
Abstract
The close interplay between hydraulic and biotic processes controls the evolution of ecosystems in large, circumboreal peatlands and the degree to which they lose, gain or sequester carbon. In peatlands, biota significantly alters surficial acid–base equilibrium and solute chemistry by releasing dissolved organic compounds into surface and pore waters, which then mix through advection and dispersion through pore water flow paths.
We report herein the results of new geochemical mixing models that incorporates organic acid dissociation constants to understand how organic acids control the acid–base chemistry in mixtures of dilute acidic bog pore waters and circum-neutral groundwater (typical of carbonate terrains) that upwells or disperses into peat deposits.
In our new mixing models, at least twice as much groundwater is required to neutralize bog water acidity when dissolved organic carbon concentrations exceed 10 mmol/L than in their absence. Although it remains uncertain how future climatic change will alter the composition of dissolved organic matter in peatlands, organic acids should remain an important factor with respect to potential neutralization of peatland waters by groundwater sources. The acid base equilibria in large peat basins not only has important implications for vegetation patterning but also for biogeochemical cycles in these globally important reservoirs for carbon.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Dissolved organic carbon in peat pore water consists mostly of humic and fulvic acids having a wide variety of functional groups (Thurman 1985). Bogs and fens in patterned northern peatlands consist of ecological communities that depend upon waterlogged conditions and different availability of solutes from both groundwater found in underlying mineral soils and precipitation. The understory of bogs in circumboreal peatlands typically consists of mostly acid-tolerant Sphagnum moss, whereas adjacent fen ecosystems contain a broad range of sedges and other species that grow better in water closer to circumneutral in pH.
Bogs ecologically transition to fens when geochemical thresholds in the near surface waters are exceeded with respect to a pH of 4.2 and calcium concentration of 0.05 mmol l−1 (Glaser 1992; Glaser et al. 1997). Where pH values rise above about 5.6 (the pH of precipitation in equilibrium with atmospheric CO2), methanogenesis is favored in deep peat (e.g., Ferry 1993), leading to observed large accumulations of free phase methane below 1 m depth in some large mires (e.g., Romanowicz et al. 1995; Glaser et al. 2004a, b, c). However, because of the complexities of acid–base chemistry in organic-rich peat pore water, it remains uncertain how much discharging groundwater is needed to neutralize acidic water in bog peat sufficiently to cause an ecological shift from bog to fen and enhance methanogenesis.
Since the mid-1980’s, wetland scientists have understood that hydraulic conditions caused by water table mounds under raised bogs cause vertical hydraulic gradients that drive surface acidic bog water downwards into the deeper bog peat. Pore water even in deep bog peat normally remains dilute because the precipitation on bogs moves downward in a pronounced recharge function (e.g., Glaser et al. 1997).
The surface water and pore water in the upper 50 cm of bog peat typically remain acidic and solute poor, because bogs only depend on atmospheric deposition for water and salts and lack sufficient base cations to neutralize organic acids released from decaying Sphagnum. The high cation-exchange capacity of the Sphagnum moss in bogs contributes to the pore water acidity, along with peat oxidation which adds organic acids and carbon dioxide (e.g., Shotyk 1988; Clymo and Hayward 1982;. Hemond 1980; Spearing 1972).
These organic acids dissociate and release H+-ions along with those released by cation exchange for base metals in the Sphagnum moss (Breemen 2003; Gorham et al. 1985). The dissociated organic acids cause charge imbalances (difference between positive charged metals and negative charged ligands) greater than +50 % because the negative charged organic acid functional groups cannot be directly measured in routine analyses (Siegel et al. 2006). Indeed, Glaser et al. (2004b) suggested that organic acids released from peat in the Hudson Bay Lowlands generated pH low enough to modify the ecosystem landscape, despite the presence of calcareous subsurface sediments having high buffering capacity.
In the Glacial Lake Agassiz Peatlands of Minnesota raised bogs tend to overlie groundwater discharge zones in the underlying mineral soils where mineralized water can, during decadal scale droughts, upwell into the peat column to move downward recharging water to the side (e.g., Glaser et al. 2004a; Glaser et al. 2004b; Siegel and Glaser 1987). Then, bog peat can be almost completely neutralized by upwelling groundwater (Siegel et al. 1995a, b).
Along with the upwelling groundwater, chemical diffusion, and transverse dispersion (mixing) under lateral flow conditions (Reeve et al. 2001) also transport bases into lower bog peat to neutralize the acids generated by the decomposition of organic matter or by carbon dioxide deposited directly by atmospheric precipitation. Siegel (1983) suggested through simple model simulations that if ~10 % of groundwater from underlying mineral soils mixes with acidic dilute bog surface water, the pH of bog surface water could rise from less than 3.2 to 5.7, (Siegel and Glaser 1987). However, these models did not include the acid load generated by humic and fulvic acids which could dissociate and release hydrogen ions, thereby diminishing inorganic buffering of pH by dissolved carbonate.
In this paper we present the results of a new series of geochemical reaction models designed specifically to address the problem of how organic acids affect the complex acid–base chemistry of peat pore waters. In our heuristic modeling experiments, we directly incorporated organic acid dissociation constants (Siegel et al. 2006) with other organic acid properties to determine the extent to which organic acids affect the neutralization process. Here we hypothesize that bog organic acids may generate sufficient hydrogen ions to necessitate much greater mixing of groundwater with bog surface waters to buffer pore waters to a pH greater than 5.7.
Background and Study Area
In our study area, the Glacial Lake Agassiz Peatlands of northern Minnesota (GLAP), peat landforms spread over 7600 km2 on a former lake plain. The peat overlies 40–60 m of unconsolidated calcareous glacial-lacustrine deposits that mantle metamorphic bedrock (Glaser 1992). The thickness of peat ranges from 3 to 5 m in bogs and ~3 m in fens (Glaser 1992). Bog pore waters at GLAP have pH < 4.2 and calcium concentration <0.05 mmol/L. Black spruce (Picea mariana) or lichens and various species of Sphagnum dominate bog vegetation. Precipitation solely sustains bog plants, which tolerate acidic pore water. Fens, in contrast, have slightly acidic to circumneutral water with pH 4.2 to 8, and higher calcium concentrations (>0.05 mmol/L). Groundwater discharge sustains fen peat vegetation, which are commonly sedges such as Carex lasiocarpa (Glaser 1992; Glaser et al. 1981).
The peat pore waters in the raised bogs of GLAP, as well as elsewhere (e.g., Hudson Bay lowlands, Reeve et al. 1996) have more than 20 % charge imbalance (Siegel et al. 2006) caused by the organic acids. Siegel et al. (2006) estimated the bulk de-protonation constants (pKa) of organic acids in GLAP bogs from charge balance considerations and a tri-protic organic acid dissociation model. The dissolved organic carbon in these bog pore waters, despite their low mole site densities of protons, behaves as a strong acid, whereas fen-generated organic acids with higher mole site densities behave as weak acids.
Methods
Field Sampling
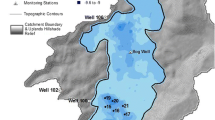
We evaluated acid–base reactions in pore water from two raised bog profiles (Red Lake II and Lost River) within GLAP (Fig. 1) by incorporating the acidity generated by humic acids. We selected Red Lake II bog because it is the largest raised bog in GLAP (>160 km2) and has landform patterns similar to that found in other large peat basins (Glaser et al. 1997, 2004a, b, c). The Red Lake II bog complex straddles the center of the local climatic gradient, which becomes progressively drier toward the west (Baker et al. 1985). The Lost River bog complex, in contrast, consists of groundwater springs sustaining a spring-fen mound adjacent to a raised bog (Siegel and Glaser 1987; Glaser et al. 1990). These two features reflect the largest contrast in vegetation and water chemistry found in peatlands (Glaser et al. 1990), and surface water chemistry from these two bogs and spring fen landforms embrace the range of that found large peat basins.

Map of Glacial Lake Agassiz Peatlands, northern Minnesota. The Red Lake II and Lost River bogs are marked by arrows. Modified from Glaser 1992
The basal peat-mineral contact occurs at a depth of 4.3 m in both bogs. We collected pore water samples in peat from open-ended, hand-installed piezometers every 0.5 m interval from the water table to the peat-mineral soil interface and used methods described in Chason and Siegel (1986). We used a portable peristaltic pump to sample pore water from the piezometers, at rate of about 30 mL/min and measured pH and specific conductivity in situ, with a Cole-Palmer WTW field pH meter and specific conductivity meter. We collected 30 mL samples for major cations and 60 mL for major anions and alkalinity and stored them in high-density polyethylene bottles. (Sentence deleted as suggested).
To characterize the groundwater end member, we therefore collected groundwater samples from ten domestic wells 15 to 20 m deep in mineral soils immediately south of the peatland and treated the samples in the same manner as the ones we obtained from the peatland.
Analysis and Quality Assurance
We filtered in the lab all samples collected for major chemical analysis through 0.45 μm filters and preserved aliquots for metals analysis by adding reagent-grade nitric acid to lower the pH to <2 units. We measured metals concentrations with a Beckman Spectrospan-V Direct Current Plasma Spectrometer. We repeated each analysis three times and the difference in our measurements was <6 %. We used a Dionex ICS-2000 IC (ion chromatography) (Deleted Mas Spectrometer) system to analyze ligands with a precision and accuracy <5 %. We used a Shimadzu Total Organic Carbon (TOC) analyzer Hto determine the DOC and dissolved Inorganic Carbon (DIC) concentrations of pore waters, respectively. The precision and accuracy of DIC analyses were ±5 % and ±3 %, respectively, and that of DOC analyses were ±1.5 %.
Geochemical Modeling
Evaluating Organic Acid Charge
We theoretically mixed geogenous ground water and acidic bog water to evaluate how the acid–base chemistry of peat pore water changes with different proportions of bog water and ground water. To do this, we first calculated charge balances as the difference between the sum of the equivalence of major positive charged species (including hydronium ion, calcium, magnesium, sodium, potassium, strontium, and iron) from major negative charged species (bicarbonate calculated from DIC, sulfate, nitrate and chloride). Organic acids, unanalyzed directly, are also major negative charge inorganic species.
To evaluate the thermodynamic controls over these organic acids, we assumed that the excess positive charge in the charge balance calculation reflected unanalyzed negative charge contributed by organic acids (e.g., Driscoll et al. 1994; Siegel et al. 2006), and then from this, estimated the organic anion charge from pH and dissolved organic carbon (DOC) concentrations, assuming a triprotic analog model with mole site density assigned for bog organic acids from Siegel et al. (2006). Using this procedure, our charge balance error for pore waters became less than ±20 %. While this error bar may seem large, adopting a smaller range of error would not be prudent given that we necessarily would have to treat the total organic acid load as a single surrogate organic acid species and force fit the solution.
Selection of End Members for Mixing Calculations
We theoretically mixed two end members in our study by changing their proportions in the mixing process. We chose for our two component mixtures the most acidic bog water, surface water at Red Lake II bog, rich in organic acids but low in dissolved solids, and the most inorganic-rich groundwater from mineral soils at a nearby domestic well, the Nelson Home well (See Supplemental Data).
Bogs normally recharge underlying mineral soils with acidic water, and therefore ground water sampled immediately below bog peat now would have inherently less buffering capacity than the pure groundwater end member deeper in the glacial till. Moreover, this groundwater end member continually changes in its chemical composition because of variable rates of recharge and dilution. Therefore, we chose to use pure groundwater from the sampled domestic well as one end member for our heuristic mixing models to methodically bias our results towards the least amount of groundwater necessary to neutralize bog pore waters. We recognize that the actual amount needed would be much higher, if more dilute ground water end members used. Our approach mimics that done before by (Siegel. 1983) who did not incorporate organic acids in his mixing model so we now can clearly compare our results to the prior model which did not incorporate organic acids.
PHREEQC Coding
We used PHREEQC (Parkhurst and Appelo 1999), a United States Geological Survey (USGS) geochemical modeling computer code, to calculate the species of ions in the waters and then mix and react them in various proportions to get a final solution. Basically, we simulated the natural ‘titration’ caused by mixing acidic bog water with more basic groundwater to determine the extent to which organic acids in peat pore water control the neutralization process.
PHREEQC calculates the charge imbalance and speciation of our end member solutions from the following criterion:
where, ε represents the error percentages in charge balance.
The program also routinely calculates the saturation state of the pore waters relative to a set of minerals, in particular, for calcite:
Ks0 is the solubility product and [x] activity of “x” species.
The program also calculates the ion activity product for calcite, the dominant mineral dissolving to condition groundwater chemistry in underlying mineral soils (e.g., Siegel and Glaser 1987).
IAP is the ion activity product of calcite dissolution and from this and the equilibrium constants the model calculates a saturation index:
where SI is the saturation index of the mineral calcite.
If SI = 0 then the solution is saturated with respect to the mineral calcite in this case. If SI is negative, the mineral is undersaturated and can dissolve. If the SI is more than 0, the mineral can precipitate. Given expected uncertainty in analyses and thermodynamic data, we considered log SI = 0 ± 0.5 within saturation limits.
Of the suite of minerals in the underlying ground water, including quartz, feldspars and other oxides, calcite fundamentally controls the geochemistry of groundwater beneath the peat because other minerals dissolve and precipitate at orders of magnitude slower rates and contribute minimally to neutralization of pore waters when calcite is present (e.g., Siegel and Glaser 1987; Langmuire 1997).
We used PHREEQC to mix the bog and groundwater solutions in various proportions to assess how much groundwater would be required to neutralize pore water in bogs. We constructed two sets of mixing models: one without incorporating triprotic organic acids (which did not attain charge balance error within 20 %) and one that included triprotic organic acids.
PHREEQC allows including species not already in the accompanying thermodynamic database in simulations as long as the added species conform to the required format. For the models incorporating organic acids, we used the pKa values and mole site densities from a generic triprotic organic acid after Siegel et al. (2006) to reflect the organic charge in modeled end member waters for the bog. Values of pKa refer to the logarithms of the equilibrium constants of organic acid deprotonation (loss of H+−ions) and using these values constitutes standard practice in geochemisty (e.g., Langmuire 1997).
We fully recognize that the organic acids we modeled may vary locally in bog pore-waters, depending on the botanical composition of the peat and variable peat decomposition rates. However, values for the bulk organic acid compositions probably do not vary by more than a factor or two.
We assigned the molecular weight of the triprotic acid similar to that of acetic acid (CH3COOH), 60.05 atomic mass units. We realize that the actual molecular weights of humic acids are larger, but in the context of this heuristic model acetic acid can be considered a reasonable organic acid surrogate.
The governing speciation equations are as follows:
where, DOCbog represents a generic functional group found in the organic acids found in bog DOC. .
We derived the following solution species equations using Eqs. 3, 4 and 5 for insertion into PHREEQC input files as new species which would then be incorporated into the equilibrium calculations governing H+ mass balance:
We simulated mixing of the two end-members in 0.5 m increments and then compared the calculated pH with measured pH (see supplemental data).
Results
Water Chemistry
Pore water chemistry is shown in Table 1. Pore waters at Red Lake II bog and Lost River bog crests have low pH (4.0 to <6.0) to depth of about 2.5 m. Below 2.5 m, the pH of pore waters rises well above 6.0 (Fig. 2a). The specific conductance profiles increase with depth from ~50 to 167 μs/cm in the Red Lake II and ~50 to 812 μs/cm in the Lost River bog respectively (Fig. 2c). All major cation concentrations were low (e.g., calcium concentrations ranged from 0.02 to 0.26 mmol/L) in the upper and middle section of bog peat and increased almost by an order of magnitude below 2.5 m depth (Fig. 2b).

Profiles of a. pH (in units), b. Calcium (mmol/L) and c. Specific Conductance (uS/cm) from Red Lake II Bog (filled boxes) and Lost River Bog (open boxes). Note the increase in all of these parameters with depth, reflected a component of groundwater mixing with acidic peat surface water
Other divalent cations (magnesium, sodium and strontium) showed similar increases in concentrations with depth below 2.5 m to the base of peat at Red Lake II and Lost River bogs (Table 1). The solute concentrations at Lost River bog were higher than Red Lake II bog at all depths throughout the peat profile.
Sulfate and nitrate were almost non-existent (below detection level 0.43 μmol/L for NO3 − and 0.26 μmol/L for SO4 2−) and are negligible with respect to charge balance, as would be expected under methanogenic anoxic conditions. The DOC concentrations of pore water in Lost River bog were an order of magnitude higher than the DOC concentrations in pore waters at Red Lake II bog. At both bogs, the DOC concentrations decrease with depth because of mixing with low DOC mineralized water from below (Fig. 3).

Profiles of a. Dissolve Organic Carbon (DOC) and b. Percent Charge Imbalance with Depth from Red Lake II Bog (filled boxes) and Lost River Bog (open boxes). Note how organic acids cause charge imbalance by contributing negative charge unaccounted for by standard analyses
Simulations
PHREEQC initially calculated the groundwater end-member charge balances to within ±5 % error and charge imbalance for the dilute acidic bog waters >+/− 50. This large charge imbalance decreased with depth without including organic acids (3) but fell to ±5 % after we included the pKa-s for organic acids in the charge balance model. PHREEQC showed that the groundwater end member was equilibrated with calcite, as expected, given the calcareous mineral soils beneath the peat. In contrast, the bog pore waters were under saturated with respect to calcite at shallow depths and were therefore capable of dissolving more calcite if it were present. However, the saturation state increased with depth to reach equilibrium with calcite towards the basal peat/mineral soil contact along with increasing concentrations of base cations and alkalinity (Fig. 4).

Saturation Indices for Calcite with Depth in Peat Pore Waters from Red Lake II Bog (filled boxes) and Lost River Bog (open boxes). Note how peat pore waters approach equilibrium (SI = 0) with respect to calcite near the peat-mineral soil interface
When organic acids were included in the simulations, more ground water was needed to neutralize acidic bog recharge water. (Table 2; Fig. 5). In general, the set of simulations for Red Lake II bog that included organic acids (DOC concentration ~6 mmol/L), needed twice as much groundwater (10 %), to raise the pH compared to mixing that did not include organic acids. Similarly, mixing simulations for Lost River bog waters that incorporated organic acids (DOC ~13 mmol/L) needed twice as much groundwater (20 %) for the neutralization process to occur to about 5.7. The charge balance error for the final mixed solution in the simulations always fell within ±5 % error.

Percentage of Ground Water Needed to Neutralize Bog Surface Water to pH > 5.7 with and without Organic Acids (OA)
Discussion and Conclusions
The availability of DIC in pore waters derived from calcium bicarbonate type groundwater mostly controls the acid neutralizing capacity in peatlands. A warming climate may substantially lower the water table in peatlands where groundwater mounds under bogs currently block upward advection of groundwater towards bog surfaces. Our results suggest that at a minimum, 10–20 % of groundwater is needed to mix with shallow pore water on bogs to raise the pH sufficiently to equilibrate to a pH of 5.7, typical of that found in precipitation in regions unaffected by industrial pollution (Berner and Berner 2012) but also rich fens of the GLAP and nearby regions (Glaser 1992; Glaser et al. 1990, 2004a, b, c). This amount of groundwater, incorporating organic acids with inorganic buffers, is twice that previously estimated by Siegel et al. (2006).
More ground water must discharge into peatlands than previously thought to generate observed pH profiles, particularly since our mixing end-member for ground water used the most concentrated groundwater sample we could find in the study area. With more dilute groundwater end-members, even more groundwater would have to mix within the peat pore waters to generate the same changes in pH.
An inherent problem in calculating how ground water and bog water mixes is the dynamic nature of peatland hydrology, wherein mineralized water in the glacial deposits under bogs may be diluted through recharge downward during moist periods, and then enriched during other times, during droughts. Then, deep groundwater in the mineral soils displaces that diluted by prior peat pore water with smaller concentrations. We also recognize that changing dynamics of methanogenesis in bogs might accompany future climate change and thereby change the mix of organic acids in peat pore water. But we feel it unlikely that the net pKa-s for bulk bog organic acids will change dramatically from what we found and that our study.
References
Baker DG, Kuehnast EL, Zandlo JA (1985) Climate of Minnesota. Part XV Normal temperatures 1951–1980 and their application. University of Minnesota Agriculture Experiment Station AD SB-2777–2985
Berner EK, Berner RA (2012) Global environment: water, air, and geochemical cycles. Princeton University Press, Princeton
Breemen NV (2003) How Sphagnum bogs down other plants. Trends Ecol Evol 10:270–275
Chason DB, Siegel DI (1986) Hydraulic conductivity and related physical-properties of peat, lost River Peatland. North Minn Soil Sci 142:91–99
Clymo RS, Hayward PM (1982).The ecology of Sphagnum. In: Smith, A.J.E. (Ed), Bryophyte Ecology, Chapman & Hall, pp. 229–289
Driscoll CT, Lehtinen MD, Sullivan TJ (1994) Modeling the acid–base chemistry of organic solutes in Adirondack, New York, lakes. Water Resour Res 30:297–306
Ferry J (ed) (1993) Methanogenesis: ecology, physiology, biochemistry, & genetics. Chapman & Hall Inc., New York
Glaser PH (1992) Vegetation and water chemistry in the patterned peatlands of Minnesota. In: Wright HE Jr, Coffin BA, Aaseng NE (eds) The patterned Peatlands of Minnesota. University of Minnesota Press, Minneapolis, pp 15–26
Glaser PH, Wheeler GA, Gorham E, Wright HE Jr (1981) The patterned peatlands of the Red Lake peatland, northern Minnesota: vegetation, water chemistry, and landforms. J Ecol 69:575–599
Glaser PH, Janssens JA, Siegel DI (1990) The response of vegetation to chemical and hydrological gradients in the Lost River Peatlands, Northern Minnesota. J Ecol 78:1021–1048
Glaser PH, Siegel DI, Shen Y, Romanowicz EA (1997) Regional linkages between raised bogs and groundwater flow systems in the Glacial Lake Agassiz region of northern Minnesota. J Ecol 85:91–99
Glaser PH, Hansen BCS, Siegel DI, Reeve AS, Morin PJ (2004a) Rates, pathways and drivers for peatland development in the Hudson Bay Lowlands, northern Ontario. Can J Ecol 92:1036–1053
Glaser PH, Siegel DI, Reeve AS, Janssens JA, Janecky DR (2004b) Tectonic drivers for vegetation patterning and landscape evolution in the Albany River region of the Hudson Bay Lowlands. J Ecol 92:1054–1070
Glaser PH, Chanton JP, Morin P, Rosenberry DO, Siegel DI, Ruud O, Chasar LI, Reeve AS (2004c) Surface deformations as indicators of deep ebullition fluxes in a large northern peatland, global Biogeochem. Cycles 18:GB1003. doi:10.1029/2003GB00206
Gorham E, Eisenreich SJ, Ford J, Santelmann MV (1985) Chemistry of bog waters. In: Stumm, W. (Ed.), The Chemical Processes in Lakes, pp. 330–363
Hemond HF (1980) Biogeochemistry of Thoreau’s Bog, Concord. Mass Ecol Monogr 50:507–526
Langmuire D (1997) Aqueous environmental geochemistry. Prentice Hall, New Jersey, p 600
Parkhurst DL, Appelo CAJ (1999) User’s guide to PHREEQC - a computer program for speciation, reaction-path, advective-transport, and inverse geochemical calculations. US Geol Surv Water Resour Investig Rep 99–4259
Reeve AS, Siegel DI, Glaser PH (1996) Geochemical controls on peatland pore water from the Hudson Bay Lowland: a multivariate statistical approach. J Hydrol 181:285–304
Reeve AS, Siegel DI, Glaser PH (2001) Simulating dispersive mixing in large peatlands. J Hydrol 242:103–114
Romanowicz EA, Siegel DI, Chanton JP, Glaser PH (1995) Temporal variations in dissolved methane deep in the Lake Agassiz peatlands, Minnesota (USA). Global Biogeochem Cycles 9:197–212
Shotyk B (1988) Review of the inorganic geochemistry of peats and peatland waters. Earth Sci Rev 25:95–176
Siegel DI (1983) Ground water and evolution of patterned mires, Glacial Lake Agassiz Peatlands, Northern Minnesota. J Ecol 71:913–921
Siegel DI, Glaser PH (1987) Groundwater flow in a bog-fen complex, Lost river peatland, northen Minnesota. J Ecol 75:743–754
Siegel DI, Reeve A, Glaser PH, Romanowicz E (1995a) Climate-driven flushing of pore water from humified peat: geochemical and ecological ramifications. Nature 374:531–533
Siegel DI, Reeve AS, Glaser PH, Romanowicz EA (1995b) Climate-driven flushing of pore-water in peatlands. Nature 374:531–533
Siegel DI, Glaser PH, So J, Janecky DR (2006) The dynamic balance between organic acids and circumneutral groundwater in a large boreal peat basin. J Hydrol 320:421–431
Spearing AM (1972) Cation-exchange capacity and glacturonic acid content of several species of Sphagnum in sandy ridge bog, central New York State. Bryol 75:1148–1154
Thurman EM (1985) Organic geochemistry of natural waters. Martinus Nitinus Nijhoff/Dr. W. Junk Publisher, Dordrecht, p 507
Acknowledgments
We thank the National Science Foundation for supporting this research effort.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 48 kb)
Rights and permissions
About this article

Cite this article
Dasgupta, S., Siegel, D.I., Zhu, C. et al. Geochemical Mixing in Peatland Waters: The Role of Organic Acids. Wetlands 35, 567–575 (2015). https://doi.org/10.1007/s13157-015-0646-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13157-015-0646-2