
Abstract
Within the family Solanaceae, the tribe Hyoscyameae comprises eight genera distributed across Eurasia. Despite a few attempts to understand the relationships within this tribe, the affiliations among most genera remain unresolved. Recently, complete chloroplast genomes from several species of Hyoscyameae were published, enabling phylogenomic inferences. We sequenced the plastome of Scopolia carniolica, the second species of the genus to be reported. Genomic comparisons across the tribe revealed identical gene content and small differences in genome length. Phylogenetic analyses confirmed that Atropa is an early-diverging lineage sister to the rest of the tribe, resolved as two clades. One includes well-supported relationships between Przewalskia, Physochlaina, and Scopolia, and these three genera are sister to Atropanthe. This clade is sister to a second clade composed of Hyoscyamus and Anisodus. The strongly supported phylogenetic affiliations of Atropanthe, Anisodus, and Hyoscyamus represent the major advancements as previous studies were not able to resolve these relationships. Interestingly, the genus Scopolia is paraphyletic in respect to Physochlaina, based on ITS2 sequences. Finally, the evolution of the mitochondrial cox1 intron is reinterpreted. Two independent horizontal acquisitions are inferred, one in the ancestor of Przewalskia, Physochlaina, and S. japonica and another in Hyoscyamus, with no intron losses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The tribe Hyoscyameae belongs to the family Solanaceae and includes eight genera distributed exclusively in Eurasia: Archihyoscyamus, Anisodus, Atropa, Atropanthe, Hyoscyamus, Physochlaina, Przewalskia, and Scopolia (Hoare & Knapp, 1997; Lu, 1997; Olmstead et al., 1999; Yuan et al., 2006). Two diversity centers can be identified in this tribe: the Mediterranean-Turanian region, where Atropa and Archihyoscyamus grow, and the Tibetan Plateau, where Przewalskia, Anisodus, and Atropanthe can be found (Tu et al., 2010). The genus Hyoscyamus has a wider distribution, with species growing in the Mediterranean Basin or Asia. Physochlaina spp. are found in Asia, and species of Scopolia are distributed disjointly in eastern Asia and southeastern Europe (Tu et al., 2010). The main characteristic of this group is the presence of tropane alkaloids that were widely used by old civilizations and are still used today (Fatur, 2020; Hoare & Knapp, 1997; Ullrich et al., 2016; Xiao & He, 1983). Many of these substances are pharmaceutical products (Grynkiewicz & Gadzikowska, 2008). The anatomy, seed and pollen morphology, cytology, floral organogenesis, and embryology of members of the tribe Hyoscyameae have been studied (D'Arcy & Zhi-yun, 1992; Hoare & Knapp, 1997; Jang et al., 2011; Kaya et al., 2016; Lu, 1997; Tu et al., 2005; Yang, Zhang, Anming, et al., 2002a; Yang, Zhang, Lu, et al., 2002b; Zhang et al., 1994; Zhang et al., 2005). However, these features were not sufficient to identify the species belonging to this tribe and distinguish among genera and species. Molecular systematics of the tribe Hyoscyameae found strong support for the monophyly of the group, but not all relationships within the tribe were resolved with strong bootstrap support (Ghahremaninejad & Riahi, 2021; Olmstead et al., 1999; Sanchez-Puerta & Abbona, 2014; Särkinen et al., 2013; Tu et al., 2010). A study based on the gene “waxy” revealed the hybrid history of the polyploid Atropa belladonna and showed that it is sister to the rest of the tribe (Yuan et al., 2006). Recently, short reports described the complete chloroplast genomes (cpDNAs) of several members of the tribe Hyoscyameae (Jiang et al., 2019; Park & Lee, 2016; Tian et al., 2020; Tong et al., 2019; Zhang et al., 2017; Zhang & Chi, 2019) opening the possibility of a phylogenomic approach to understanding the relationships within the Hyoscyameae. A few of these studies included preliminary trees based on a subset of the taxa available, suggesting that chloroplast genome comparisons promise to resolve relationships among genera of the tribe (Jiang et al., 2019; Tian et al., 2020; Tong et al., 2019; Zhang & Chi, 2019). Up-to-date, complete chloroplast genomes of at least one species of each genus of the tribe Hyoscyameae (except for Archihyoscyamus) have been published: Atropa belladonna L. (Schmitz-Linneweber et al., 2002), Anisodus acutangulus C. Y. Wu & C. Chen (Tian et al., 2020), Anisodus tanguticus (Maxim.) Pascher (Zhang & Chi, 2019), Atropanthe sinensis (Hemsl.) Pascher (Jiang et al., 2019), Hyoscyamus niger L. (Sanchez-Puerta & Abbona, 2014), Physochlaina orientalis (Bieb.) G. Don (Gandini et al., 2019), Physochlaina physaloides (L.) G. Don (Tong et al., 2019), Przewalskia tangutica Maxim. (Zhang et al., 2017), and Scopolia parviflora (Dunn) Nakai (Park & Lee, 2016). In addition, a single nuclear marker, the internal transcribed spacer 2 (ITS2), has been reported for several species and genera of the tribe. Phylogenies based on this marker denote its usefulness to distinguish among species of Hyoscyameae (Chen et al., 2017; Hajrasouliha et al., 2014; Kim et al., 2003; Liu et al., 2017; Meiklejohn et al., 2019; Miller, 2002), but a comprehensive study of the phylogeny based on this nuclear marker is still missing.
An earlier evolutionary study of the tribe Hyoscyameae identified a highly mobile intron in the mitochondrial gene cox1 (cytochrome oxidase subunit 1) of 14 species of the tribe and two other lineages of the family Solanaceae (Sanchez-Puerta et al., 2011). The number of independent intron acquisitions through horizontal gene transfer (HGT) events could not be determined given the unresolved phylogenetic relationships within the tribe Hyoscyameae (Sanchez-Puerta & Abbona, 2014; Sanchez-Puerta et al., 2011).
In the present study, we assembled the complete chloroplast genome sequence of Scopolia carniolica Jacq., the second species of the genus to be reported, and performed genomic comparisons and phylogenetic inferences of the tribe Hyoscyameae. The goals of this study were (1) to examine the phylogenetic relationships of the Hyoscyameae based on full-length chloroplast genome alignments and nuclear ITS2 sequences, (2) to identify highly variable intergenic regions that could be used as a source for future phylogenetic analyses of species within the tribe, and (3) to re-evaluate the evolution of the cox1 intron within the Hyoscyameae.
Materials and methods
Plant material, DNA extraction, and sequencing
Plants of Scopolia carniolica from southeastern Europe were obtained from the Plantsman’s Preference (Norfolk, England, https://www.plantpref.co.uk/). Total genomic DNA was extracted from fresh leaf tissue following Dellaporta et al. (1983) and sequenced at BGI Group (formerly Beijing Genomics Institute) using the Illumina HiSeq 4000 sequencing platform. A total of ~94.9 M clean paired-end reads, 150 bp in length, were generated from a 250 bp insert library.
Genome assembly
Genomic reads were assembled de novo using Velvet v1.2.10 with a k-mer value of 131 and ABySS v4.2.1 and SOAPdenovo v2.04 with a k-mer value of 127. Contigs were considered plastidial if they had BLAST hits against a database of chloroplast genomes from Solanaceae at the NCBI GenBank repository. Selected plastid contigs from the three assemblies were combined based on overlapping sequences to eliminate redundancy using the Geneious assembler v.11.1.5 (Kearse et al., 2012) with custom parameters (minimum overlap 300 bp, minimum overlap identity 100%). The five plastid contigs, ranging from 1151 to 92,284 bp, were extended individually to reveal possible repeats at contig ends using SSAKE v3.8.5. Finally, contigs were manually joined and curated using Consed v.29.0 (Gordon et al., 1998), reaching a final assembly of a circular molecule of 156,316 bp.
The mitochondrial gene cox1 from S. carniolica was extracted from the Velvet assembly using all genomic reads and deposited in GenBank (accession number MW079308). Also, S. carniolica, H. niger, and P. orientalis nuclear internal transcribed spacer (ITS) sequences were assembled de novo with Velvet v1.2.10 assembler using a subset of the Illumina reads. The subset was generated by mapping each species read data (this study, SRX647921, and SRX5830318, respectively) over a custom ITS database using Bowtie2 local alignment with standard presets (Langmead & Salzberg, 2012). The three ITS sequences were deposited in GenBank (accession numbers MW079311, MW079309, and MW079310, respectively).
Genome annotation
The cpDNA of S. carniolica was annotated using Geneious R11 by transferring the annotations from the P. orientalis plastome (GenBank accession NC_044154.1). Annotations were manually curated (start and stop codons and exon/intron boundaries) by inspection of gene alignments. The S. carniolica cpDNA was deposited in GenBank under the accession number MT937171. The chloroplast genome map was plotted using OGDRAW.
Genome comparisons and phylogenetic analysis
The complete cpDNA of S. carniolica was compared to nine other Hyoscyameae genomes available at GenBank: Anisodus acutangulus, Anisodus tanguticus, Atropa belladonna, Atropanthe sinensis, Hyoscyamus niger, Physochlaina orientalis, Physochlaina physaloides, Przewalskia tangutica, and Scopolia parviflora. Pairwise comparisons and whole-genome identity between S. carniolica and the nine Hyoscyameae cpDNAs were performed with the mVISTA program (Frazer et al., 2004) in Shuffle-LAGAN and with a custom python script.
To accurately compare the IR junction regions between S. carniolica cpDNA and nine other Hyoscyameae species, we first re-annotated all the analyzed species as most genomes had missing annotations (Suppl. Data 1). In general, the LSC and SSC regions were not annotated, and the fragmented genes at the SSC-IRb and IRa-LSC junctions were mislabeled or missing. The newly annotated genomes were visually inspected using Geneious R11 (Kearse et al., 2012), and IR junctions were illustrated using Adobe Illustrator 2019.
To examine the evolutionary relationships among seven of the eight genera that compose the tribe Hyoscyameae, phylogenetic analyses based on the complete plastome sequences were performed. Lycium chinense, Nicotiana tabacum, and Solanum tuberosum were used as outgroups. Plastome sequences were aligned using MAFFT v. 1.3.7 (Katoh & Standley, 2013). The last inverted repeat region was removed to avoid sequence overrepresentation giving a total alignment length of 134,327 positions (Suppl. Data 2). Two short inversions were identified by inspecting the alignment and were removed from the alignment prior to the phylogenetic analyses. One inversion (trnC-psbM) had been described before (Sanchez-Puerta & Abbona, 2014). The other inversion (rps16-trnK) was examined here in detail. BLAST searches permitted inspecting the occurrence of the latter inversion in other plastomes. Mfold (Zuker, 2003) with default parameters was used to examine the secondary structure of the inversion and flanking sequences.
Maximum likelihood analyses were performed with RAxML v.8.0.0 (Stamatakis, 2014) with 1000 rapid bootstrapping replicates. Plastid and nuclear ITS sequences were analyzed independently. The best-fitting nucleotide substitution models (GTR+I+G and HKY85, respectively) were estimated by MODELTEST version 3.7 using the Akaike information criterion (Posada & Crandall, 1998). Besides, Maximum parsimony analyses were done using PAUP* (Swofford, 2002) with 1000 bootstrap replicates.
Results and discussion
General organization of Scopolia carniolica chloroplast genome
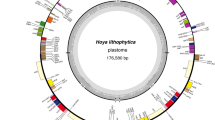
The cpDNA of S. carniolica was assembled into a single circular molecule of 156,316 bp in length. Like most land plant plastomes, it presents a quadripartite structure (Fig. 1). The long and small single-copy regions (LSC and SSC) are 86,479 bp and 18,031 bp long, separated by two inverted repeats (IRa and IRb) of 25,903 bp each. The overall GC content is 37.6% (IR, 42.9%; LSC, 35.7%; SSC, 31.9%), resembling other Hyoscyameae species (Gandini et al., 2019; Park & Lee, 2016; Sanchez-Puerta & Abbona, 2014; Zhang & Chi, 2019). The cpDNA presents 115 unique genes, including 80 protein-coding genes or conserved hypothetical chloroplast ORFs, four ribosomal RNAs, and 30 transfer RNAs (Table S1). Fourteen genes have one cis-spliced intron whereas three have two (clpP, rps12, ycf3). The rps12 gene is trans-spliced, with one exon in the LSC region and the other two within the IR. Like in other Solanaceae, the gene infA is a pseudogene, and truncated copies of the rps19 and ycf1 genes are located over the IRa-LSC and IRb-SSC junctions, respectively. Seventeen genes and the two first exons of the rps12 gene are included in the IR and, therefore, are duplicated (Table S1). Four genes present abnormal start codons: ndhD and psbL start with ACG and rps19 and ycf15 start with GTG. The non-canonical start codons ACG and GTG have been previously described in other plant species (Raubeson et al., 2007). The ACG start codons are likely converted to AUG by RNA editing (C to U), as shown for other Solanaceae species (Amiryousefi et al., 2018; Sasaki et al., 2003).

The chloroplast genome of Scopolia carniolica. Large and small single-copy regions (LSC and SSC, respectively) and inverted repeats (IR) are indicated. Genes inside and outside the circle indicate clockwise and counterclockwise transcription directions, respectively. Gene positions are color-coded according to functional categories of genes. The GC content across the cpDNA is shown in the internal circle, and the 50% GC content is depicted with a thin line. Genes with introns are in boldface. The photograph shows the individual of S. carniolica studied here (courtesy of Tim Fuller)
Genome-wide comparisons among the plastomes of the Hyoscyameae
We compared the plastome of Scopolia carniolica with nine other complete cpDNAs available at NCBI databases (Fig. 2). The Hyoscyameae chloroplast genomes are highly conserved, with identical gene content, gene order, and no variation in the start and stop codons (Suppl. Data 3). The majority of the genes show low divergence; the sequence identity is 99.5% on average for all genes across the tribe (Table S1, Fig. 2). The small plastid RNA gene (sprA) is the most divergent with identity of 95%. In addition, the ψrps19 and ψycf1 also present lower identities of 96.6% and 93.6%, respectively. This is expected for pseudogenes with no selection pressure. Chloroplast genome sizes within the tribe range from 155,570 bp in Przewalskia tangutica (Zhang et al., 2017) to 156,687 bp in Atropa belladonna (Schmitz-Linneweber et al., 2002). This length variation can be attributed to differences in non-coding regions, as all genes are almost identical in size, and to the contraction/expansion of the inverted repeats. The mean pairwise identity of S. carniolica cpDNA and the rest of the genera of the tribe is 98% (Fig. 2). Non-coding regions harbor higher levels of divergence, with a mean identity of 97.4%. The overall identity was much higher (99.5%) between the two species of Scopolia (Fig. 2).

Similarity plot comparing Scopolia carniolica chloroplast genome to other members of the tribe Hyoscyameae. The Y-axis represents the sequence identity (within the range 70–100%) from pairwise comparisons between the chloroplast genomes of Scopolia carniolica and other species of the tribe using the VISTA program. Coding and non-coding regions are in green and gray colors, respectively. The percent identities across the whole genome, including all coding and all non-coding regions of S. carniolica, and each species of the tribe are shown below species’ names. Purple and orange bars indicate previously reported variable and more conserved plastid regions, respectively. Novel variable regions described in this study are in blue
Expansion and contraction of IR regions within Hyoscyameae
Contractions and expansions at the IR regions’ borders represent the main reasons for the size variation of chloroplast genomes (Kim & Lee, 2004) and may reflect the phylogenetic history. IR lengths vary slightly within Hyoscyameae, from 25,282 bp in P. tangutica to 25,906 bp in A. acutangulus. Comparisons of the boundaries among the four chloroplast regions (LSC, IRb, SSC, IRa) between the tribe Hyoscyameae and N. tabacum show that all species, except P. tangutica, exhibit a larger IR region as reported for other Solanoideae (Amiryousefi et al., 2018) (Figure S1). The larger IR includes 59 to 75 bp of the rps19 gene creating a truncated ψrps19 copy at the IRa-LSC junction. In contrast, the IR region in P. tangutica was slightly shorter to expand the LSC region over the rpl2 gene creating instead a truncated ψrpl2 at the IRa-LSC junction. On the other end of the inverted repeat, and in agreement with most angiosperm chloroplasts, a complete copy of the ycf1 gene spans over the SSC-IRa junction duplicating the 5’end at the IRb-SSC junction. In nine of the ten Hyoscyameae analyzed species, the IR also spanned over a greater region (~450 bp) of the ycf1 gene creating a larger ψycf1 than N. tabacum and other Solanoideae. In contrast, a contraction in the P. tangutica plastome resulted in a shorter ψycf1. No obvious differences in the length of the IR have been observed between species of the same genus (Fig. 2). Nonetheless, a study of the IRa-LSC region in 13 Nicotiana species showed that the IR region suffered contractions and expansions during the evolution of the genus but found IR to be identical in four cultivars of Nicotiana tabacum (Goulding et al., 1996).
Short inversions in the plastomes of Hyoscyameae
Small inversions are common among chloroplast genomes (Kelchner & Wendel, 1996; Kim & Lee, 2005). In general, they are located downstream the 3’ends of genes, they present inverted repeat sequences at their ends, and they form stem-loop hairpin structures that are thought to stabilize the corresponding mRNA (Kim & Lee, 2005). A previously described 10-nt inversion in the trnC-psbM spacer (Sanchez-Puerta & Abbona, 2014) was observed in the two species of Anisodus and in P. tangutica. During alignment screening, we also detected a 46-nt polymorphic stretch in the Scopolia carniolica cpDNA, which was the result of an inversion in the trnK-rps16 intergenic region (Figure S2). This inversion is not present in Scopolia parviflora. By analyzing the surrounding sequence with Mfold (Zuker, 2003), we detected a stem-loop hairpin structure with identical inverted repeats of 15 bp flanking the inversion and relatively high free energy (Figure S2). This inversion is located within the plastid spacer rps16-trnK, 27 bp downstream the 3’end of the rps16 gene. A BLASTn search in the nucleotide NCBI databases revealed that this inversion is also present in other Solanaceae, such as Markea huilensis, M. lopezii, M. epifita, Trianaea neovisae, and T. speciosa from the tribe Juanulloeae (Figure S2). A study of the phylogeny of the Juanulloeae using this molecular marker recognized this homoplasious inversion and excluded it from the data set (Orejuela et al., 2017). The absence of this inversion in all other Hyoscyameae species and the variable presence in species of Markea and Trianaea indicate that this short inversion can be generated within a short time by intramolecular recombination between the inverted flanking sequences, as reported for other inversions in plant chloroplast genomes (Kim & Lee, 2005).
Phylogenetic relationships within the Hyoscyameae based on complete plastomes
We examined the phylogenetic relationships among seven of the eight genera that compose the tribe based on complete chloroplast genome alignments. Three other Solanaceae species, Lycium chinense, Nicotiana tabacum, and Solanum tuberosum were included as outgroups. The identified short inversions in the cpDNA disrupted the site-wise homology and were excluded from the alignment before phylogenetic inference. The maximum likelihood (ML) and maximum parsimony (MP) phylogenetic analyses found a highly resolved tree in which all branches showed high bootstrap support values (Fig. 3A). S. carniolica is closely related to S. parviflora and to a clade formed by the two species of Physochlaina and Przewalskia tangutica. This relationship is supported by seed characters, plastid markers, and a nuclear gene (Olmstead et al., 2008; Sanchez-Puerta & Abbona, 2014; Tu et al., 2010; Zhang et al., 2005), and it differs slightly from trees based on complete cpDNAs of fewer taxa (Tian et al., 2020; Tong et al., 2019). This clade is placed as a sister group to Atropanthe sinensis with strong support (97–100%). The second clade included Anisodus tanguticus and Anisodus acutangulus as sister to Hyoscyamus niger (Fig. 3A). The strongly supported phylogenetic affiliations of Atropanthe, Anisodus, and Hyoscyamus represent the major contributions of this work as previous studies based on less than ten plastid markers (Olmstead et al., 2008; Sanchez-Puerta & Abbona, 2014; Tu et al., 2010; Zhang et al., 2005) or a nuclear gene (Yuan et al., 2006) were not able to resolve these relationships. Finally, Atropa belladonna is sister to the rest of the tribe, as clearly demonstrated analyzing a retrotransposon inserted in the waxy gene (Yuan et al., 2006).

Maximum likelihood phylogenetic trees of the tribe Hyoscyameae. A Tree based on the complete chloroplast genomes. The last inverted repeat region was removed to avoid sequence overrepresentation. The alignment length is 134,327 nt. B Tree based on nuclear ITS2 sequences. The seven individuals of Atropa belladonna (KX674979, MF096061, MF348980, MG217536, MK895642, KX166233, AY028147) were identical and are represented with a triangle to simplify the display of the tree. Numbers above and below the branches represent support values based on 1000 bootstrap replicates of ML and MP analyses, respectively
Plastid markers at the species level in Hyoscyameae
Despite a much clearer picture of the relationships within the tribe Hyoscyameae, the number and delimitation of species within the multispecific genera of the tribe and their phylogenetic relationships remain poorly understood given very limited molecular data from species of the tribe. Here, we examined variable plastid regions as potential molecular markers for the identification and distinguishing species of Hyoscyameae. Within the tribe, the most variable regions larger than 100 bp include trnH-psbA, trnK-rps16, rps16-trnQ, trnC-psbM, trnE-trnT, ycf3-rps4, petA-psbJ, petD-rpoA, ψinfA-rps8, ndhF-rpl32, sprA-trnL, and ndhE-ndhG (Fig. 2, Table 1). No variable loci were located within the IR regions consistent with the reduced rate of synonymous substitutions that characterize these regions (Mower & Vickrey, 2018; Zhu et al., 2015). Some of the variable regions, along with other more conserved ones, have been previously used for taxonomic purposes in the family Solanaceae, particularly for the tribe Hyoscyameae (Table 1, Fig. 2). However, phylogenetic trees of the Hyoscyameae based on those markers were not fully resolved. In particular, the phylogenetic affiliations of Atropanthe, Anisodus, and Hyoscyamus were not strongly supported (Olmstead et al., 2008; Sanchez-Puerta & Abbona, 2014; Tu et al., 2010), and the monophyly of the genus Scopolia was questioned (Olmstead et al., 2008). We compared variable regions’ identity between species of the same genus to evaluate their potential value as taxonomic tools at the species level (Table 1). The number of polymorphisms between the same genus species was very low indicating that these regions are not sufficiently informative to assess phylogenetic relationships among species within the tribe Hyoscyameae. Even when the whole cpDNA is considered, a total of 201 polymorphic sites on average differentiate species of the same genus (Table 1). We conclude that only full-length chloroplast genomes may be useful for phylogenetic inference at the species level. Alternatively, a more promising approach would be developing nuclear molecular markers, which are more variable and thus may be more informative for species of the tribe Hyoscyameae.
Phylogenetic inference based on nuclear ITS sequences
Sequences from a single nuclear marker (i.e., ITS2) with broad taxon sampling for meaningful phylogenetic analyses of the tribe Hyoscyameae are available in public databases. Highly variable regions such as ITS can help to compare species and closely related genera (Baldwin et al., 1995). We performed phylogenetic analyses based on ITS2 sequences from all genera of the tribe Hyoscyameae (except for Przewalskia) along with that of the individual of S. carniolica studied here. The ITS2 sequence of the individual of S. carniolica under study is highly similar to that of other individuals of this species (99.8%) confirming its taxonomic identification. Even though the backbone of the ITS2 tree is poorly resolved in general (Fig. 3B), it recovers all genera as monophyletic, except for Scopolia. The single species of Archihyoscyamus is sister to the monophyletic genus Hyoscyamus, in agreement with previous studies that support its segregation based on its unique habitat (Lu, 1997), seed morphology (Zhang et al., 2005), and recent molecular analysis (Ghahremaninejad & Riahi, 2021).
The ITS2 sequences from all three samples of S. japonica cluster with Physochlaina with moderate bootstrap support (79%) in the ML tree, while those of S. carniolica, S. parviflora, and S. lutescens form a monophyletic group sister to the clade formed by Physochlaina spp. and S. japonica (Fig. 3B). The paraphyly of the genus Scopolia, specifically due to the unexpected position of S. japonica, had been previously noticed based on plastid markers (Olmstead et al., 2008). However, another study using plastid markers but with different sampled individuals suggested that Scopolia was monophyletic (Tu et al., 2010). In this study, we analyzed ITS2 sequences from three individuals of S. japonica generated independently by different groups (Chen et al., 2017; Kim et al., 2003; Liu et al., 2017). Unfortunately, no ITS data are available for the individuals of S. japonica analyzed by Olmstead et al. (2008) or Tu et al. (2010) to assess the discrepancies.
The number and delimitation of species within the genus Scopolia remain unclear (http://www.theplantlist.org; Jang et al., 2011, Festi, 1996). S. carniolica is a type of species that has a wide distribution across central and southeast Europe (Festi, 1996). Two species grow in Korea, S. parviflora and S. lutescens, while S. japonica is found in Japan. The individuals of S. parviflora and S. lutescens have been included within the species S. japonica due to the lack of distinct morphological differences (Festi, 1996; Jang et al., 2011). However, molecular comparisons of ITS sequences from these four species distinguished S. carniolica and S. japonica from the Korean species, which had identical ITS sequences (Kim et al., 2003). The lack of diagnostic features and identical ITS2 sequences indicates that S. parviflora and S. lutescens are synonyms (Kim et al., 2003). The sample of S. japonica studied by Tu et al. (2010) was obtained from South Korea, while the sample analyzed by Olmstead et al. (2008) comes from a Japanese collection. The genus Scopolia may be paraphyletic due to the odd phylogenetic relationships of Japanese samples of S. japonica observed with plastid (Olmstead et al., 2008) and ITS2 (Fig. 3B) sequences, while Korean samples are closely related to S. carniolica. Detailed and comparative studies based on cytoplasmic and nuclear markers of different samples of Scopolia obtained from Japan, Korea, and Europe are needed to test this hypothesis further and to determine the number of species of the genus.
Multiple acquisitions of the cox1 intron within the tribe Hyoscyameae
The cox1 intron is known as highly mobile among plant mitochondria, with more than 80 described horizontal transfers during angiosperm evolution (Cho et al., 1998; Sanchez-Puerta et al., 2008). A signature sequence generally accompanies intron acquisition, termed co-conversion tract (CCT), which replaces a short region of the second exon’s 5’ end. Within the family Solanaceae, three horizontal transfers of the cox1 intron (in Brunfelsia, Mandragora, and the tribe Hyoscyameae) were described, while 426 species of Solanaceae that belong to 70 genera lack this intron (Sanchez-Puerta et al., 2011). Brunfelsia jamaicensis obtained its cox1 intron in an independent event and from a different source than the other two intron-containing Solanaceae lineages. On the other hand, intrafamilial transfers between Mandragora spp. and the tribe Hyoscyameae explain highly similar introns in these two unrelated lineages of Solanoideae (Sanchez-Puerta et al., 2011). Within the Hyoscyameae, four genera have the cox1 intron (and the CCT): Hyoscyamus (nine species), Physochlaina (three species), Przewalskia (one species, P. tangutica), and Scopolia (one species, S. japonica; Sanchez-Puerta & Abbona, 2014 made a mistake in Fig. 3 stating that S. carniolica [instead of S. japonica] has the cox1 intron). In contrast, the other three genera of the tribe (Anisodus, Atropa, and Atropanthe) lack the intron and the CCT, indicating that they never had the cox1 intron (Sanchez-Puerta & Abbona, 2014; Sanchez-Puerta et al., 2011). The partially resolved phylogeny of the tribe based on 10 plastid markers leads Sanchez-Puerta and Abbona (2014) to propose two alternative evolutionary scenarios explaining the distribution of the cox1 intron (and the CCT) in Hyoscyameae. One hypothesis suggested a single cox1 intron acquisition in the ancestor of a clade formed by Hyoscyamus, Physochlaina, Przewalskia, and Scopolia, and the second hypothesis inferred two independent intron gains: one in Hyoscyamus and a separate one for the other three genera.
We assembled the cox1 gene of Scopolia carniolica and found that it lacks the intron and the co-conversion tract (Suppl. Data 4). This result was somewhat surprising because S. japonica presents the cox1 intron and the CCT (Sanchez-Puerta et al., 2011). Noticeably, the sample of S. japonica with the cox1 intron studied by Sanchez-Puerta et al. (2011) was closely related to Przewalskia and not to S. carniolica based on two plastid markers (Olmstead et al., 2008). Again, it is unfortunate that only fragmentary data are available and that the cox1 intron status of other species of Scopolia and in particular of those samples of Scopolia studied by Tu et al. (2010) is unknown.
According to the plastid (Olmstead et al., 2008) and ITS2 (Fig. 3B) phylogenies of the Hyoscyameae, the genus Scopolia may be paraphyletic in respect to Physochlaina and Przewalskia. Under this evolutionary scenario of the tribe Hyoscyameae, two intron acquisitions could explain the presence of the cox1 intron in distinct lineages of the tribe (Fig. 4), as suggested by Sanchez-Puerta and Abbona (2014). However, the timing of the cox1 intron gain is different here, as it is proposed to have taken place in the ancestor of Physochlaina, Przewalskia, and S. japonica, while all other species of Scopolia may not present the intron nor the CCT (Fig. 4). Overall, two and four horizontal transfer events of the cox1 intron (and no intron losses) took place during the evolution of the tribe Hyoscyameae and the family Solanaceae, respectively. Three of them were intrafamilial HGT events and include intron transfers between Mandragora, Hyoscyamus, and the ancestor of Physochlaina, Przewalskia, and S. japonica. This agrees with the observation that intrafamilial transfers are frequent in other angiosperm lineages (Sanchez-Puerta et al., 2008).

An evolutionary scenario for the cox1 intron acquisitions in the tribe Hyoscyameae. Purple dots indicate mitochondrial cox1 intron acquisitions by horizontal gene transfer. The paraphyly of the genus Scopolia is supported by plastid and nuclear markers (see text and Fig. 3)
References
Amiryousefi, A., Hyvönen, J., & Poczai, P. (2018). The chloroplast genome sequence of bittersweet (Solanum dulcamara): Plastid genome structure evolution in Solanaceae. PLoS One, 13(4), e0196069. https://doi.org/10.1371/journal.pone.0196069.
Baldwin, B. G., Sanderson, M. J., Porter, J. M., Wojciechowski, M. F., Campbell, C. S., & Donoghue, M. J. (1995). The ITS region of nuclear ribosomal DNA: A valuable source of evidence on angiosperm phylogeny. Annals of the Missouri Botanical Garden, 82, 247–277.
Chen, X., Xiang, L., Shi, L., Li, G., Yao, H., Han, J., Lin, Y., Song, J., & Chen, S. (2017). Identification of crude drugs in the Japanese pharmacopoeia using a DNA barcoding system. Scientific Reports, 7, 1–7. https://doi.org/10.1038/srep42325.
Cho, Y., Qiu, Y. L., Kuhlman, P., & Palmer, J. D. (1998). Explosive invasion of plant mitochondria by a group I intron. Proceedings of the National Academy of Sciences of the United States of America, 95(24), 14244–14249. https://doi.org/10.1073/pnas.95.24.14244.
D'Arcy, W. G., & Zhi-yun, Z. (1992). Notes on the Solanaceae of China and neighboring areas. Novon, 2, 124–128.
Dellaporta, S. L., Wood, J., & Hicks, J. B. (1983). A rapid method for DNA extraction from plant tissue. Plant Molecular Biology, 1, 19–21.
Fatur, K. (2020). “Hexing herbs” in ethnobotanical perspective: A historical review of the uses of anticholinergic Solanaceae plants in Europe. Economic Botany, 74(2), 140–158. https://doi.org/10.1007/s12231-020-09498-w.
Festi, F. (1996). Scopolia carniolica Jacq. Eleusis, 5(34), 34–45.
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., & Dubchak, I. (2004). VISTA: Computational tools for comparative genomics. Nucleic Acids Research, 32(Web Server), W273–W279. https://doi.org/10.1093/nar/gkh458.
Gandini, C. L., García, L. E., Abbona, C. C., & Sanchez-Puerta, M. V. (2019). The complete organelle genomes of Physochlaina orientalis: Insights into short sequence repeats across seed plant mitochondrial genomes. Molecular Phylogenetics and Evolution, 137, 274–284. https://doi.org/10.1016/j.ympev.2019.05.012.
Ghahremaninejad, F., & Riahi, M. (2021). The systematic position of Archihyoscyamus A.M.Lu (Solanaceae Juss.): An analysis based on nuclear and plastid sequences. Botany Letters, 168(1), 96–101. https://doi.org/10.1080/23818107.2020.1824806.
Gordon, D., Abajian, C., & Green, P. (1998). Consed: A graphical tool for sequence finishing. Genome Research, 8(3), 195–202. https://doi.org/10.1101/gr.8.3.195.
Goulding, S. E., Wolfe, K. H., Olmstead, R. G., & Morden, C. W. (1996). Ebb and flow of the chloroplast inverted repeat. Molecular and General Genetics, 252(1-2), 195–206. https://doi.org/10.1007/BF02173220.
Grynkiewicz, G., & Gadzikowska, M. (2008). Tropane alkaloids as medicinally useful natural products and their synthetic derivatives as new drugs. Pharmacological Reports, 60(4), 439–463. https://doi.org/10.3390/molecules24040796.
Hajrasouliha, S., Massoumi, A. A., TaherNejadSattar, S. M., Hamdi, M., & Mehregan, I. (2014). A phylogenetic analysis of Hyoscyamus L. (Solanaceae) species from Iran based on ITS and trnL-F sequence data. Jbes, 5(1), 647–654.
Hoare, A. L., & Knapp, S. (1997). A phylogenetic conspectus of the tribe Hyoscyameae (Solanaceae). Bulletin of the Natural History Museum, Botany Series, 27, 11–29.
Jang, C. G., Park, M. S., Nam, B. M., Pak, H. H., Eom, J. A., & Chung, G. Y. (2011). Distribution and morphological characteristics of Scopolia japonica Maxim in Korea, Korean Journal of Plant Taxonomy., 41(3), 271–279. https://doi.org/10.11110/kjpt.2011.41.3.271.
Jiang, Z., Yusupov, Z., Huang, X., Makhmudjanov, D., Tojibaev, K., & Deng, T. (2019). Characterization of the complete plastome of Atropanthe sinensis (Solanaceae). Mitochondrial DNA Part B, 4(2), 2932–2933. https://doi.org/10.1080/23802359.2019.1662742.
Katoh, K., & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772–780. https://doi.org/10.1093/molbev/mst010.
Kaya, A., Satıl, F., & Aslan, M. K. (2016). Seed morphology of the genus Hyoscyamus L. in Turkey and its systematic significance. Acta Microscópica, 25(2), 48–55.
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., Buxton, S., Cooper, A., Markowitz, S., Duran, C., & Thierer, T. (2012). Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics, 28(12), 1647–1649. https://doi.org/10.1093/bioinformatics/bts199.
Kelchner, S. A., & Wendel, J. F. (1996). Hairpins create minute inversions in non-coding regions of chloroplast DNA. Current Genetics, 30(3), 259–262. https://doi.org/10.1007/s002940050130.
Kim, K. J., & Lee, H. L. (2004). Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Research, 11(4), 247–261. https://doi.org/10.1093/dnares/11.4.247.
Kim, K. J., & Lee, H. L. (2005). Widespread occurrence of small inversions in the chloroplast genomes of land plants. Molecules and Cells, 19(1), 104–113.
Kim, Y. D., Paik, J. H., Kim, S. H., & Hong, S. P. (2003). Phylogeny of Scopolia Jacq. s. str. based on ITS sequences. Korean Journal of Plant Taxonomy, 33(4), 373–386. https://doi.org/10.11110/kjpt.2003.33.4.373.
Langmead, B., & Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods, 9(4), 357–359. https://doi.org/10.1038/nmeth.1923.
Liu, J., Shi, L., Song, J., Sun, W., Han, J., Liu, X., Hou, D., Yao, H., Li, M., & Chen, S. (2017). BOKP: A DNA barcode reference library for monitoring herbal drugs in the Korean Pharmacopoeia. Frontiers in Pharmacology, 8, 3–9. https://doi.org/10.3389/fphar.2017.00931.
Lu, A. M. (1997). Archihyoscyamus: A new genus of Solanaceae from Western Asia. Adansonia, 19, 135–138.
Meiklejohn, K. A., Damaso, N., & Robertson, J. M. (2019). Assessment of BOLD and GenBank – Their accuracy and reliability for the identification of biological materials. PLoS One, 14(6), e0217084–e0217014. https://doi.org/10.1371/journal.pone.0217084.
Miller, J. S. (2002). Phylogenetic relationships and the evolution of gender dimorphism in Lycium (Solanaceae). Systematic Botany, 27(2), 416–428.
Mower, J. P., & Vickrey, T. L. (2018). Structural diversity among plastid genomes of land plants. In Advances in Botanical Research, 85, 263–292. https://doi.org/10.1016/bs.abr.2017.11.013.
Olmstead, R. G., Sweere, J. A., Spangler, R. E., Bohs, L., & Palmer, J. D. (1999). Phylogeny and provisional classification of the Solanaceae based on chloroplast DNA. Solanaceae, IV, 111–137.
Olmstead, R. G., Bohs, L., Migid, H. A., Santiago-Valentin, E., Garcia, V. F., & Collier, S. M. (2008). A molecular phylogeny of the Solanaceae. Taxon, 57(4), 1159–1181.
Orejuela, A., Wahlert, G., Orozco, C. I., Barboza, G., & Bohs, L. (2017). Phylogeny of the tribes Juanulloeae and Solandreae (Solanaceae). Taxon, 66(2), 379–392. https://doi.org/10.12705/662.6.
Park, J. H., & Lee, J. (2016). The complete plastid genome of Scopolia parviflora (Dunn.) Nakai (Solanaceae). Korean Journal of Plant Taxonomy, 46(1), 60–64. https://doi.org/10.11110/kjpt.2016.46.1.60.
Posada, D., & Crandall, K. A. (1998). MODELTEST: Testing the model of DNA substitution. Bioinformatics, 14(9), 817–818. https://doi.org/10.1093/bioinformatics/14.9.817.
Raubeson, L. A., Peery, R., Chumley, T. W., Dziubek, C., Fourcade, H. M., Boore, J. L., & Jansen, R. K. (2007). Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics, 8(1), 174–127. https://doi.org/10.1186/1471-2164-8-174.
Sanchez-Puerta, M. V., & Abbona, C. C. (2014). The chloroplast genome of Hyoscyamus niger and a phylogenetic study of the tribe Hyoscyameae (Solanaceae). PLoS One, 9(5), e98353. https://doi.org/10.1371/journal.pone.0098353.
Sanchez-Puerta, M. V., Cho, Y., Mower, J. P., Alverson, A. J., & Palmer, J. D. (2008). Frequent, phylogenetically local horizontal transfer of the cox1 group I Intron in flowering plant mitochondria. Molecular Biology and Evolution, 25(8), 1762–1777. https://doi.org/10.1093/molbev/msn129.
Sanchez-Puerta, M. V., Abbona, C. C., Zhuo, S., Tepe, E. J., Bohs, L., Olmstead, R. G., & Palmer, J. D. (2011). Multiple recent horizontal transfers of the cox1 intron in Solanaceae and extended co-conversion of flanking exons. BMC Evolutionary Biology, 11(1), 277. https://doi.org/10.1186/1471-2148-11-277.
Särkinen, T., Bohs, L., Olmstead, R. G., & Knapp, S. (2013). A phylogenetic framework for evolutionary study of the nightshades (Solanaceae): A dated 1000-tip tree. BMC Evolutionary Biology, 13(1), 214. https://doi.org/10.1186/1471-2148-13-214.
Sasaki, T., Yukawa, Y., Miyamoto, T., Obokata, J., & Sugiura, M. (2003). Identification of RNA editing sites in chloroplast transcripts from the maternal and paternal progenitors of tobacco (Nicotiana tabacum): Comparative analysis shows the involvement of distinct trans-factors for ndhB editing. Molecular Biology and Evolution, 20(7), 1028–1035. https://doi.org/10.1093/molbev/msg098.
Schmitz-Linneweber, C., Regel, R., Du, T. G., Hupfer, H., Herrmann, R. G., & Maier, R. M. (2002). The plastid chromosome of Atropa belladonna and its comparison with that of Nicotiana tabacum: The role of RNA editing in generating divergence in the process of plant speciation. Molecular Biology and Evolution, 19(9), 1602–1612. https://doi.org/10.1093/oxfordjournals.molbev.a004222.
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30(9), 1312–1313. https://doi.org/10.1093/bioinformatics/btu033.
Swofford, D. (2002). PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4.0 (Vol. 4). Sinauer Associates.
Tian, X., Bai, J. Q., Yang, C. W., Zhang, Y.-M., & Li, G. D. (2020). Characterization of the complete chloroplast genome sequence of Anisodus acutangulus (Solanaceae). Mitochondrial DNA Part B, 5(1), 893–894. https://doi.org/10.1080/23802359.2020.1717387.
Tong, L., Zhu, Y. X., Lei, F. W., Shen, X. L., & Mu, X. Y. (2019). The complete chloroplast genome of Physochlaina physaloides (Solanaceae), an important medicinal plant. Mitochondrial DNA Part B, 4(2), 3427–3428. https://doi.org/10.1080/23802359.2019.1674730.
Tu, T. Y., Sun, H., Gu, Z. J., & Yue, J. P. (2005). Cytological studies on the Sino-Himalayan endemic Anisodus and four related genera from the tribe Hyoscyameae (Solanaceae) and their systematic and evolutionary implications. Botanical Journal of the Linnean Society, 147(4), 457–468.
Tu, T., Volis, S., Dillon, M. O., Sun, H., & Wen, J. (2010). Dispersals of Hyoscyameae and Mandragoreae (Solanaceae) from the New World to Eurasia in the early Miocene and their biogeographic diversification within Eurasia. Molecular Phylogenetics and Evolution, 57(3), 1226–1237. https://doi.org/10.1016/j.ympev.2010.09.007.
Ullrich, S. F., Hagels, H., & Kayser, O. (2016). Scopolamine: A journey from the field to clinics. Phytochemistry Reviews, 16(2), 333–353. https://doi.org/10.1007/s11101-016-9477-x.
Xiao, P. G., & He, L. Y. (1983). Ethnopharmacologic investigation on tropane-containing drugs in Chinese solanaceous plants. Journal of Ethnopharmacology, 8(1), 1–18. https://doi.org/10.1016/0378-8741(83)90086-7.
Yang, D. Z., Zhang, Z. Y., Anming, L., Kun, S., & Liu, J. Q. (2002a). Floral organogenesis and development of two taxa in tribe Hyoscyameae (Solanaceae) -- Przewalskia tangutica and Hyoscyamus niger. Journal of Integrative Plant Biology, 44(8), 889–894.
Yang, D. Z., Zhang, Z. Y., Lu, A. M., Sun, K., & Liu, J. Q. (2002b). Floral organogenesis and development of two taxa of the Solanaceae -- Anisodus tanguticus and Atropa belladonna. Israel Journal of Plant Sciences, 50(2), 127–134. https://doi.org/10.1560/9J1P-6GCQ-M375-51P4.
Yuan, Y. W., Zhang, Z. Y., Chen, Z. D., & Olmstead, R. G. (2006). Tracking ancient polyploids: A retroposon insertion reveals an extinct diploid ancestor in the polyploid origin of Belladonna. Molecular Biology and Evolution, 23(12), 2263–2267. https://doi.org/10.1093/molbev/msl099.
Zhang, G., & Chi, X. (2019). The complete chloroplast genome of Anisodus tanguticus, a threatened plant endemic to the Qinghai-Tibetan Plateau. Mitochondrial DNA Part B, 4(1), 1191–1192. https://doi.org/10.1080/23802359.2019.1591209.
Zhang, Z. Y., Lu, A. M., & D'Arcy, W. G. (1994). Solanaceae. Flora of China, 17, 300–332.
Zhang, Z. Y., Yang, D. Z., Lu, A. M., & Knapp, S. (2005). Seed morphology of the tribe Hyoscyameae (Solanaceae). Taxon, 54(1), 71–83. https://doi.org/10.2307/25065303.
Zhang, D., Fan, L., Lei, Z., Wang, T., Guo, X., & Hu, Q. (2017). Characterization of the complete chloroplast genome of the endangered Przewalskia tangutica Maxim. Conservation Genetics Resources, 9(3), 409–413. https://doi.org/10.1007/s12686-017-0696-2.
Zhu, A., Guo, W., Gupta, S., Fan, W., & Mower, J. P. (2015). Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytologist, 209(4), 1747–1756. https://doi.org/10.1111/nph.13743.
Zuker, M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Research, 31(13), 3406–3415. https://doi.org/10.1093/nar/gkg595.
Acknowledgements
We thank Tim Fuller for sharing his photograph of Scopolia carniolica. This work used the Toko Cluster from FCEN-UNCuyo, which forms part of the SNCAD-MinCyT, Argentina.
Availability of data and material
The assembled plastome and gene sequences were deposited in GenBank.
Code availability
Not applicable.
Funding
This work was supported by Agencia Nacional de Promoción Científica y Tecnológica (PICT-2017-0691) and Universidad Nacional de Cuyo (06/A724) through funding to M.V.S-P.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by MZ, CLG, and VNI. The manuscript was written by CLG, VNI, and MVSP and edited by MZ. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
All authors have read and approved this manuscript.
Consent for publication
All authors have read and approved this manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
C. L. Gandini and V. N. Ibañez are co-first authors.
Rights and permissions
About this article

Cite this article
Gandini, C.L., Ibañez, V.N., Zubko, M.K. et al. Complete plastome phylogeny and an update on cox1 intron evolution of Hyoscyameae (Solanaceae). Org Divers Evol 21, 521–532 (2021). https://doi.org/10.1007/s13127-021-00501-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13127-021-00501-3