
Abstract
Tachykinins (TKs) include an evolutionarily conserved group of small bio-active peptides which possess a common carboxyl-terminal sequence, Phe-X-Gly-Leu-Met-NH2. TKs also have been shown to have implications in different steps of carcinogenesis, such as angiogenesis, mitogenesis, metastasis, and other growth-related events. The biological actions of substance P (SP), as the most important member of the TK family, are mainly mediated through a G protein-coupled receptor named neurokinin-1 receptor (NK1R). More recently, it has become clear that SP/NK1R system is involved in the initiation and activation of signaling pathways involved in cancer development and progression. Therefore, SP may contribute to triggering a variety of effector mechanisms including protein synthesis and a number of transcription factors that modulate the expression of genes involved in these processes. The overwhelming insights into the blockage of NK1R using specific antagonists could suggest a therapeutic approach in cancer therapy. In this review, we focus on evidence supporting an association between the signaling pathways of the SP/NK1R system and cancer cell proliferation and development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer is a chronic disease that is caused by faulty genome surveillance and signal transduction mechanisms [17, 33]. Recently, researchers have shown that mutations in genes that encode different components of various signaling pathways occur at high frequency in cancers [27, 56]. Therefore, understanding the underlying mechanisms involved in cancer progression may contribute to develop novel diagnostic and therapeutic approaches for the detection and treatment of cancers.
Tachykinins (TKs) include an evolutionarily conserved family of small bio-active peptides including substance P (SP), neurokinin A (NKA), and B (NKB) [46]. The mammalian TKs possess a common carboxyl-terminal sequence, represented by Phe-X-Gly-Leu-Met-NH2, where X is either a branched aliphatic or aromatic amino acid [53]. These peptides are widely distributed in mammalian peripheral and central nervous systems and participate in the regulation of physiological functions like inflammatory processes, hematopoiesis, wound healing, leukocyte trafficking, microvasculature permeability, and cell survival [13, 31, 38, 44, 68]. TKs also have been shown to have implications in different steps of carcinogenesis, such as angiogenesis, mitogenesis, metastasis, and other growth-related events [8, 45].
SP, as the most important member of the mammalian TK peptides, is derived from a single preprotachykinin A gene. The biological functions of SP are mainly mediated through a G protein-coupled receptor (GPCR) named neurokinin-1 receptor (NK1R) [36, 57]. It has been identified that both SP and NK1R are overexpressed in some cancer cells including breast, ovarian, pancreas, thyroid, prostate, glioblastoma, and astrocytoma [4, 7, 9, 22, 39, 44]. Additionally, the binding of SP to NK1R mediates a major tumor progression pathway through initiation and activation of phosphoinositide 3-kinase (PI3K), mitogen-activated protein kinases (MAPKs), and other signaling pathways. Therefore, SP may contribute to triggering a variety of effector mechanisms including protein synthesis and progression of the eukaryotic cell cycle and a number of transcription factors that modulate the expression of genes involved in these processes [34, 37].
In this review, the signaling functions of SP/NK1R and its effect on the pathogenesis of cancer progression are summarized. Understanding this can lead to a more effective therapeutic approach or improve diagnostic procedures that are used for cancer therapy/diagnosis.
Tachykinins and canonical Wnt signaling transduction pathway
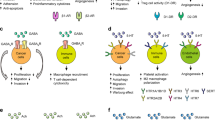
The canonical Wnt signaling cascade is an important transduction pathway that regulates cell survival, proliferation, progression, and metastasis through the activation of β-catenin [47, 49]. There are also several reports that Wnt ligands (such as Wnt 1, Wnt 2, and Wnt 3a) are a large family of secreted glycoproteins that are cysteine-rich and highly hydrophobic. Wnts are produced as precursor proteins that contain a short N-terminal signal sequence and a mature segment that varies in length from approximately 320 to 400 amino acids [19, 58, 64]. Accordingly, in the absence of Wnt ligands, β-catenin is targeted for degradation through the interaction with Axin, adenomatous polyposis coli (APC), and the protein kinase GSK-3. On the other hand, when cells are stimulated by Wnts, the degradation complex is dissociated and β-catenin translocates to the nucleus. It results in β-catenin accumulation and association with members of the lymphoid enhancer-binding factor 1/T cell-specific transcription factor (LEF/TCF) protein family, leading to increased expression of several genes involved in cell proliferation including cyclin D1 and c-Myc (summarized in Fig. 1) [26, 29].

The SP-NK1R signaling pathways. The SP-occupied neurokinin-1 receptor is a substrate for phosphorylation by G protein-coupled receptor kinase (GRK). Phosphorylation enhances the interaction of NK1R with β-arrestin, thereby translocating to the plasma membrane and mediating G protein uncoupling and receptor desensitization. SP bind to NK1R, followed by activation of Ras/Raf/MAPK and/or PI3K/Akt/mTOR. Raf, MAPK, Akt, and mTOR are classified as serine/threonine-specific protein kinases, which leads to stimulation of downstream proteins involved in the initiation of protein synthesis, resulting in cellular growth, invasion, and metastasis. In addition, administration of NK1R antagonist mediated suppression of the P38 MAPK signaling pathway, which results in NK-κB downregulation and consequent reduction of proinflammatory cytokines synthesis. On the other hand, Dickkopf1(DKK1) is an inhibitor of the canonical Wnt signaling pathway, which acts to interdict the interaction of LRP5/6 and Frizzled with Wnt ligands. Administration of NK1R antagonist is able to downregulate Wnt pathway-associated proteins through upregulation of Dkk1
Several studies have been published to report a cross talk between administration of NK1R antagonist and the suppression of the Wnt signaling pathway (summarized in Fig. 1) [21, 67]. For instance, Garnier et al. reported that the administration of aprepitant (< 40 μM), as a highly selective human NK1R antagonist, downregulates Wnt pathway-associated proteins including cyclin D1, c-Myc, and LEF-1, which thereby leads to G2 cell cycle arrest and apoptosis in colon cancer cell lines [14]. Similarly, Niu et al. indicated that the administration of another NK1R antagonist called NKP608 (10 μM for 24 h) has the potential to reduce colorectal cancer cell invasion and migration through blocking the ability of Wnt3a to stimulate cyclin D1, β-catenin, and vascular endothelial growth factor (VEGF) signaling activity [40]. Consistently, another in vitro study showed that aprepitant (40 μM) exerts an anti-tumorigenic effect via impaired interaction of Forkhead Box M1 protein with β-catenin that results in inhibiting canonical Wnt signaling pathway in human hepatoblastoma cell lines [21]. Taken together, these results conclusively suggest that pharmacological inhibition of NK1R is detected as a promising therapeutic target in reducing tumor growth in many types of cancer.
Tachykinins and MAPK signaling pathway
Mitogen-activated protein kinase (MAPK) cascades are ubiquitous and well-defined signal transduction modules found in eukaryotic cells which play crucial roles in regulating various essential cellular processes including migration, proliferation, differentiation, and programmed cell death. Activation of MAPKs occurs through dual phosphorylation of both regulatory threonine and tyrosine residues, in response to a wide variety of extracellular signals including growth, hormones, neurotrophic factors, and cytokines.
Phosphorylated forms of ERK and p38 are two major components of the MAPK family, which have been found to participate in tumor invasiveness and progression (summarized in Fig. 1) [10, 48, 51]. In support of a critical role of NK1R in tumor differentiation, the data presented by Yamaguchi et al. indicated that NK1R is required for the induction of c-Myc protein expression and cell growth through the activation of the ERK1/2 pathway in human astrocytoma cells [61]. In line with this, another in vitro study revealed that administration of rottlerin (20 μM) and PP2 (20 μM), a protein kinase C delta inhibitor and sarcoma kinase (Src) inhibitor respectively, could attenuate the SP-dependent ERK1/2 phosphorylation and thereby inhibiting the growth of human glioblastoma cells. These results clearly suggest that Src and PKC delta are candidate molecules that serve as signal transducers between SP stimuli and ERK1/2 activation in human glioblastoma cells [62].
Beta-arrestin1 (ARRB1) is a scaffold protein that contributes to GPCR desensitization and has also been associated with NK1R-mediated SP actions in tumor cell proliferation (summarized in Fig. 1) [16, 42, 66]. For example, Zhang et al. found that ARRB1 knockdown could induce apoptosis and G2/M cell cycle arrest via suppressing the ERK1/2 and Akt–NK1R interaction in glioblastoma cells [66].
Matrix metalloproteinases (MMPs) are a major group of degradative enzymes that act as effectors of extracellular matrix invasion and metastasis in cancer cells. In line with this, Li et al. showed that SP (< 1000 nM for 60 min) induces MMP-2 and MMP-14 expression through enhanced activation of ERK1/2, JNK, and Akt signaling pathways, thereby leading to breast cancer cell proliferation and invasion [25]. Moreover, another study indicated that SP significantly enhanced the MMP-9 expression in endometrial adenocarcinoma, which in turn promotes tumor invasion, angiogenesis, and metastasis. Taken together, these data also suggest that inactivation of NK1R by L733.060, an NK1R antagonist, could reverse the SP-induced effects and may serve as an ideal target for novel therapeutic strategies designed to enhance the treatment of endometrial adenocarcinoma [28]. Human hemokinin-1 (hHK-1) is a member of the mammalian tachykinin family, which is overexpressed in tumor cells and has the greatest affinity for NK1R [59]. In line with this, an in vitro study indicated that hHK1 (< 1000 nM) could increase the MMP2 expression and then promote the melanoma cell migration through overactivation of the MAPK pathway [28]. These findings support the idea that NK1R is critical for cell proliferation and development, which may be developed a promising therapeutic strategy to improve clinical outcomes in the treatment of cancers.
The p38 MAPK is a signal transduction component that mediates various inflammatory cellular responses in human tumors (summarized in Fig. 1) [52]. Consistently, Fiebich et al. presented evidence showing that SP leads to induced accumulation of IL-6 in the human astrocytoma cell line by upregulating p38 MAPK signaling independent of NF-κB activation [12]. To further support the hypothesis that SP enhanced activation of key proinflammatory signal transduction pathways, Yamaguchi et al. showed that SP (100 nM) may also participate in IL-6 and IL-8 expression through p38 MAPK-mediated NF-κB activation in the human astrocytoma cell line. Moreover, the administration of ketamine (< 1 mM), as one of the NK1R antagonists, could reduce the SP-enhanced proinflammatory cytokines synthesis through inactivation of signaling effectors such as ERK, P38MAPK, and NF-κB involved in astrocytoma cell proliferation and metastasis [63]. These data clearly support the role of SP/NK1R system in regulating inflammatory processes and provide further evidence that the targeting NK1R by specific inhibitors is potential to serve as a leading agent for the development of therapeutic strategies against various malignancies.
Tachykinins and PI3K/AKT signaling pathway
The phosphoinositide 3-kinase (PI3K)/Akt signaling axis plays crucial roles in mediating both cell proliferation and development [30]. Recently, researchers have shown that the balance between PI3K/Akt and p53 pathways determines the cancer cells sensitivity to apoptosis [1, 5]. Furthermore, several studies provide new evidence of positive cross talk between the NK1R overexpression and PI3K/Akt-mediated cell proliferation and apoptosis resistance (summarized in Fig. 1) [2, 3].
For example, treating pre-B ALL cells with aprepitant at 20 μM for 1 day effectively reduces the phospho/total Akt ratio. These findings suggest that the inhibitory effect of aprepitant on Akt phosphorylation leads to an increased expression of P21 and P27 cell cycle regulatory proteins, which in turn induces G1/S cell cycle arrest and programmed cell death [3]. Consistent with the pro-apoptotic effect of NK1 receptor pharmacological inhibitors on cancer cells, Akazawa et al. demonstrated that 10 μM L-733,060, as a specific NK1R antagonist, could dissociate Akt-NK1R interaction, and results in dephosphorylation and inactivation of Akt. It has been shown that L-733,060 induces apoptotic cell death in glioblastomas [2]. These results clearly support the hypothesis that the blockage of NK1R with specific antagonists could counteract the SP-induced cell growth and may serve as a promising therapeutic target for cancer prevention.
Tachykinins and mTOR signaling pathway
The mammalian target of rapamycin (mTOR) signaling pathway is considered to be one of the most important molecular mechanisms of activating transducer proliferation signals in tumor cells [23, 24]. The mTOR kinase exists in two functionally separate multiprotein complexes, mTORC1 and mTORC2. The mTORC1 mediates the activation of p70 S6 kinase (p70S6k) and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), whereas relatively little is known concerning the biological roles and functions of mTORC2. Although the signaling mechanisms that connect mTORC2 activity to malignancy are currently unclear, it has been shown that mTORC2 overactivation is associated with cell proliferation, survival, and differentiation [18, 41].
Rapamycin is a potent cytostatic agent which selectively inhibits the mTOR function and thereby blocks cell cycle progression at the G1/S phase [6]. Several lines of studies have shown that mTOR signaling axis is also activated by SP/NK1R in cancer cells (summarized in Fig. 1). Additionally, the blockage of mTOR activity by rapamycin could attenuate the phosphorylation of p70S6k and 4E-BP1 proteins, as well as the SP-induced cell proliferation and survival [21, 54]. For example, Sharif et al. indicated that SP (100 nM) enhances cell growth and metastasis by induced phosphorylation and activation of p70s6K and 4E-BP1 in human astrocytoma cell lines. These results also suggest that the use of rapamycin at low concentration (< 1 nM) is able to induce a gain-of-function complex with FK506 binding protein 12. This interaction significantly suppresses the SP-mediated phosphorylation of mTOR axis proteins and could be used as an effective therapeutic agent for the management of human malignancies [55].
Consistently, Ilmer et al. demonstrated that the administration of aprepitant (< 40 μM) attenuates mTORC1 activation and thereby reduces the phosphorylation of its downstream effectors including 4E-BP1/2, p70S6K, and S6, leading to a decrease in transcript synthesis of ribosomal proteins which promotes protein synthesis levels during hepatoblastoma cell proliferation [21]. It is becoming clear that the inhibition of NK1R with specific antagonists may contribute to the development of future anticancer strategies in cancer therapy.
Antitumor properties of NK1R antagonists against cancers
Major advances have been made towards identifying the specific agonists and antagonists for NK1R in order to provide the ideal tool for understanding the interactions in numerous biological and pathobiological processes. It has been reported that NK1R is capable of modifying the emotional behavior, stress, emesis, anxiety, depression, migraine, and cellular mechanisms of neurodegeneration in the central nervous system. In the peripheral nervous system, NK1R overexpression could lead to several inflammatory diseases such as asthma, chronic obstructive pulmonary disease (COPD), inflammatory bowel disease (IBD), and rheumatoid arthritis (RA) [43].
Recently, several studies demonstrated that NK1R antagonists elicit an antitumor response in various human tumor cell lines, including acute myeloid leukemia, endometrial cancer, glioma, and neuroblastoma [15, 20, 32, 65]. It should be noted that such NK1R antagonists could reduce the basal level of Akt phosphorylation, which is also associated with enhanced caspase-3-dependent apoptotic cell death in human tumors [3]. In addition, tumor cell invasion is critical as it plays a major role and the first step in cancer metastasis which is the feature of malignant tumors resulting in dissemination of primary tumor cells to distant organs [11, 60]. In line with this, it has also recently been suggested that the same NK1R antagonists are able to suppress the SP-induced metastatic progression, cell migration, and invasion in various cancer cells. It seems this effect is mediated through the dissociation of the β-arrestin-containing complex and thereby the inactivation of the MAPK pathway [66]. Besides, acting as a highly potent antitumor agent, NK1R antagonists have an effective antiangiogenic action by suppressing hypoxia-inducible factor (HIF-1α) and VEGF expression as well as endothelial cell growth [35, 50]. Therefore, taken together, these findings clearly suggest that NK1R antagonists completely inhibit the SP-mediated proliferation and metastasis of tumor cells, which in turn could be considered as a novel therapeutic target for cancer treatment.
Conclusions and future perspectives
The findings described in this review article strongly support the hypothesis that a network formed by SP and NK1 receptor can exert cancer-enhancing effects in cellular and animal models. In fact, the SP/NK1R may act as an integrated system to regulate critical molecular mechanisms involved in tumor growth, differentiation, migration, and apoptosis resistance.
Pharmacological inhibition of NK1R might, therefore, be considered as a novel therapeutic procedure against malignancies. However, the carcinogenic activity of SP/NK1R signaling pathways have been intensively studied, and the underlying mechanisms of these functions are poorly understood. A deeper comprehension of NK1R signaling processes could lead to understanding or the addition of novel agents to regulate pathological responses and therefore improved management of cancers. More in vivo studies are required to illuminate the role of SP/NK1R system in cancer and their potential role in improving the chemotherapeutic effects of NK1R antagonist.
References
Abraham AG, O’Neill E (2014) PI3K/Akt-mediated regulation of p53 in cancer. Portland Press Limited, London
Akazawa T, Kwatra SG, Goldsmith LE, Richardson MD, Cox EA, Sampson JH, Kwatra MM (2009) A constitutively active form of neurokinin 1 receptor and neurokinin 1 receptor-mediated apoptosis in glioblastomas. J Neurochem 109:1079–1086
Bayati S, Bashash D, Ahmadian S, Safaroghli-Azar A, Alimoghaddam K, Ghavamzadeh A, Ghaffari SH (2016) Inhibition of tachykinin NK 1 receptor using aprepitant induces apoptotic cell death and G1 arrest through Akt/p53 axis in pre-B acute lymphoblastic leukemia cells. Eur J Pharmacol 791:274–283
Bigioni M, Benzo A, Irrissuto C, Maggi CA, Goso C (2005) Role of NK-1 and NK-2 tachykinin receptor antagonism on the growth of human breast carcinoma cell line MDA-MB-231. Anti-Cancer Drugs 16:1083–1089
Catasus L, Gallardo A, Cuatrecasas M, Prat J (2009) Concomitant PI3K–AKT and p53 alterations in endometrial carcinomas are associated with poor prognosis. Mod Pathol 22:522–529
Chan S (2004) Targeting the mammalian target of rapamycin (mTOR): a new approach to treating cancer. Br J Cancer 91:1420–1424
Cremins JD, Michel J, Farah JM, Krause JE (1992) Characterization of substance P-like immunoreactivity and tachykinin-encoding mRNAs in rat medullary thyroid carcinoma cell lines. J Neurochem 58:817–825
Davoodian M, Boroumand N, Mehrabi Bahar M, Jafarian AH, Asadi M, Hashemy SI (2019) Evaluation of serum level of substance P and tissue distribution of NK-1 receptor in breast cancer. Mol Biol Rep 46:1285–1293. https://doi.org/10.1007/s11033-019-04599-9
Debeljuk L (2006) Tachykinins and ovarian function in mammals. Peptides 27:736–742
Farooq A, Zhou M-M (2004) Structure and regulation of MAPK phosphatases. Cell Signal 16:769–779
Fidler IJ (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3:453–458
Fiebich BL, Schleicher S, Butcher RD, Craig A, Lieb K (2000) The neuropeptide substance P activates p38 mitogen-activated protein kinase resulting in IL-6 expression independently from NF-kappa B. J Immunol (Baltimore, MD : 1950) 165:5606–5611
Fukuhara S, Shimizu M, Matsushima H, Mukai H, Munekata E (1998) Signaling pathways via NK1 receptors and their desensitization in an AR42J cell line. Peptides 19:1349–1357
Garnier A, Vykoukal J, Hubertus J, Alt E, von Schweinitz D, Kappler R, Berger M, Ilmer M (2015) Targeting the neurokinin-1 receptor inhibits growth of human colon cancer cells. Int J Oncol 47:151–160. https://doi.org/10.3892/ijo.2015.3016
Gharaee N, Pourali L, Jafarian AH, Hashemy SI (2018) Evaluation of serum level of substance P and tissue distribution of NK-1 receptor in endometrial cancer. Mol Biol Rep:1–6
Gurevich VV, Gurevich EV (2006) The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol Ther 110:465–502
Hahn WC, Weinberg RA (2002) Rules for making human tumor cells. N Engl J Med 347:1593–1603
Hassanian SM, Dinarvand P, Smith SA, Rezaie AR (2015) Inorganic polyphosphate elicits pro-inflammatory responses through activation of the mammalian target of rapamycin complexes 1 and 2 in vascular endothelial cells. J Thromb Haemost 13:860–871
Hausmann G, Bänziger C, Basler K (2007) Helping wingless take flight: how WNT proteins are secreted. Nat Rev Mol Cell Biol 8:331–336
Henssen AG, Odersky A, Szymansky A, Seiler M, Althoff K, Beckers A, Speleman F, Schäfers S, De Preter K, Astrahanseff K (2017) Targeting tachykinin receptors in neuroblastoma. Oncotarget 8:430
Ilmer M, Garnier A, Vykoukal J, Alt E, von Schweinitz D, Kappler R, Berger M (2015) Targeting the neurokinin-1 receptor compromises canonical Wnt signaling in hepatoblastoma. Mol Cancer Ther 14:2712–2721. https://doi.org/10.1158/1535-7163.mct-15-0206
Iwamura M, Egawa S, Uchida T, Koshiba K, Cockett AT, Gershagen S (1998) Suppression of the growth and invasiveness of human prostate cancer cells in vitro by neuropeptide antagonist substance P analogues. In: Urologic oncology: seminars and original investigations, vol 1. Elsevier, Amsterdam, pp 24–28
Javid H, Soltani A, Mohammadi F, Hashemy SI (2019) Emerging roles of microRNAs in regulating the mTOR signaling pathway during tumorigenesis. J Cell Biochem 120:10874–10883. https://doi.org/10.1002/jcb.28401
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293
Li J, Zeng Q, Zhang Y, Li X, Hu H, Miao X, Yang W, Zhang W, Song X, Mou L, Wang R (2016) Neurokinin-1 receptor mediated breast cancer cell migration by increased expression of MMP-2 and MMP-14. Eur J Cell Biol 95:368–377. https://doi.org/10.1016/j.ejcb.2016.07.005
Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20:781–810
Luo J, Manning BD, Cantley LC (2003) Targeting the PI3K-Akt pathway in human cancer. Cancer Cell 4:257–262
Ma J, Yuan S, Cheng J, Kang S, Zhao W, Zhang J (2016) Substance P promotes the progression of endometrial adenocarcinoma. Int J Gynecol Cancer 26:845–850. https://doi.org/10.1097/igc.0000000000000683
Mao J, Wang J, Liu B, Pan W, Farr GH III, Flynn C, Yuan H, Takada S, Kimelman D, Li L (2001) Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol Cell 7:801–809
Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E (2014) PI3K/AKT signaling pathway and cancer: an updated review. Ann Med 46:372–383
Mayordomo C, García-Recio S, Ametller E, Fernández-Nogueira P, Pastor-Arroyo EM, Vinyals L, Casas I, Gascón P, Almendro V (2012) Targeting of substance P induces cancer cell death and decreases the steady state of EGFR and Her2. J Cell Physiol 227:1358–1366
Molinos-Quintana A, Trujillo-Hacha P, Piruat J, Bejarano-García J, García-Guerrero E, Pérez-Simón J, Muñoz M (2018) Human acute myeloid leukemia cells express Neurokinin-1 receptor, which is involved in the antileukemic effect of neurokinin-1 receptor antagonists. Investig New Drugs 1–10
Moraes M, Neto J, Menck C (2012) DNA repair mechanisms protect our genome from carcinogenesis. Front Biosci 17:1362–1388
Mou L, Xing Y, Kong Z, Zhou Y, Chen Z, Wang R (2011) The N-terminal domain of human hemokinin-1 influences functional selectivity property for tachykinin receptor neurokinin-1. Biochem Pharmacol 81:661–668
Munoz M, Covenas R (2018) Glioma and neurokinin-1 receptor antagonists: a new therapeutic approach. Anti Cancer Agents Med Chem
Munoz M, Rosso M, Covenas R (2011) The NK-1 receptor: a new target in cancer therapy. Curr Drug Targets 12:909–921
Munoz M, Covenas R, Esteban F, Redondo M (2015) The substance P/NK-1 receptor system: NK-1 receptor antagonists as anti-cancer drugs. J Biosci 40:441–463
Murthy RG, Reddy BY, Ruggiero JE, Rameshwar P (2007) Tachykinins and hematopoietic stem cell functions: implications in clinical disorders and tissue regeneration. Front Biosci 12:4779–4787
Newton S, Walker A, Page N (2009) Stimulation of PKC [beta] II-dependent ERK1/2 signalling by endokinin B and substance P may cause gene transcription via the tachykinin NK1 receptor in astrocytoma cells
Niu X-L, Hou J-F, Li J-X (2018) The NK1 receptor antagonist NKP608 inhibits proliferation of human colorectal cancer cells via Wnt signaling pathway. Biol Res 51:14
Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K-I, Hara K, Tanaka N, Avruch J, Yonezawa K (2003) The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem 278:15461–15464
Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS (2000) Differential affinities of visual arrestin, βarrestin1, and βarrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem 275:17201–17210
Öztürk Ö, Aki-Yalcin E, Ertan-Bolelli T, Bolelli K, Nur-Hidayat A, Bingol-Ozakpinar O, Ozdemir F, Yalcin I (2017) Possible mechanism of action of neurokinin-1 receptors (NK1R) antagonists. J Pharm Pharmacol 5:787–797
Palma C (2006) Tachykinins and their receptors in human malignancies. Curr Drug Targets 7:1043–1052
Patacchini R, Maggi CA (2001) Peripheral tachykinin receptors as targets for new drugs. Eur J Pharmacol 429:13–21
Pennefather JN, Lecci A, Candenas ML, Patak E, Pinto FM, Maggi CA (2004) Tachykinins and tachykinin receptors: a growing family. Life Sci 74:1445–1463
Rahmani F, Avan A, Hashemy SI, Hassanian SM (2018) Role of Wnt/beta-catenin signaling regulatory microRNAs in the pathogenesis of colorectal cancer. J Cell Physiol 233:811–817. https://doi.org/10.1002/jcp.25897
Raman M, Chen W, Cobb M (2007) Differential regulation and properties of MAPKs. Oncogene 26:3100–3112
Reya T, Clevers H (2005) Wnt signalling in stem cells and cancer. Nature 434:843–850. https://doi.org/10.1038/nature03319
Rosso M, Munoz M, Berger M (2012) The role of neurokinin-1 receptor in the microenvironment of inflammation and cancer. Sci World J 2012
Roux PP, Blenis J (2004) ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68:320–344
Schieven GL (2005) The biology of p38 kinase: a central role in inflammation. Curr Top Med Chem 5:921–928
Severini C, Improta G, Falconieri-Erspamer G, Salvadori S, Erspamer V (2002) The tachykinin peptide family. Pharmacol Rev 54:285–322
Sharif M (1998) Mitogenic signaling by substance P and bombesin-like neuropeptide receptors in astrocytic/glial brain tumor-derived cell lines. Int J Oncol 12:273–286
Sharif M, Sharif T, Dilling M, Hosoi H, Lawrence J, Houghton P (1997) Rapamycin inhibits substance P-induced protein synthesis and phosphorylation of PHAS-I (4E-BP1) and p70 S6 kinase (p70 (S6K)) in human astrocytoma cells. Int J Oncol 11:797–805
Shaw RJ, Cantley LC (2006) Ras, PI(3) K and mTOR signalling controls tumour cell growth. Nature 441:424–430. https://doi.org/10.1038/nature04869
Singh D, Joshi DD, Hameed M, Qian J, Gascón P, Maloof PB, Mosenthal A, Rameshwar P (2000) Increased expression of preprotachykinin-I and neurokinin receptors in human breast cancer cells: implications for bone marrow metastasis. Proc Natl Acad Sci 97:388–393
Smolich BD, McMahon JA, McMahon AP, Papkoff J (1993) Wnt family proteins are secreted and associated with the cell surface. Mol Biol Cell 4:1267–1275
Song H, Yin W, Zeng Q, Jia H, Lin L, Liu X, Mu L, Wang R (2012) Hemokinins modulate endothelium function and promote angiogenesis through neurokinin-1 receptor. Int J Biochem Cell Biol 44:1410–1421
Weinberg R (2007) Moving out: invasion and metastasis. Biol Cancer 1:587–654
Yamaguchi K, Kugimiya T, Miyazaki T (2005) Substance P receptor in U373 MG human astrocytoma cells activates mitogen-activated protein kinases ERK1/2 through Src. Brain Tumor Pathol 22:1–8
Yamaguchi K, Richardson MD, Bigner DD, Kwatra MM (2005) Signal transduction through substance P receptor in human glioblastoma cells: roles for Src and PKCdelta. Cancer Chemother Pharmacol 56:585–593. https://doi.org/10.1007/s00280-005-1030-3
Yamaguchi K, Kumakura S, Murakami T, Someya A, Inada E, Nagaoka I (2017) Ketamine suppresses the substance P-induced production of IL-6 and IL-8 by human U373MG glioblastoma/astrocytoma cells. Int J Mol Med 39:687–692. https://doi.org/10.3892/ijmm.2017.2875
Zhan T, Rindtorff N, Boutros M (2017) Wnt signaling in cancer. Oncogene 36:1461–1473
Zhang Y-X, Li X-F, Yuan G-Q, Hu H, Song X-Y, Li J-Y, Miao X-K, Zhou T-X, Yang W-L, Zhang X-W (2017) β-Arrestin 1 has an essential role in neurokinin-1 receptor-mediated glioblastoma cell proliferation and G2/M phase transition. J Biol Chem M116:770420
Zhang YX, Li XF, Yuan GQ, Hu H, Song XY, Li JY, Miao XK, Zhou TX, Yang WL, Zhang XW, Mou LY, Wang R (2017) Beta-arrestin 1 has an essential role in neurokinin-1 receptor-mediated glioblastoma cell proliferation and G2/M phase transition. J Biol Chem 292:8933–8947. https://doi.org/10.1074/jbc.M116.770420
Zhou J, Ling J, Song H, Lv B, Wang L, Shang J, Wang Y, Chang C, Ping F, Qian J (2016) Neurokinin-1 receptor is a novel positive regulator of Wnt/β-catenin signaling in melanogenesis. Oncotarget 7:81268
Zhuo H, Helke C (1993) Neurokinin B peptide-2 neurons project from the hypothalamus to the thoracolumbar spinal cord of the rat. Neuroscience 52:1019–1028
Author information
Authors and Affiliations
Contributions
All authors contributed to the gathering of data and writing the manuscript; HJ was responsible to coordinate with the authors, and SIH finalized and submitted the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key points
• Tachykinins have implications in different steps of carcinogenesis.
• SP/NK1R system activates signaling pathways involved in tumorigenesis.
• The blockage of NK1R could suggest a therapeutic approach in cancer therapy.
Rights and permissions
About this article

Cite this article
Javid, H., Mohammadi, F., Zahiri, E. et al. The emerging role of substance P/neurokinin-1 receptor signaling pathways in growth and development of tumor cells. J Physiol Biochem 75, 415–421 (2019). https://doi.org/10.1007/s13105-019-00697-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-019-00697-1