
Abstract
Lamin A/C is the major architectural protein of cell nucleus in charge of the nuclear mechanosensing. By integrating extracellular mechanical and biochemical signals, lamin A/C regulates multiple intracellular events including mesenchymal stem cell (MSC) fate determination. Herein, we review the recent findings about the effects and mechanisms of lamin A/C in governing MSC lineage commitment, with a special focus on osteogenesis and adipogenesis. Better understanding of MSC differentiation regulated by lamin A/C could provide insights into pathogenesis of age-related osteoporosis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Age-related bone loss, increasing the fracture risk of the elderly individuals, has become increasingly prevalent and has caused enormous health care costs. Approximately 13% of the individuals would experience osteoporosis in China [64], and about 20% of the patients suffering from osteoporotic fractures died in 1 year because of related complications [11]. The disease has been found to be characterized by decreasing bone formation and increasing adipose tissue infiltration in bone marrow, which is the result of the switch of mesenchymal stem cell (MSC) differentiation from osteogenesis to adipogenesis [19]. However, the mechanisms of the switch remain unclear.
One possible mediator of MSC fate is lamin A/C, a kind of lamin protein in the nucleus contributing to nuclear mechanics [13] and mechanotransduction [50]. Lamin A/C levels decreased in osteoblasts during aging [16], indicating participation of lamin A/C in the development of senile skeletal phenotypes. In vivo evidence showed that mutation of LMNA caused Hutchinson-Gilford progeria syndrome (HGPS), the disease characterized by age-related bone loss [39]. Animals with mutations in LMNA or in genes encoding lamin A/C post-transcriptional processing proteins display osteoporosis phenotypes including spontaneous fractures, declined bone density, loss of trabecular and cortical bone, decrease of bone forming cells, and accumulation of adipocytes in the bone marrow [42, 56, 68]. The experimental evidence supported the involvement of lamin A/C in regulating MSC fate in age-related bone loss, but the mechanisms are not fully understood.
Mechanical cues have recently emerged as key regulators of MSC differentiation [34, 57], and our previous work has found that external forces could affect the maintenance and lineage specification of MSCs [33, 69]. As a key structure in the direct mechanical force transmission to the nucleus [37], lamin A/C could serve as one of the mediators by which mechanical stimulations exert influence on differentiation of MSCs. Additionally, mechanical homeostasis, an inherent property of tissues, also participates in the regulation of MSC fate [65]. Furthermore, lamin A/C also involves in regulating MSC differentiation merely under biochemical stimulations.
To understand the role of lamin A/C in regulating differentiation of MSCs, we summarize the state-of-the-art findings of lamin A/C in MSC differentiation under mechanical stimulations or merely induced by biochemical signals.
Lamin A/C in nucleus
Lamins lie in the inner nuclear membrane as a meshwork [36], and they have a tripartite structure with α-helical domain, amino-terminal and carboxy-terminal domains [14, 58, 59]. They are classified into type A and type B. Unlike type B lamin, which is expressed ubiquitously in all cells, type A lamin is only expressed in differentiated cells [25] and its expression levels are varied in cells from different tissues. Type A lamin, encoded by LMNA, consists of two major isoforms—lamin A and lamin C.
The maturation of lamin A/C requires several steps of post-translational modifications, consisting of isoprenylation of cysteine in the CAAX box, cleavage of the AAX motif, methylation of the isoprenylated cysteine, and the removal of carboxy-terminal 18 amino acids by endoprotease Zmpste24 [20]. After maturation, the assembly of lamin A/C polymers involves dimerization through their α-helical rod domain and subsequent multimerization through head-to-tail overlapping between dimers [27].
Lamin A/C structurally connects with cytoskeleton via linkers of nucleoskeleton and cytoskeleton (LINC) complexes [12, 58, 59]. LINC complexes are composed of Sad1/UNC-84 (SUN) domain proteins and Klarsicht/ANC-1/Syne homology (KASH) domain proteins. SUN domain proteins are located at the inner nuclear membrane and bind with lamin A/C. KASH domain proteins mainly consist of Nesprins (Nesprin 1, 2, and 3) and reside at the outer nuclear membrane, connecting SUN domain with all sorts of cytoskeletons, including actin filaments, tubulin, and intermediate filaments (such as desmin and vimentin). Therefore, LINC complexes mediate force transmission from cytoskeleton to lamins, and disruption in them was found to block cellular mechano-transduction from cytoskeleton to lamins [58, 59].
As the main components of nuclear lamina and mediator of nuclear-cytoskeletal coupling, lamin A/C determines nuclear mechanics and is involved in mechanotransduction. Its anomalies lead to pathological changes of cell functions and behaviors, and gives rise to a group of diseases known as laminopathies [14, 58, 59]. For example, Emery-Dreifuss muscular dystrophy, manifested by joint contractures, muscular weakness, and cardiomyopathy, is evidenced to be related with impaired cellular mechanotransduction [6].
Lamin A/C in the differentiation of MSCs under mechanical cues
MSC differentiation potentials are extensively controlled by mechanical signals. MSCs can sense the extracellular mechanical signals via a plethora of mechanosensitive elements and translate them into biochemical responses in cytoplasm and nucleus, resulting in the switch of their differentiation state. Traditionally, it is believed that activated mechanosensors on cytoplasm membrane, such as mechanosensitive ion channels and integrins, activate or suppress key molecules in mechanosensitive signaling cascades in cytoplasm and change their subcellular localization. This subsequently leads to the alteration of gene expression in the nucleus. In the past two decades, the nucleus has been found to be another key mechanosensor, and direct transmission of mechanical signals from the cytoplasm membrane, via cytoskeleton to the nucleus has been identified as another mechanotransduction pathway [37]. Lamin A/C, connecting with cytoskeleton through LINC complexes, participates in the direct force transmission [10, 29] and affects the mechano-induced differentiation of MSCs.
Changes of lamin A/C in MSC under mechanical cues
Lamin A/C in MSCs could respond to applied forces, two-dimensional substrate, and three-dimensional microenvironments by changing its expression, localization, and structure. These changes of lamin A/C would in return affect MSC differentiation state.
Applied forces are among the mechanical cues that could evidence the mechanosensitive property of lamin A/C in MSC differentiation. It was found that both soluble factor–induced and stretch loading–stimulated MSC differentiation were accompanied by increased expression levels and nuclear periphery translocation of lamin A/C [23]. Interestingly, the reactions of lamin A/C under stretch loading was faster than those under soluble factors [23], implying that direct force transmission system to lamin A/C was more efficient than biochemical factors in MSC differentiation induction. The alterations of lamin A/C, in return, led to the enhanced MSC mechanosensitivity as a response to applied stretch forces [23], suggesting the role of lamin A/C in mediating mechanical cue transmission in MSCs.
Recent studies have also focused on lamin A/C sensing two-dimensional mechanical cues and its role in MSC differentiation under the signals. The most widely studied two-dimensional mechanical signal is substrate stiffness. Compared with soft substrate, stiff substrate was found to result in increased amount [18, 60] and decreased phosphorylation level [9] of lamin A/C in MSCs. Substrate stiffness can also alter MSC differentiation states, in which MSCs favor osteogenesis on stiff substrate while adipogenesis on soft substrate [60, 67]. Responses of lamin A/C to different substrate stiffness reciprocally participate in the substrate stiffness–induced MSC osteogenesis and adipogenesis. Overexpression of lamin A/C could reinforce the stiff substrate–induced osteogenesis [60]. On the contrary, LMNA knockdown was found to enhance the soft-substrate-promoted adipogenic differentiation [60]. Additionally, it has been recently reported that lamin A/C was enriched in the apical and lateral surfaces of nuclei, exclusively forming a dome-like ultrastructure [30], and matrix stiffness could change its polarized distribution. Culturing cells on softer substrate led to the conversion of distribution of lamin A/C from asymmetric dome to isotropic shell, while stiffer substrate caused the opposite outcome [26]. Lamin A/C also exhibited isotropic distribution in MSCs during adipogenesis and asymmetric distribution during osteogenesis [26]. However, the link between lamin A/C polarization and cell differentiation induced by extracellular matrix (ECM) still needs elucidation.
Lamin A/C can also mediate the mechanotransduction of cells growing on substrate with topographical changes. The substrate with 350-nm width grating could lead to the down-regulation of lamin C in MSCs, along with decreased adipogenesis and increased osteogenesis [31]. Furthermore, when compared to circle-shape micro-pattern substrate, stem cells grown on micro-pattern substrate with triangular morphology exhibited reduced lamin A/C labeling, triangularly deformed and softer nuclei [61]. Similarly, compared to cells cultured on flat and concave surface, MSCs on convex surface showed the increased expression of lamin A/C along with enhanced osteogenesis [66].
Crosstalk between lamin A/C and other mechanosensitive signaling pathways—mechanism of lamin A/C regulating MSC differentiation
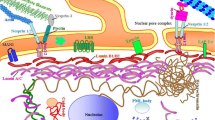
As a nucleus protein, lamin A/C can interact extensively with other proteins and get involved in multiple signaling events. Growing evidence implies a crosstalk existing between lamin A/C and other mechanosensitive signaling pathways, which emerges as a new molecular mechanism of how lamin A/C senses mechanical cues and mediates MSC differentiation (Fig. 1). One of the focused models to study mechanical signals is substrate with different rigidity. As evidenced by multiple studies, MSC cultured on stiff substrate favor osteogenic differentiation whereas MSC cultured on soft substrate turn towards adipogenic lineage [60, 67].

Crosstalk between lamin A/C and other mechanosensitive signaling pathways. (a) Lamin A/C might have crosstalk with YAP/TAZ through regulating actin filaments. (b) Lamin A/C regulates MKL1 nuclear import through actin filaments. (c) Lamin A/C increases nuclear import of RARG
RA pathway
Retinoic acid (RA) pathway has been found to intercommunicate with mechano-transduction pathways mediated by lamin A/C. The transduction of RA pathway is dependent on retinoic acid receptors (RAR), a group of nuclear receptor superfamily to activate the transcription of target genes. RARs mainly consist of RARA, RARB, and RARG. RARG has been found to participate in MSC lineage specification. Its antagonists and agonists could respectively promote and inhibit osteogenesis [21].
Swift et al. [60] firstly found the crosstalk between RARG and lamin A/C, reporting that RARG has corresponding binding sites, RA-responsive elements (RARE), on the promoter of LMNA to regulate the expression of lamin A/C. Agonist of RARG, RA, and suppressed lamin A expression, while antagonist AGN enhanced lamin A expression. Interestingly, the effects of RARG on lamin A/C only existed when cells were cultured on stiff substrate and were abrogated on soft substrate, indicating that substrate stiffness was the upstream signal of the crosstalk between RARG and lamin A/C. Reciprocally, lamin A/C could regulate RARG. Overexpression of lamin A/C was found to promote the nuclear translocation of RARG, and lamin A/C deficiency could abolish MSC osteogenesis promoted by RARG antagonists [28]. Therefore, lamin A and RA pathway form a feed-forward loop in MSC sensing substrate stiffness and differentiation.
YAP/TAZ
Co-activators YAP (Yes-associated protein) and TAZ (transcriptional co-activators with PDZ-binding motif) are downstream effectors of canonical Hippo signaling pathway and are the key regulators in cell perceiving their microenvironments [15]. Stiff substrate could lead to increased expression and nuclear aggregation of YAP/TAZ in MSCs [15, 18]. Combined with their transcriptional coeffector TEAD, YAP/TAZ promotes osteogenesis and impairs adipogenesis of MSCs in response to stiff substrate [15].
Rather than relying on canonical Hippo signaling, YAP/TAZ was also the downstream of the assembly and contractibility of actin filaments in mechanotransduction [15]. As lamin A/C could regulate actin filament assembly [41, 47], lamin A/C might have crosstalk with YAP/TAZ mediated by actin filaments as evidence suggested. The nuclear aggregation of YAP induced by substrate stiffness has been found to be abolished in LMNA mutated myoblasts [5]. However, a non-monotonic relationship between YAP1 and lamin A level has also been presented: the knockdown of LMNA resulted in decreased YAP1 levels and lamin A up-regulation by stiff matrix led to nuclear aggregation of YAP1, but LMNA overexpression in MSCs on stiff matrix also resulted in the decrease of total YAP1 levels and nuclear localization [60]. Therefore, the relationship between lamin A/C and YAP/RAZ has not reached a consensus, and the results could be impacted by different sources of cells, experiment protocols etc.
MKL1-SRF pathway
Megakaryoblastic leukemia 1 (MKL1) acts as the intermediary between actin filaments and serum response factor (SRF). It senses mechanical signals via actin polymerization. The mechano-induced polymerization of G-actin results in the release of MKL1 from monomeric G-actin and its nuclear translocation, eliciting subsequent activation of SRF and its target genes encoding proteins that regulate cytoskeletal architecture and dynamics [40, 41, 44, 62]. Thus, the actin-MKL-SRF circuit can bridge the cytoplasmic actin to nuclear genome to modulate cell differentiation [61]. It was found that MKL1 could inhibit cell adipogenic differentiation by suppressing PPARγ [43, 54].
Lamin A/C has been reported to play a key role in actin-MKL1/SRF signaling pathway. Embryonic stem cells presented strong positive correlation between the levels of lamin A/C and nuclear translocation of MKL1 [61]. LMNA knockdown and LMNA N195K mutant fibroblasts exhibited decreased nuclear import and increased export of MKL1 by impairing actin polymerization [24]. LMNA knockdown could further suppress the transcription of several components of the SRF pathway and enhance MSC adipogenesis [60]. Collectively, lamin A/C and MKL1 form a feed-forward loop in MSC sensing mechanical cues, and they could synergistically promote MSC osteogenesis while inhibiting adipogenesis induced by mechanical signals.
Lamin A/C in the differentiation of MSCs under biochemical cues
Lamin A/C can directly regulate the expression of genes by interacting with promoter regions of some transcription factors on chromatin [7, 22], or change the activity and localization of key transcription factors in signaling cascades by directly interacting with them [70]. Therefore, in addition to the role in mediating the transmission of mechanical cues, studies have also investigated the involvement of lamin A/C in the activity of differentiation-related signaling pathways and transcription factors during osteogenesis and adipogenesis of MSCs merely under biochemical induction. The interactions between lamin A/C and differentiation-related transcription factors under biochemical cues have been comprehensively reviewed (refer to [48, 63]), so we make a brief summary and supplement some latest findings below (Table 1).
During osteogenesis of MSCs, lamin A/C could activate WNT/β-catenin signaling by physically interacting with and facilitating the nuclear translocation of β-catenin [4]. Lamin A/C overexpression was also found to promote osteogenesis by activating Notch pathway [55]. However, another study reported lamin A/C downregulation could increase Notch signaling through affecting emerin localization to nuclear envelope [32]. In addition, lamin A/C was also directly related with Runx2 activity during osteogenesis. Deficiency of lamin A/C led to either significantly lower levels of Runx2 [51] or a reduction of its binding activity with its targeted gene [1, 35]. Moreover, Runx2 and Smads could be anchored by the integral inner nuclear membrane protein MAN-1, which hinders their interactions with other effectors [2, 46]. Lamin A/C deficiency led to higher levels of MAN-1 and subsequently increased nuclear periphery colocalization of Runx2 [35].
On the contrary, lamin A/C could exert repressive effect on MSC adipogenic differentiation by deactivating PPARγ or regulating Notch signaling [4, 60]. The lack of lamin A/C could facilitate the release of PPARγ, which is normally trapped at the nuclear periphery by lamin A/C and its associated proteins [45], and promote its activation [8]. Moreover, lamin A/C overexpression could inhibit adipogenesis by activating Notch signaling [55], but LMNA R482L mutation was reported to inhibit cardiac MSC adipogenesis by counteracting the activation of Notch signaling [49].
The accumulation of immature lamin A/C caused by disruption of post-translational processing has also been reported to alter MSC lineage commitment. Prelamin A farnesylation is one of the most significant post-translational processing of lamin A/C, and the inhibition of farnesylation caused the accumulation of prelamin A in nucleus and the loss of osteogenic capability of MSCs [17]. Meanwhile, the inhibition of prelamin A farnesylation could also arrest adipogenic differentiation of MSCs possibly via the inhibition of PPARγ expression and activation [52]. Therefore, the accumulation of prelamin A did not have an opposite effect on osteogenic and adipogenic differentiation. The reason might be that accumulation of prelamin impairs the ability of mature lamin A/C to enhance the activity of osteogenic transcription factors, while prelamin A itself could sequestrate and inhibit transcription factors related to adipogenesis. In addition, prelamin A is also the substrate of endoprotease Zmpste24, which is responsible for the final cleavage in post-translation of prelamin A [38]. Zmpste24−/− mice exhibited spontaneous bone fractures [3], decreased number of osteoblasts, and increased level of fat infiltration in bone marrow [53]. In vitro settings, bone marrow cells from those deficient mice had impaired osteogenic potential and enhanced adipogenic ability [53]. Collectively, lamin A/C with mature structure is critical in MSC differentiation, and the disruption of the process would impact MSC osteogenesis and adipogenesis context-dependently.
Conclusion
Over the past two decades, strong evidence has suggested that lamin A/C extensively participates in regulating lineage specification of MSCs. Two mechanisms are primarily involved in the regulation. First, via linking with cytoskeleton, lamin A/C can sense different mechanical cues in the microenvironment, and thereby enhances osteogenesis while inhibits adipogenesis of MSCs. Secondly, lamin A/C can also regulate the response of MSCs to biochemical cues by directly interacting with multiple signaling pathways and key transcription factors. However, we are still at the preliminary stage of the exploration of these mechanisms. For example, as recent evidence showed asymmetric distribution of lamin A/C in nucleus, will this spatial pattern affect the link between lamin A/C with the cytoskeleton? How would it consequently influence mechano-induced lineage determination of the MSCs? Further studies are expected to answer these questions. Moreover, although the crosstalk between lamin A/C and some mechanosensitive signaling pathways is elucidated, the involvement of other classical mechanosensitive signaling pathways, such as Notch signaling, still awaits further exploration. To summarize, more studies are anticipated to enrich the evidence on the detailed mechanisms of lamin A/C in regulation of MSC lineage determination.
Treatments of age-related bone loss require detailed knowledge of cellular changes in bone marrow. Age-related alterations in lamin A/C might be one vital biological process that leads to adipose infiltration in bone marrow. With extensive adipose accumulation that softens MSC substrate within the bone marrow, lamin A/C might be reciprocally tuned and lead to preferred switch towards adipogenesis over osteogenesis in the MSCs. Therefore, better understanding of lamin A/C in MSC niche might provide novel insights into pathogenic mechanism of age-related osteoporosis and potential approach to break the vicious cycle and improve the prognosis of the disease.
Abbreviations
- ECM:
-
Extracellular matrix
- HGPS:
-
Hutchinson-Gilford progeria syndrome
- KASH:
-
Klarsicht/ANC-1/Syne homology
- LINC:
-
Linkers of nucleoskeleton and cytoskeleton
- MKL1:
-
Megakaryoblastic leukemia 1
- MSC:
-
Mesenchymal stem cell
- RA:
-
Retinoic acid
- RAR:
-
Retinoic acid receptors
- RARE:
-
RA-responsive elements
- SRF:
-
Serum response factor
- SUN:
-
Sad1/UNC-84
- YAP:
-
Yes-associated protein
- TAZ:
-
transcriptional co-activators with PDZ-binding motif
References
Akter R, Rivas D, Geneau G, Drissi H, Duque G (2009) Effect of lamin A/C knockdown on osteoblast differentiation and function. J Bone Miner Res 24:283–293. https://doi.org/10.1359/jbmr.081010
Bengtsson L (2007) What MAN1 does to the Smads. TGFbeta/BMP signaling and the nuclear envelope. FEBS J 274:1374–1382
Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, Wisner ER, Van Bruggen N, Carano RA, Michaelis S, Griffey SM, Young SG (2002) Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A 99:13049–13054. https://doi.org/10.1073/pnas.192460799
Bermeo S, Vidal C, Zhou H, Duque G (2015) Lamin A/C acts as an essential factor in mesenchymal stem cell differentiation through the regulation of the dynamics of the Wnt/beta-catenin pathway. J Cell Biochem 116:2344–2353. https://doi.org/10.1002/jcb.25185
Bertrand AT, Ziaei S, Ehret C, Duchemin H, Mamchaoui K, Bigot A, Mayer M, Quijano-Roy S, Desguerre I, Laine J, Ben Yaou R, Bonne G, Coirault C (2014) Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J Cell Sci 127:2873–2884. https://doi.org/10.1242/jcs.144907
Bianchi A, Manti PG, Lucini F, Lanzuolo C (2018) Mechanotransduction, nuclear architecture and epigenetics in Emery Dreifuss muscular dystrophy: tous pour un, un pour Tous. Nucleus 9:276–290. https://doi.org/10.1080/19491034.2018.1460044
Bickmore WA, van Steensel B (2013) Genome architecture: domain organization of interphase chromosomes. Cell 152:1270–1284. https://doi.org/10.1016/j.cell.2013.02.001
Boguslavsky RL, Stewart CL, Worman HJ (2006) Nuclear lamin A inhibits adipocyte differentiation: implications for Dunnigan-type familial partial lipodystrophy. Hum Mol Genet 15:653–663. https://doi.org/10.1093/hmg/ddi480
Buxboim A, Swift J, Irianto J, Spinler KR, Dingal PC, Athirasala A, Kao YR, Cho S, Harada T, Shin JW, Discher DE (2014) Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr Biol 24:1909–1917. https://doi.org/10.1016/j.cub.2014.07.001
Chambliss AB, Khatau SB, Erdenberger N, Robinson DK, Hodzic D, Longmore GD, Wirtz D (2013) The LINC-anchored actin cap connects the extracellular milieu to the nucleus for ultrafast mechanotransduction. Sci Rep 3:1087. https://doi.org/10.1038/srep01087
Cosman F, de Beur SJ, LeBoff MS, Lewiecki EM, Tanner B, Randall S, Lindsay R (2014) Clinician's guide to prevention and treatment of osteoporosis. Osteoporos Int 25:2359–2381. https://doi.org/10.1007/s00198-014-2794-2
Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D (2006) Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol 172:41–53. https://doi.org/10.1083/jcb.200509124
Dahl KN, Kalinowski A (2011) Nucleoskeleton mechanics at a glance. J Cell Sci 124:675–678. https://doi.org/10.1242/jcs.069096
Dittmer TA, Misteli T (2011) The lamin protein family. Genome Biol 12:222. https://doi.org/10.1186/gb-2011-12-5-222
Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S (2011) Role of YAP/TAZ in mechanotransduction. Nature 474:179–183. https://doi.org/10.1038/nature10137
Duque G, Rivas D (2006) Age-related changes in lamin A/C expression in the osteoarticular system: laminopathies as a potential new aging mechanism. Mech Ageing Dev 127:378–383. https://doi.org/10.1016/j.mad.2005.12.007
Duque G, Vidal C, Rivas D (2011) Protein isoprenylation regulates osteogenic differentiation of mesenchymal stem cells: effect of alendronate, and farnesyl and geranylgeranyl transferase inhibitors. Br J Pharmacol 162:1109–1118. https://doi.org/10.1111/j.1476-5381.2010.01111.x
Galarza Torre A, Shaw JE, Wood A, Gilbert HTJ, Dobre O, Genever P, Brennan K, Richardson SM, Swift J (2018) An immortalised mesenchymal stem cell line maintains mechano-responsive behaviour and can be used as a reporter of substrate stiffness. Sci Rep 8:8981. https://doi.org/10.1038/s41598-018-27346-9
Gimble JM, Zvonic S, Floyd ZE, Kassem M, Nuttall ME (2006) Playing with bone and fat. J Cell Biochem 98:251–266. https://doi.org/10.1002/jcb.20777
Goldberg MW, Huttenlauch I, Hutchison CJ, Stick R (2008) Filaments made from A- and B-type lamins differ in structure and organization. J Cell Sci 121:215–225. https://doi.org/10.1242/jcs.022020
Green AC, Kocovski P, Jovic T, Walia MK, Chandraratna RAS, Martin TJ, Baker EK, Purton LE (2017) Retinoic acid receptor signalling directly regulates osteoblast and adipocyte differentiation from mesenchymal progenitor cells. Exp Cell Res 350:284–297. https://doi.org/10.1016/j.yexcr.2016.12.007
Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B (2008) Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453:948–951. https://doi.org/10.1038/nature06947
Heo SJ, Driscoll TP, Thorpe SD (2016) Differentiation alters stem cell nuclear architecture, mechanics, and mechano-sensitivity. Elife 5. https://doi.org/10.7554/eLife.18207
Ho CY, Jaalouk DE, Vartiainen MK, Lammerding J (2013) Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 497:507–511. https://doi.org/10.1038/nature12105
Hutchison CJ, Worman HJ (2004) A-type lamins: guardians of the soma? Nat Cell Biol 6:1062–1067. https://doi.org/10.1038/ncb1104-1062
Ihalainen TO, Aires L, Herzog FA, Schwartlander R, Moeller J, Vogel V (2015) Differential basal-to-apical accessibility of lamin A/C epitopes in the nuclear lamina regulated by changes in cytoskeletal tension. Nat Mater 14:1252–1261. https://doi.org/10.1038/nmat4389
Isobe K, Gohara R, Ueda T, Takasaki Y, Ando S (2007) The last twenty residues in the head domain of mouse lamin A contain important structural elements for formation of head-to-tail polymers in vitro. Biosci Biotechnol Biochem 71:1252–1259. https://doi.org/10.1271/bbb.60674
Ivanovska IL, Swift J, Spinler K, Dingal D, Cho S, Discher DE (2017) Cross-linked matrix rigidity and soluble retinoids synergize in nuclear lamina regulation of stem cell differentiation. Mol Biol Cell 28:2010–2022. https://doi.org/10.1091/mbc.E17-01-0010
Kim DH, Khatau SB, Feng Y, Walcott S, Sun SX, Longmore GD, Wirtz D (2012) Actin cap associated focal adhesions and their distinct role in cellular mechanosensing. Sci Rep 2:555. https://doi.org/10.1038/srep00555
Kim DH, Wirtz D (2015) Cytoskeletal tension induces the polarized architecture of the nucleus. Biomaterials 48:161–172. https://doi.org/10.1016/j.biomaterials.2015.01.023
Kulangara K, Yang J, Chellappan M, Yang Y, Leong KW (2014) Nanotopography alters nuclear protein expression, proliferation and differentiation of human mesenchymal stem/stromal cells. PLoS One 9:e114698. https://doi.org/10.1371/journal.pone.0114698
Lee B, Lee TH, Shim J (2017) Emerin suppresses notch signaling by restricting the notch intracellular domain to the nuclear membrane. Biochim Biophys Acta Mol Cell Res 1864:303–313. https://doi.org/10.1016/j.bbamcr.2016.11.013
Li J, Wang J, Zou Y, Zhang Y, Long D, Lei L, Tan L, Ye R, Wang X, Zhao Z (2012) The influence of delayed compressive stress on TGF-beta1-induced chondrogenic differentiation of rat BMSCs through Smad-dependent and Smad-independent pathways. Biomaterials 33:8395–8405. https://doi.org/10.1016/j.biomaterials.2012.08.019
Li R, Liang L, Dou Y, Huang Z, Mo H, Wang Y, Yu B (2015) Mechanical strain regulates osteogenic and adipogenic differentiation of bone marrow mesenchymal stem cells. Biomed Res Int 2015:873251. doi:https://doi.org/10.1155/2015/873251, 1, 10
Li W, Yeo LS, Vidal C, McCorquodale T, Herrmann M, Fatkin D, Duque G (2011) Decreased bone formation and osteopenia in lamin a/c-deficient mice. PLoS One 6:e19313. https://doi.org/10.1371/journal.pone.0019313
Lombardi ML, Lammerding J (2011) Keeping the LINC: the importance of nucleocytoskeletal coupling in intracellular force transmission and cellular function. Biochem Soc Trans 39:1729–1734. https://doi.org/10.1042/bst20110686
Maniotis AJ, Chen CS, Ingber DE (1997) Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci U S A 94:849–854
Maraldi NM, Capanni C, Cenni V, Fini M, Lattanzi G (2011) Laminopathies and lamin-associated signaling pathways. J Cell Biochem 112:979–992. https://doi.org/10.1002/jcb.22992
Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, Brooks BP, Gerber LH, Turner ML, Domingo DL, Hart TC, Graf J, Reynolds JC, Gropman A, Yanovski JA, Gerhard-Herman M, Collins FS, Nabel EG, Cannon RO 3rd, Gahl WA, Introne WJ (2008) Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med 358:592–604. https://doi.org/10.1056/NEJMoa0706898
Miralles F, Posern G, Zaromytidou AI, Treisman R (2003) Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113:329–342
Mouilleron S, Guettler S, Langer CA, Treisman R, McDonald NQ (2008) Molecular basis for G-actin binding to RPEL motifs from the serum response factor coactivator MAL. EMBO J 27:3198–3208. https://doi.org/10.1038/emboj.2008.235
Mounkes LC, Kozlov S, Hernandez L, Sullivan T, Stewart CL (2003) A progeroid syndrome in mice is caused by defects in A-type lamins. Nature 423:298–301. https://doi.org/10.1038/nature01631
Nobusue H, Onishi N, Shimizu T, Sugihara E, Oki Y, Sumikawa Y, Chiyoda T, Akashi K, Saya H, Kano K (2014) Regulation of MKL1 via actin cytoskeleton dynamics drives adipocyte differentiation. Nat Commun 5:3368. https://doi.org/10.1038/ncomms4368
Olson EN, Nordheim A (2010) Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 11:353–365. https://doi.org/10.1038/nrm2890
Osmanagic-Myers S, Dechat T, Foisner R (2015) Lamins at the crossroads of mechanosignaling. Genes Dev 29:225–237. https://doi.org/10.1101/gad.255968.114
Pan D, Estevez-Salmeron LD, Stroschein SL, Zhu X, He J, Zhou S, Luo K (2005) The integral inner nuclear membrane protein MAN1 physically interacts with the R-Smad proteins to repress signaling by the transforming growth factor-{beta} superfamily of cytokines. J Biol Chem 280:15992–16001. https://doi.org/10.1074/jbc.M411234200
Parmacek MS (2007) Myocardin-related transcription factors: critical coactivators regulating cardiovascular development and adaptation. Circ Res 100:633–644. https://doi.org/10.1161/01.RES.0000259563.61091.e8
Pekovic V, Hutchison CJ (2008) Adult stem cell maintenance and tissue regeneration in the ageing context: the role for A-type lamins as intrinsic modulators of ageing in adult stem cells and their niches. J Anat 213:5–25. https://doi.org/10.1111/j.1469-7580.2008.00928.x
Perepelina K, Dmitrieva R, Ignatieva E, Borodkina A, Kostareva A, Malashicheva A (2018) Lamin A/C mutation associated with lipodystrophy influences adipogenic differentiation of stem cells through interaction with notch signaling. Biochem Cell Biol 96:342–348. https://doi.org/10.1139/bcb-2017-0210
Philip JT, Dahl KN (2008) Nuclear mechanotransduction: response of the lamina to extracellular stress with implications in aging. J Biomech 41:3164–3170. https://doi.org/10.1016/j.jbiomech.2008.08.024
Rauner M, Sipos W, Goettsch C, Wutzl A, Foisner R, Pietschmann P, Hofbauer LC (2009) Inhibition of lamin A/C attenuates osteoblast differentiation and enhances RANKL-dependent osteoclastogenesis. J Bone Miner Res 24:78–86. https://doi.org/10.1359/jbmr.080902
Rivas D, Akter R, Duque G (2007) Inhibition of protein Farnesylation arrests adipogenesis and affects PPARgamma expression and activation in differentiating mesenchymal stem cells. PPAR Res 2007:81654–81657. https://doi.org/10.1155/2007/81654
Rivas D, Li W, Akter R, Henderson JE, Duque G (2009) Accelerated features of age-related bone loss in zmpste24 metalloproteinase-deficient mice. J Gerontol A Biol Sci Med Sci 64:1015–1024. https://doi.org/10.1093/gerona/glp089
Rosenwald M, Efthymiou V, Opitz L, Wolfrum C (2017) SRF and MKL1 independently inhibit Brown adipogenesis. PLoS One 12:e0170643. https://doi.org/10.1371/journal.pone.0170643
Scaffidi P, Misteli T (2008) Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol 10:452–459. https://doi.org/10.1038/ncb1708
Schmidt E, Nilsson O, Koskela A, Tuukkanen J, Ohlsson C, Rozell B, Eriksson M (2012) Expression of the Hutchinson-Gilford progeria mutation during osteoblast development results in loss of osteocytes, irregular mineralization, and poor biomechanical properties. J Biol Chem 287:33512–33522. https://doi.org/10.1074/jbc.M112.366450
Song F, Jiang D, Wang T, Wang Y, Lou Y, Zhang Y, Ma H, Kang Y (2017) Mechanical stress regulates osteogenesis and adipogenesis of rat mesenchymal stem cells through PI3K/Akt/GSK-3beta/beta-catenin signaling pathway. Biomed Res Int 2017:6027402. doi:https://doi.org/10.1155/2017/6027402, 1, 10
Stroud MJ (2018) Linker of nucleoskeleton and cytoskeleton complex proteins in cardiomyopathy. Biophys Rev 10:1033–1051. https://doi.org/10.1007/s12551-018-0431-6
Stroud MJ, Banerjee I, Veevers J, Chen J (2014) Linker of nucleoskeleton and cytoskeleton complex proteins in cardiac structure, function, and disease. Circ Res 114:538–548. https://doi.org/10.1161/circresaha.114.301236
Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, Rehfeldt F, Speicher DW, Discher DE (2013) Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341:1240104. https://doi.org/10.1126/science.1240104
Talwar S, Jain N, Shivashankar GV (2014) The regulation of gene expression during onset of differentiation by nuclear mechanical heterogeneity. Biomaterials 35:2411–2419. https://doi.org/10.1016/j.biomaterials.2013.12.010
Vartiainen MK, Guettler S, Larijani B, Treisman R (2007) Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 316:1749–1752. https://doi.org/10.1126/science.1141084
Vidal C, Bermeo S, Fatkin D, Duque G (2012) Role of the nuclear envelope in the pathogenesis of age-related bone loss and osteoporosis. BoneKEy reports 1:62. https://doi.org/10.1038/bonekey.2012.62
Wang Y, Tao Y, Hyman ME, Li J, Chen Y (2009) Osteoporosis in China. Osteoporos Int 20:1651–1662. https://doi.org/10.1007/s00198-009-0925-y
Wen JH, Vincent LG, Fuhrmann A, Choi YS, Hribar KC, Taylor-Weiner H, Chen S, Engler AJ (2014) Interplay of matrix stiffness and protein tethering in stem cell differentiation. Nat Mater 13:979–987. https://doi.org/10.1038/nmat4051
Werner M, Blanquer SB, Haimi SP, Korus G, Dunlop JW, Duda GN, Grijpma DW, Petersen A (2017) Surface curvature differentially regulates stem cell migration and differentiation via altered attachment morphology and nuclear deformation. Adv Sci (Weinh) 4:1600347. https://doi.org/10.1002/advs.201600347
Xie J, Zhang D, Zhou C, Yuan Q, Ye L, Zhou X (2018) Substrate elasticity regulates adipose-derived stromal cell differentiation towards osteogenesis and adipogenesis through beta-catenin transduction. Acta Biomater 79:83–95. https://doi.org/10.1016/j.actbio.2018.08.018
Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, Majumdar S, Bergo MO, Young SG, Fong LG (2006) A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest 116:2115–2121. https://doi.org/10.1172/jci28968
Ye R, Hao J, Song J, Zhao Z, Fang S, Wang Y, Li J (2014) Microenvironment is involved in cellular response to hydrostatic pressures during chondrogenesis of mesenchymal stem cells. J Cell Biochem 115:1089–1096. https://doi.org/10.1002/jcb.24743
Zastrow MS, Vlcek S, Wilson KL (2004) Proteins that bind A-type lamins: integrating isolated clues. J Cell Sci 117:979–987. https://doi.org/10.1242/jcs.01102
Funding
This work was supported by grants from the National Natural Science Foundation of China (31470904, Zhihe Zhao; 81400522, Rui Ye).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, B., Yang, Y., Keyimu, R. et al. The role of lamin A/C in mesenchymal stem cell differentiation. J Physiol Biochem 75, 11–18 (2019). https://doi.org/10.1007/s13105-019-00661-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-019-00661-z