recommended by the manufacturer. After quantitation, the amplicons from each reaction mixture were pooled in equimolar ratios based on concentration and subjected to emulsion PCR to generate amplicon libraries, as recommended by 454 Life Sciences (Branford, CT, USA). Amplicon pyrosequencing was performed from the A-end using a 454 Roche GS FLX + Sequencing Method Manual_XLR70 kit on a Roche Genome Sequencer GS FLX Titanium platform (Roche, NJ, USA) at Majorbio Bio-pharm Technology Co., Ltd, Shanghai, China.
Data analysis
Before the 454 pyrosequencing and microbial analysis, 11 samples from the same treatment at each sampling time were pooled together for one mixed soil sample. Thus, there were four sets of mixed samples (RE-D, RE-J, SU-D and SU-J) for the comparative analysis of bacterial communities. Three repeated tests were performed for each set and the resulting data were averaged. The data sets generated from pyrosequencing were analyzed with a comprehensive bioinformatics software package, MOTHUR (http://schloss.micro.umass.edu/mothur/Main_Page), for pre-processing, identification of operational taxonomic units (OTUs), taxonomic assignment and statistical analysis (Schloss et al. 2009). To normalize our data, the data were subsampled from the original dataset before the assessment of OTUs. Before analysis of the pyrosequencing data, all failed sequence reads, sequences shorter than 200 bp, and sequences containing ambiguous bases, low-quality sequence ends and tags were removed, and the remaining sequences were divested of any nonbacterial ribosome sequences and chimeras using the UCHIME algorithm (Edgar et al. 2011). For taxonomy-based analysis, the RDP Classifier of the Ribosomal Database Project (RDP) was used at a confidence threshold of 80 % (Wang et al. 2007). For the determination of OTUs, sequences were assigned to phylotype clusters at cutoff levels of 3, 5, and 10 %, and the sequences from each OTU were taxonomically assigned to a bacterial 16S rRNA Silva reference alignment using a naïve Bayesian classifier. The clusters were constructed at a 3 % dissimilarity cut-off and served as OTUs for determining richness and diversity indices, ACE, Chao (Chao and Bunge 2002), and Good’s coverage using MOTHUR (Schloss et al. 2009).
Statistical significance was determined by one-way analysis of variance (ANOVA) followed by Dunnett’s T3 post hoc test. A two-way ANOVA was applied to analyze the effects of season and reclamation treatment on microbial communities. To compare bacterial community structures across all samples based on the relative abundance of bacterial phyla and proteobacterial classes, principal components analysis (PCA) was performed using CANOCO for Windows (ter Braak and Smilauer 2002). Correlations among physicochemical and microbiological characteristics were conducted by Spearman rank correlation testing. Graphing and data analysis were performed with SPSS BASE ver.11.5 statistical software (SPSS, Chicago, IL, USA) and OriginLab Origin Pro software (version 9.0; OriginLab, Northampton, MA, USA).
16S rRNA gene amplicon pyrosequencing data accession number
The pyrosequencing-generated sequences of the soil bacteria reported in this paper have been deposited in the NCBI Sequence Read Archive (SRA) database under project Accession Number SRA091276.
Results
General characteristics of the pyrosequencing-derived data set
To characterize the bacterial lineages present in the four groups of mine soil samples collected in winter and summer (RE-D, RE-J, SU-D, SU-J), pyrosequencing of the V2–V3 region of the 16S rRNA genes was performed. A dataset consisting of 37,611 high quality sequences with a read length of 500 bp across all samples was generated. A total of 12,382, 12,130, 6,142, and 6,957 high-quality bacterial 16S rRNA gene sequences were obtained from each sample taken from regions RE-D, SU-D, RE-J, and SU-J, respectively (Table S1). We were able to assign 32,363 sequences to the domain Bacteria and to classify 31,758 (98.13 %) of these sequences below the domain level. The Good’s Coverage estimator revealed that 76.8–92.9 % of the estimated taxonomic richness was covered by the sequencing effort.
Chemical characteristics of reclaimed and subsided soils
There were relevant differences between the reclamation and subsidence regions with respect to edaphic properties. In both seasons, the amounts of SOM, TN, and AK were significantly higher in reclamation-treated soil compared with the unreclaimed post-mining soil (P < 0.05) (Table 1), although the amounts of total phosphorus and pH were similar between the reclaimed and subsided soils. These results were expected given the long-term application of organic amendments during the reclamation practice process on this area.
Differences in bacterial diversity
Rarefaction curves depicting the effect of dissimilarity on the number of OTUs showed distinct patterns in bacterial diversity across the four sampling groups (Fig. 2). As suggested by Roesch et al. (2007), bacterial diversity was evaluated using rarefaction curves for 3 (0.03), 5 (0.05), and 10 % (0.10) sequence dissimilarities. Accurate taxonomic assignments and Alpha-diversity estimations based on OTU picker data were made of the species present in the soil at 3 % dissimilarity level (Table 2). The shapes of the rarefaction curves confirmed that the reclaimed soil (RE) were more diverse than the subsided soil (SU). The following trend was found in terms of species richness at 3 % dissimilarity: RE-D > RE-J > SU-J > SU-D. As shown in Table 2, among samples collected in December, the maximum predicted OTUs at 3 % dissimilarity was 2997 for RE and 1433 for SU. For the samples collected in July, the maximum predicted OTUs was 2263 for RE and 2206 for SU. The non-parametric analysis of diversity indexes (ACE, Chao1, Shannon) showed similar trends at 3 % genetic distance, with higher values in RE. For example, in the winter, the ACE estimators of diversity were 7992 for RE and 3643 for SU; the Shannon indices were 7.16 for RE and 5.74 for SU. The data obtained clearly demonstrated that reclaimed soil had significantly more diversity than subsided soil.

Rarefaction curves representing the numbers of operational taxonomic units (OTUs) versus the number of tags sampled from pyrosequencing data at distances of 0.03, 0.05, and 0.10. (a, b) Rarefaction curves of reclaimed soils sampled in winter (RE-D) and in summer (RE-J); (c, d) Rarefaction curves of subsided soils sampled in winter (SU-D) and in summer (SU-J)
Difference in bacterial community structure between reclaimed and subsided soils
The composition of bacterial taxa differed between reclaimed and unreclaimed post-mining soil (Fig. 3). The dominant taxa in reclaimed soils were Gemmatimonadetes (15.15 %), Chloroflexi (14.70 %), Actinobacteria (11.84 %) and Nitrospirae (11.56 %), whereas in subsided soils the predominant phylogenetic groups were Actinobacteria (27.43 %), Acidobacteria (16.78 %) and Planctomycetes (15.90 %).

Relative abundance of the dominant bacteria phyla (proteobacterial classes) based on the individual sample and the whole dataset. Sequences not classified to any known phylum are included as “No rank”. Phylogenetic groups accounting for <1 % of all classified sequences are summarized in the artificial group “others”. RE-D reclaimed soil sampled in winter, RE-J reclaimed soil sampled in summer, SU-D subsided soil sampled in winter, SU-J subsided soil sampled in summer
Classes Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, and Deltaproteobacteria, all part of the most dominant phylum Proteobacteria, had a markedly different abundance between the two soil systems (Fig. 3). Betaproteobacteria was the largest sub-group of Proteobacteria in RE, and Gammaproteobacteria was the largest sub-group in SU. The relative abundance of Gammaproteobacteria among the four sampling groups was significantly different, and SU-D had the highest abundance (26.28 %), followed by RE-D (5.76 %), RE-J (13.32 %), and SU-J (7.38 %). Principal component analysis (PCA) based on the relative abundances of the different bacterial phyla and proteobacterial classes confirmed that the bacterial communities in reclaimed soils differed from communities in subsided soils (Fig. 4). The results showed that first two axes of the PCA explained 42.1 and 35.2 %, respectively, of the total variation in the data. Bacterial communities from the same sampling site tended to group together, with stronger clustering between samples from similar soil systems than from similar sampling seasons. As shown in Fig. 4, profiles of bacterial communities from reclaimed soils (RE-D and RE-J) formed a cluster, and were separated from subsided soils of different sampling seasons (SU-D and SU-J). We observed significantly higher relative abundances of Actinobacteria, Bacteroidetes, Planctomycetes, Alphaproteobacteria and Gammaproteobacteria in subsided soils than in reclaimed soils whereas Betaproteobacteria, Firmicutes, Gemmatimonadetes and Nitrospirae showed the opposite pattern (Figs. 3, 4).

Principal component analysis (PCA) of bacterial communities as affected by reclamation and season, based on the relative abundance of bacterial phyla and proteobacterial classes. PCA1 explained 42.1 % and PCA2, 35.2 % of variability. Every vector points to the direction of increase for a given variable so that soil samples with similar bacterial communities are localized in similar positions in the diagram. Circle and Diamond symbol types respectively represent the reclaimed soil (RE) and subsided soil (SU) (different soil systems). White and black respectively represent soils sampled in December (-D) and July (-J) (different seasons). Acido Acidobacteria, Actino Actinobacteria Bactero Bacteroidetes, Gemmati Gemmatimonadetes, Chloro Chloroflexi, Firmi Firmicutes, Nitro Nitrospirae, Plancto Planctomycetes, Verruco Verrucomicrobia, Alpha-pr Alphaproteobacteria, Beta-pr Betaproteobacteria, Gamma-pr Gammaproteobacteria, Delta-pr Deltaproteobacteria. SOM soil organic matter, TN total nitrogen, TP total phosphorus, AK available potassium
At the genus level, the microbial composition also differed greatly between reclaimed and subsided soils. To compare the distribution of bacterial genera within sites form reclaimed and subsided soils, the respective abundances of 16 predominant genera in different sites were examined (Fig. 5). Regardless of different seasons, the relative abundances of Nitrospiraceae_Nitrospira (5.15 % for RE-D, 2.67 % for RE-J), Gemmatimonas (4.63 % for RE-D, 0.36 % for RE-J), Chloracidobacterium (1.83 % for RE-D, 0.11 % for RE-J) and Pseudomonas (0.22 % for RE-D, 2.44 % for RE-J) were higher in reclaimed soils than in subsided soils. The relative abundances of Nitriliruptor, Lysobacter, Roseiflexus, and Arthrobacter were higher in subsided compared with reclaimed soils (8.94 vs. 0.09 %, 2.03 vs. 0.59 %, 1.22 vs. 0.47 %, and 1.20 vs. 0.17 %, respectively).

Relative abundance of the predominant bacterial genera in reclaimed and subsided soil collected in two sampling seasons. The abundance is presented in terms of a percentage of the total number of sequences in a sample. RE-D reclaimed soil sampled in winter, RE-J reclaimed soil sampled in summer, SU-D subsided soil sampled in winter, SU-J subsided soil sampled in summer
Seasonal variations in bacterial community composition
The change in dominant groups of bacterial communities with seasons was also noted. The relative abundance of Acidobacteria and Bacteroidetes was higher in samples collected in July (RE-J and SU-J) than in those collected in December (RE-D and SU-D) (24.76 vs. 2.78 % and 13.32 vs. 6.77 %, respectively). Relevant differences were also found in the proportions of four Proteobacteria sub-groups: Gammaproteobacteria were more abundant in December soil samples than in July samples (32.04 vs. 20.7 %), whereas Deltaproteobacteria were more abundant in July soil samples than in December ones (19.83 vs. 9.16 %). 16S rRNA gene surveys showed that bacteria belonging to Acidobacteria and Nitrospirae were rare or absent from the December samples of subsided soil (SU-D). Even within the same region, bacterial community structures differed between the time of sampling, and the species evenness of July samples was greater than that of December ones. Thus, the shifts in soil bacterial community composition correlated with a change from winter to summer.
Impact of soil properties on the relative abundances of bacterial taxa
We found a significant association between relative abundances of popular phyla (proteobacterial classes) and physicochemical properties in the soils. In particular, SOM and AK showed the highest correlation with the Bray-Curtis dissimilarities of bacterial taxa composition in both sampling periods. As is seen from Fig. 4, abundant phyla of RE samples were more alike and correlated with higher SOM and TN contents, as shown by their close grouping and by the vectors. On the other hand, bacterial communities of SU formed a separate group associated with higher AK contents and lower TN and SOM contents. We found that dominant bacterial phyla (proteobacterial classes) such as Chloroflexi, Firmicutes, Gammaproteobacteria, Betaproteobacteria and Nitrospirae were spread in quadrant II. The relative abundances of those abundant phyla were positively correlated with SOM, TN and TP. We also found strong correlations between AK and relative abundances of bacterial groups. The abundances of Alphaproteobacteria and Gammaproteobacteria were negatively correlated with soil AK, whereas the abundances of Acidobacteria, Deltaproteobacteria and Planctomycetes were positively correlated with AK. Overall, relative abundances of the most abundant phyla were significantly correlated with AK, SOM, and TN. Soil pH and TP were also environmental factors affecting microbial communities.
Relationships between soil chemical properties and microbiological parameters
There was a positive correlation between microbial diversity parameters and the contents of SOM, TN, TP, AK, and pH (r = 0.583–0.782, P < 0.05 or P < 0.01) (Table 3). The correlation analysis indicated highly significant positive correlations between SOM and Chao, Shannon index, and ACE (P < 0.01). Highly significant positive correlations were also observed between soil AK and those microbiological parameters (Chao, Shannon index, and ACE) (P < 0.01). These results suggest that SOM and AK were the main factors influencing soil microorganisms. In addition, bacterial diversity parameters also showed significant correlations with the concentrations of TN and pH (P < 0.05), but were not significantly related to TP.
Discussion
This work represents the implementation of 454 high-throughput sequencing technology for comparative analysis of soil microbial communities in China. The 16S rRNA pyrosequencing survey can provide more sequence information on soil bacterial communities because of its capacity to identify greater numbers of bacteria than conventional molecular biology techniques (Dimitriu et al. 2010; Rastogi et al. 2010). We were able to classify 31,758 (98.13 %) of the 32,363 quality sequences below the domain level. The total number of analyzed sequences and the percentage of classified 16S rRNA gene sequences exceeded those of other pyrosequencing-based studies of soil bacterial communities in mining areas (Bastida et al. 2013; Poncelet et al. 2014). This work confirmed that this technology is suitable for quick and comprehensive evaluations of soil microflora.
Proteobacteria and Actinobacteria were in relatively high abundance in our sequence libraries, which is in agreement with previous studies (Lauber et al. 2009; Rastogi et al. 2010; Qiu et al. 2012). Regardless of seasons or regions, Proteobacteria was a ubiquitous and common group in soil (Kuffner et al. 2012; Bastida et al. 2013; Chen et al. 2013). Of the 4 sub-groups of the Proteobacteria phyla, Gammaproteobacteria was the most prevalent, which was different from other studies (Roesch et al. 2007; Bastida et al. 2013; Poncelet et al. 2014). Interesting is the fact that the populations of Pseudomonas or Pseudomonadales affiliated with the Gammaproteobacteria were observed in significant proportions in our samples, which has been reported previously by other studies in mining environments (Chen et al. 2013). Pseudomonas play a vital role in soil nitrogen cycling and participate in significant ecological processes (Rich and Myrold 2004; Keil et al. 2011). This finding may indicate a significant role for this nitrogen-cycling bacterial functional group in mine ecological systems (Ye and Thomas 2001). Within the N-cycle, nitrifying and denitrifying communities are responsible for N-losses through nitrate (NO3 −) leaching or greenhouse gas emissions in the form of nitrous oxide (N2O) (Philippot et al. 2007). It has been reported that Pseudomonas denitrificans and Pseudomonas fluorescens are the main denitrifying bacteria (Ghiglione et al.1999; Enwall et al. 2005). Pseudomonas fluorescens was found capable of ammonification in relation to soil nitrogen cycling (Holguin and Bashan 1996). In addition, Actinobacteria was widely distributed in both terrestrial and aquatic ecosystems, especially in soil (Ventura et al. 2007). Similar to the findings recently reported by Poncelet et al. (2014), our study showed that Actinobacteria-affiliated phylotypes were predominantly numerically dominated by Solirubrobacteria spp. (order Solirubrobacterales). However, their ecological significance remains poorly understood (Ishak et al. 2011; Poncelet et al. 2014).
Changes in the composition and diversity of soil bacterial communities examined with pyrosequencing after reclamation and re-vegetation of abandoned mine land were also in agreement with distinct variation in bacterial communities after reclamation and planting examined with traditional extraction of DNA and cloning by He et al. (2012). In general, bacterial community shifts across those reclaimed and subsided soil systems followed specific trends, such that (a) an increase in abundance of Firmicutes, Gemmatimonadetes and Nitrospirae was shown in reclaimed sites, (b) a decrease in abundance of Actinobacteria, Bacteroidetes, and Planctomycetes was shown in reclaimed sites, and (c) no consistent trend was shown across systems (Acidobacteria, Proteobacteria, and Chloroflexi) (Fig. 3). Similar to our study, recent reports by Lewis et al. (2012) and Banning et al. (2011) have shown a significant increase of Firmicutes in post-mined soils undergoing reclamation for 18 or 20 years after mining. However, inconsistent with the earlier work by Lewis et al. (2012), the Actinobacteria group was more abundant in subsided soil (SU) than in reclaimed mine soil (RE) in this study and, thus, suggests that the number of Actinobacteria sequences responded negatively to reclamation and restoration. These findings suggest a strong likelihood of functional role(s) played by bacteria belonging to Firmicutes, Proteobacteria and Actinobacteria in soils undergoing post-mining secondary succession (Banning et al. 2011; Lewis et al. 2012).
The present findings indicate that soils in reclaimed sites showed higher bacterial diversity indices and differing community compositions than subsided soils. Soil bacterial communities were more liable to be influenced by sampling season, reclamation and their interaction (Table S3). However, soil microbial diversity was more affected by reclamation than by season (Table S3). Microbial communities from similar soil systems clustered more strongly together (RE or SU) than those from similar sampling seasons (-D or -J) (Fig. 4). These results suggest that reclamation had greater impacts on the microbial communities than seasonal fluctuations. The chemical and microbiological data point to a strong impact of reclamation on the quality of soil. SOM, AK and TN may be key factors controlling soil bacterial diversity and community composition. There are two possible explanations for the differences in bacterial communities between reclaimed and subsided soils: (1) reclamation with long-term organic fertilizer application showed a significant influence on the physicochemical properties of the soil, which in turn affected bacterial community diversity, such that different microbial microenvironments harbored different microbial communities (Zhang et al. 2010). (2) The aboveground plant species can affect the quality and quantity of microbial metabolic substrates, which may initiate changes in the bacterial community and so affect function.
Previous research has shown that organic fertilizers can generally improve soil physical and chemical properties and that they contain nutrients beneficial to microorganisms and plants (Islam et al. 2009). Ge et al. (2008) found long-term fertilization of soil in eastern China with organic manure to yield distinct community structures with more richness and diversity. Similarly, Chu et al. (2007) found that the use of organic supplements could cause significant increases in species richness and Shannon–Weaver indices over untreated plots. It is therefore reasonable to surmise that the high microbial diversity observed in the reclaimed soil might be driven by its high concentrations of SOM or TN, which could be attributed to the effectiveness of applying organic fertilizers.
In large part, the mixed sowing of legumes with gramineous grass could also be responsible for the increased bacterial diversity in reclaimed soils. Medicago sativa and Trifolium repens have been widely planted in the East China Plain because of their strong tolerance to drought and barren soil and their outstanding heat and cold resistance (Wu et al. 2009). The community coexistence mechanisms of deep legume roots and shallow grass roots can give rise to growth of the biological nitrogen fixing bacterium, Rhizobium (Xu et al. 2010). This is highly conducive to microbial growth and reproduction. In this way, the higher OTUs and diversity indices (Chao and ACE) obtained in reclaimed soils are of ecological significance, as demonstrated by the positive impact on bacterial diversity in areas with a history of diverse crop residues and rooting systems sown with a mixture of gramineae–leguminosae herbage. The higher bacterial diversity of reclaimed soils highlights an ecological reaction of plant-cover development in reclamation soils. The production of litter and root exudate may create conditions that foster eco-diversity and stable microbial communities. On the basis of these findings, it was concluded that both vegetation cover and fertilizer treatment affected soil bacterial communities, with soil characteristics controlling or mediating biogeochemical processes and ecosystem functionality.
Conclusions
The results shown here confirm the hypothesis that soil bacterial diversity and composition were significantly affected by the 13-years land reclamation program. The analysis of bacterial 16S rRNA-based datasets from soils revealed statistically significant differences in soil bacterial diversity and community structure between the reclaimed and subsided land. Stronger clustering of bacterial communities from the same soil system indicated that reclamation practices may have a greater influence on soil microbial communities than seasonal variation. The analysis of effects of soil properties on bacterial community structure revealed that SOM, AK and TN had significant influence on bacterial community structure and diversity. Long-term additions of organic amendments and mixed sowing of different herbage have significant effects on microbial communities and facilitate the improvement of bacterial community diversity in reclaimed soils. Overall, this work provides valuable insight into the response of terrestrial ecosystems to soil reclamation of abandoned mine land and demonstrates the considerable potential of pyrosequencing in characterizing soil microbial communities.
References
Banning NC, Gleeson DB, Grigg AH, Grant CD, Andersen GL, Brodie EL, Murphy DV (2011) Soil microbial community successional patterns during forest ecosystem restoration. Appl Environ Microbiol 77:6158–6164
Bao SD (2008) Soil agricultural chemistry analysis. China Agriculture Press, Beijing
Bastida F, Hernández T, Albaladejo J, García C (2013) Phylogenetic and functional changes in the microbial community of long-term restored soils under semiarid climate. Soil Biol Biochem 65:12–21
Bremner JM, Mulvaney RL (1978) Urease activity in soils. In: Burns RG (ed) Soil Enzymes. Academic Press, New York, pp 149–196
Cabrera-Rubio R, Collado MC, Laitinen K, Salminen S, Isolauri E, Mira A (2012) The human milk microbiome changes over lactation and is shaped by maternal weight and mode of delivery. Am J Clin Nutr 96:544–551
Chao A, Bunge J (2002) Estimating the number of species in a stochastic abundance model. Biometrics 58:531–539
Chen LX, Li JT, Chen YT, Huang LN, Hua ZS, Hu M, Shu WS (2013) Shifts in microbial community composition and function in the acidification of a lead/zinc mine tailings. Environ Microbiol 15:2431–2444
Chu HY, Lin XG, Fujii T, Morimoto S, Yagib K, Hua JL, Zhang JB (2007) Soil microbial biomass, dehydrogenase activity, bacterial community structure in response to long-term fertilizer management. Soil Biol Biochem 39:2971–2976
Dimitriu PA, Prescott CE, Quideau SA, Grayston SJ (2010) Impact of reclamation of surface-mined boreal forest soils on microbial community composition and function. Soil Biol Biochem 42:2289–2297
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Enwall K, Philippot L, Hallin S (2005) Activity and composition of the denitrifying bacterial community respond differently to long-term fertilization. Appl Environ Microbiol 71:8335–8343
Fan WH, Bai ZK, Li HF, Qiao JY, Xu JW (2011) Effects of different vegetation restoration patterns and reclamation years on microbes in reclaimed soil. Trans CSAE 27:330–336
García-Orenes F, Morugán-Coronado A, Zornoza R, Scow K (2013) Changes in soil microbial community structure influenced by agricultural management practices in a Mediterranean agro-ecosystem. PLoS ONE 8:e80522
Ge Y, Zhang JB, Zhang LM, Yang M, He JZ (2008) Long-term fertilization regimes affect bacterial community structure and diversity of an agricultural soil in northern China. J Soil Sediment 8:43–50
Ghiglione JF, Philippot L, Normand P, Lensi R, Potier P (1999) Disruption of narG, the gene encoding the catalytic subunit of respiratory nitrate reductase, also affects nitrite respiration in Pseudomonas fluorescens YT101. J Bacteriol 181:5099–5102
Hahn AS, Quideau SA (2013) Long-term effects of organic amendments on the recovery of plant and soil microbial communities following disturbance in the Canadian boreal forest. Plant Soil 363:331–344
He XY, Su YR, Liang YM, Chen XB, Zhu HH, Wang KL (2012) Land reclamation and short-term cultivation change soil microbial communities and bacterial metabolic profiles. J Sci Food Agric 92:1103–1111
Holguin G, Bashan Y (1996) Nitrogen-fixing by Azospirillum brasilense Cd is promoted when co-cultured with a mangrove rhizosphere bacterium (Staphylococcus sp.). Soil Biol Biochem 28:1651–1660
Ingram LJ, Schuman GE, Stahl PD, Spackman LK (2005) Microbial respiration and organic carbon indicate nutrient cycling recovery in reclaimed soils. Soil Sci Soc Am J 69:1737–1745
Ishak HD, Miller JL, Sen R, Dowd SE, Meyer E, Mueller UG (2011) Microbiomes of ant castes implicate new microbial roles in the fungus-growing ant Trachymyrmex septentrionalis. Sci Rep 1:204
Islam MR, Trivedi P, Palaniappan P, Reddy MS, Sa T (2009) Evaluating the effect of fertilizer application on soil microbial community structure in rice based cropping system using fatty acid methyl esters (FAME) analysis. World J Microb Biot 25:1115–1117
Kaschuk G, Alberton O, Hungria M (2010) Three decades of soil microbial biomass studies in Brazilian ecosystems: lessons learned about soil quality and indications for improving sustainability. Soil Biol Biochem 42:11–13
Keil D, Meyer A, Berner D, Poll C, Schützenmeister A, Piepho H-P, Vlasenko A, Philippot L, Schloter M, Kandeler E, Marhan S (2011) Influence of land-use intensity on the spatial distribution of N-cycling microorganisms in grassland soils. FEMS Microbiol Ecol 77:95–106
Kuffner M, Hai B, Rattei T, Melodelima C, Schloter M, Zechmeister-Boltenstern S, Jandl R, Schindlbacher A, Sessitsch A (2012) Effects of season and experimental warming on the bacterial community in a temperate mountain forest soil assessed by 16S rRNA gene pyrosequencing. FEMS Microbiol Ecol 82:551–562
Lauber CL, Hamady M, Knight R, Fierer N (2009) Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75:5111–5120
Lewis DE, Chauhan A, White JR, Overholt W, Green SJ, Jasrotia P, Wafula D, Jagoe C (2012) Microbial and geochemical assessment of bauxitic un-mined and post-mined chronosequence soils from Mocho Mountains, Jamaica. Microb Ecol 64:738–749
Mummey DL, Stahl PD, Buyer JS (2002) Soil microbiological properties 20 years after surface mine reclamation spatial analysis of reclaimed and undisturbed sites. Soil Biol Biochem 34:1717–1725
Pan P, Kang Q, Li X (2003) Determination of total phosphorus in soil by ammonium molybdate spectrophotometry. Chin J Spectrosc Lab 20:697–699
Philippot L, Hallin S, Schloter M (2007) Ecology of denitrifying prokaryotes in agricultural soil. Adv Agron 96:249–305
Poncelet DM, Cavender N, Cutright TJ, Senko JM (2014) An assessment of microbial communities associated with surface mining-disturbed overburden. Environ Monit Assess 186:1917–1929
Qiu MH, Zhang RF, Xue C, Zhang SS, Li SQ, Zhang N, Shen Q (2012) Application of bio-organic fertilizer can control Fusarium wilt of cucumber plants by regulating microbial community of rhizosphere soil. Biol Fertil Soils 48:807–816
Rastogi G, Osman S, Vaishampayan PA, Andersen GL, Stetler LD, Sani RK (2010) Microbial diversity in uranium mining-impacted soils as revealed by high-density 16S microarray and clone library. Microb Ecol 59:94–108
Rich JJ, Myrold DD (2004) Community composition and activities of denitrifying bacteria from adjacent agricultural soil, riparian soil, and creek sediment in Oregon, USA. Soil Biol Biochem 36:1431–1441
Roesch LF, Fulthrope RR, Riva A, Casella G, Hadwin AKM, Angela DK, Daroub SH, Camargo FAO, Farmerie WG, Triplett EW (2007) Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1:283–290
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Horn DJV, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Sheoran V, Sheoran AS, Poonia P (2010) Soil reclamation of abandoned mine land by revegetation: a review. Int J Soil Sediment Water 3:1–4
ter Braak CJF, Smilauer P (2002) CANOCO reference manual and CanoDraw for windows user’s guide: software for canonical community Ordination (version 4.5). Ithaca, NY, USA: Microcomputer Power
Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald GF, Chater KF, van Sinderen D (2007) Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev 71:495–548
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Wilson DW, Sander LE (1996) Total carbon, organic carbon, and organic matter. In: Sparks DL (ed) Methods of soil analysis: part 3-Chemical methods. Soil Science Society of America, Madison, pp 1002–1005
Wu RP, Li H, Cao P (2009) Amelioration of weathered coal on soil physical, chemical properties and enzyme activities with vegetation restoration. J Agro-Environ Sci 28:1855–1861
Xu QF, Jiang PK, Wang HL (2010) Improvement of biochemical and biological properties of eroded red soil by artificial revegetation. J Soils Sediments 10:255–262
Ye RW, Thomas SM (2001) Microbial nitrogen cycles: physiology, genomics and applications. Curr Opin Microbiol 4:307–312
Zhang B, Deng H, Wang HL, Yin R, Hallett PD, Griffiths BS, Daniell TJ (2010) Does microbial habitat or community structure drive the functional stability of microbes to stresses following re-vegetation of a severely degraded soil? Soil Biol Biochem 42:850–859
Acknowledgments
This work was supported by the National Natural Science Foundation of China (51174207) and the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions (SZBF2011-6-B35). The authors are grateful to Majorbio Biotech Co., Ltd (Shanghai, China) for their help in sample sequencing and analysis. The authors would like to thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Li, Y., Chen, L. & Wen, H. Changes in the composition and diversity of bacterial communities 13 years after soil reclamation of abandoned mine land in eastern China. Ecol Res 30, 357–366 (2015). https://doi.org/10.1007/s11284-014-1230-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11284-014-1230-6