
Abstract
A series of protective responses could be evoked to achieve compensatory adaptation once cardiomyocytes are subjected to chronic hypoxia. MLK3/JNK/c-jun signaling pathway was previously demonstrated to be involved in this process. In the present study, we aim to further examine the performance of MLK3 in hypoxic H9C2 cells and potential mechanism. Myocardial samples of patients with congenital heart disease (CHD) were collected. H9C2 cells were cultured in hypoxic conditions for various durations. MLK3 was silenced by transfection of shRNA to evaluate its role in cell viability. We found expression of MLK3 protein was lower in patients with cyanotic CHD. In hypoxic H9C2 cells, its expression was gradually decreased in a time-dependent manner. However, there was no significant difference about expression of MLK3 mRNA. According to the results of MTT, LDH, and TUNEL, faster cell growth curve, lower death rate, and less apoptotic cells could be observed in MLK-shRNA group compared with scramble-shRNA group. Silencing of MLK3 significantly reduced expression of cleaved caspase-3, cleaved PARP, Bad, and Bax, together with increased expression of Bcl-2 and ration of Bcl-2/Bax. Both ratio of phospho-JNK/total JNK and ratio of phospho-c-jun/total c-jun were significantly decreased once MLK3 was silenced. At various reoxygenation time, MLK3 shRNA could significantly promote cell survival and decrease cell death according to MTT and LDH. Our results suggested that chronic hypoxia could reduce MLK3 expression in a posttranscriptional regulatory manner. Downregulation of MLK3 protects H9C2 cells from hypoxia-induced apoptosis and H/R injury via blocking the activation of JNK and c-jun.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congenital heart disease (CHD) is one of the most common congenital malformations in infants. Most of the complex CHDs are cyanotic, caused by right-to-left shunt in any level of the heart, in which chronic hypoxia is the basic pathophysiological process. Preliminary studies found that patients with cyanotic CHD are associated with few side effects and rarely progress to heart failure [15], although they have been in the condition of low-oxygen blood perfusion, suggesting that chronic hypoxia could probably generate compensatory adaptation. It has also been shown that chronic hypoxia confers cardiac protection against ischemia-reperfusion (I/R) and hypoxia/reoxygenation (H/R) injury [9, 17]. Therefore, it is urgently required to study the pathophysiological changes of cyanotic CHD and the underlying mechanisms of myocardial adaptation to chronic hypoxia, which will help to improve strategies for cardioprotection and enhance efficiency of clinical therapies.
Until now, limited evidence exists for elucidation of this adaptive mechanism. We have done a lot of efforts in this field. In our previous research, microRNA-138 (miR-138) was found to be increased in myocardial samples of patients with cyanotic CHD. By targeting mixed lineage kinase 3 (MLK3), upregulated miR-138 could significantly attenuate apoptosis induced by chronic hypoxia [7]. MLK3 is an important member of mitogen-activated protein kinase (MAPK) family, which plays an essential effect on cell survival and apoptosis. MLK3 is capable of phosphorylating c-jun N terminal kinase (JNK), and activated JNK could specifically bind to the amino terminal region of c-jun, thus enhancing the activity of transcription factors and promoting the expression of some apoptosis-related proteins. In cardiomyocytes, MLK3 expression was closely related to postnatal development with 7-fold increase in rat heart ventricle [12]. Ola and colleagues demonstrated that CEP-11004, a special inhibitor of MLK3, could block the activation of critical nuclear effectors (GATA-4 and activator protein 1) in the process of myocardial hypertrophy. In addition, CEP-11004 could also alleviate the agonist-induced atrial natriuretic peptide (ANP) secretion and activation of B-type natriuretic peptide (BNP) gene transcription [19]. All these results suggest suppressed activity of MLK3 may exert a protective effect on cardiomyocytes, which needs to be further investigated.
In the present study, we collected myocardial samples of patients with CHD and established in vitro chronically hypoxic cell model to detect the expression of MLK3. By using shRNA transfection, we aim to explore the role of MLK3 in cell apoptosis subjected to chronic hypoxia and clarify the potential mechanism of compensatory adaptation.
Materials and methods
Patient studied
The patients in cyanotic group should meet the inclusion criterion that the clinical diagnosis is cyanotic CHD and blood oxygen saturation is less than 85%, while the inclusion criterion in acyanotic group is that the clinical diagnosis is acyanotic CHD and blood oxygen saturation is greater than 95%. A total of 35 patients admitted to the Department of Cardiovascular Surgery, Chengdu Military General Hospital with CHD were eligible for inclusion in the study, of which 19 were acyonotic and 16 were cyanotic. All procedures were approved by hospital’s Human Ethical Committee and were performed in accordance with the Declaration of Helsinki. Informed consent was obtained from all individual participants included in the study.
Myocardial samples
Standardized anesthesia and surgical procedures were performed routinely as previously described. Muscle bundles were resected from right ventricular outflow tracts during operation and divided into two halves. Part of the sample was immediately snap frozen in liquid nitrogen and stored at −70 °C intended for Western blot or quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) analysis, while the other part was fixed in paraformaldehyde, embedded in paraffin and cut into sections intended for immunohistochemistry.
Cell culture and stimulation
Embryonic rat heart derived H9C2 cells were purchased from American Type Culture Collection (ATCC, USA) and were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, USA) with 10% fetal bovine serum (FBS; Gibco, USA). After starvation in serum-free medium overnight, cells in chronic hypoxia group were exposed to a gaseous mixture of 94% N2, 5% CO2, and 1% O2 and incubated in an Invivo200 cultivator (Ruskin Technology Ltd., UK) at 37 °C for 12, 24, 48, and 72 h, respectively. After hypoxic incubation for 72 h, cells were transferred to a cultivator containing 21% O2 for durations of 30 min, 1, 2, and 4 h to establish H/R model. Cells in control group were always maintained in normal condition (21% O2).
shRNA transfection
shRNA is a kind of small interfering RNAs that contain short hairpin structure. In the present research, shRNA was systhesized and cloned into a DNA vector named GV102 by GeneChem Co. Ltd. (Shanghai, China) to silence the expression of MLK3. MLK3 shRNA agents and Lipofectamine 2000 (Invitrogen, USA) were solved with serum-free Opti-MEM medium respectively for 5 min. Subsequently, the above reagents were mixed together and incubated at room temperature for 20 min. The mixtures were then added into each well and further cultured at 37 °C for 4 h. Transfection medium was removed followed by incubation with normal medium for 72 h. The constructed shRNA was encoded with green fluorescence protein (GFP) for determining the transfection efficiency. Transfection agent information and efficiency detection can be found in supplementary Fig. 1.
qRT-PCR analysis
Trizol reagent (Invitrogen, USA) was used to extract total RNA from cells. First strand cDNA synthesis was created by using PrimeScript RT reagent Kit purchased from Takara (Japan). SYBR Premix Ex Taq GC kit (Takara, Japan) was used to perform PCR analysis on Applied Biosystems 7500 Sequence Detection system (ABI, USA) according to manufacturer’s instructions. The primers of MLK3 and β-actin were synthesized by Sango (China) as follows: MLK3 (Homo), 5′- GTTCCGAGCCATCCAGTTG -3′ (forward), 5′- GACCTTCTCCTCCCATTCTG -3′ (reverse); MLK3 (Rat), 5′- GCTGTGGGAACTGCTGACTG -3′ (forward), 5′- TGGATGGGATGGGTAATGTT -3′ (reverse); β-actin (Homo), 5′- CAACTCCATCATGAAGTGTAAC -3′ (forward), 5′- CCACCACGGAGTACTTGCGCTC -3′ (reverse); β-actin (Rat), 5′- CTTAGACTATAGGCATGGACCT -3′ (forward), 5′- GCTAACGTTGCACGGTACGGAC -3′ (reverse). All reactions were run in triplicate. Results were calculated by fold changes relative to control subjects using 2-ΔΔCt methods.
Immunohistochemistry analysis
Myocardial samples were paraffin-embedded and cut at a thickness of 4 μm. Non-specific staining was blocked by using 10% normal goat serum for 20 min at room temperature. Sections were then incubated with primary antibody against human MLK3 (SantaCruz, USA) overnight at 4 °C. Subsequently, the slides were washed with phosphate-buffered saline (PBS) solution plus 0.1% polysorbate for three times followed by incubation for 1 h with the secondary antibody. A 3, 3′-diaminobenzidine (DAB) Kit (ZSGB-BIO, China) was used to develop the color reaction. Sections were then counterstained with Myer’s hematoxylin for 2 min. Images were captured using a microscope.
Western blotting
Total proteins were extracted by using sodium dodecylsulfate (SDS) lysis buffer (Beyotime, China), containing 50 mM Tris (pH 8.1), 1% SDS, sodium pyrophosphate, β-glycerophosphate, leupeptin, and other protease inhibitors, and then separated by SDS-PAGE gel electrophoresis in equal amounts. After the proteins were transferred to a polyvinylidene difluoride membrane (PVDF; Millipore, USA), the membranes were probed with the appropriated primary antibodies at 4 °C overnight. The following primary antibodies were used in the present study: polyclonal MLK3 (SantaCruz, USA) and caspase-3, poly ADP-ribose polymerase (PARP), Bad, Bcl-2, Bax, JNK, phospho-JNK, c-jun, phospho-c-jun (CST, USA). The phosphorylation site of c-jun is Ser73, while phospho-JNK antibody detects endogenous levels of p46 and p54 JNK dually phosphorylated at Thr183 and Tyr185. Polyclonal anti-β-actin (Bioss, China) was used as an internal control antibody. Subsequently, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies, and the bands were detected by ECL. Analysis was conducted by using Image J software.
MTT assay
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay was used to assess cell survival. Equal amounts of H9C2 cells were seeded in 96-well plates. After incubation for various hours, MTT (5 mg/ml; Sigma, USA) was added to each well. Subsequently, the plates were placed in incubator for further 4 h. Dimethyl sulfoxide (DMSO; Sigma, USA) was later added to solubilize the formazan crystals. The absorbance was measured at a wavelength of 490 nm with a microplate spectrophotometer.
LDH assay
The release of lactate dehydrogenase (LDH), a cytosolic enzyme, reflected a loss of membrane integrity in the damaged cells. The LDH assay kit (Beyotime, China) was used to detect cell death. After H9C2 cells were cultured in hypoxic conditions for 72 h, they were lysed by using LDH release reagents. LDH detection solution were then added into the supernatants and incubated in dark place at room temperature for 30 min. Absorbance was measured at 490 nm. The results were divided by the value of maximal LDH release in each group, which were expressed as cell death rate.
TUNEL assay
Cell apoptosis was quantified using a TdT-mediated dUTP biotin nick-end labeling (TUNEL) kit (Beyotime, China) labeled by Cy3 fluorescence. Cells seeded in slides were washed by PBS for three times, and then incubated in dark place at 4 °C for 60 min with TUNEL solution. The samples were further counterstained with Hoechst 33,528 (Beyotime, China) for 5 min at room temperature and examined by a fluorescent microscope. Six non-overlapping fields of vision were captured in each group. Apoptotic percentage was the ratio of TUNEL-positive nuclei to the total cell nuclei counterstaining by Hoechst.
Flow cytometry
Apoptosis was also detected by using annexin V kit (KeyGEN, China). H9C2 cells were collected, washed, and suspended in binding buffer. Phycoerythrin (PE)-conjugated annexin V and 7-amino-actinomycin D (7-AAD) were subsequently added to the cells. After incubation, binding buffer was added, and cells were analyzed by flow cytometry. Upper right quadrant reflects the proportion of necrotic cells whereas apoptotic cells appear in lower right quadrant.
Statistics
All statistical analyses were performed using SPSS 18.0 software. Data were presented as mean ± SEM. One-way analysis of variance (ANOVA) or t test was used to compare the difference between various groups. All values were obtained from at least three repeated experiments. All of the p values were two-tailed, and p < 0.05 was considered to be statistically significant.
Results
Characteristics of enrolled patients
All clinical information of enrolled patients was listed in Table 1. A total of 35 myocardial samples from 19 acyanotic and 16 cyanotic CHD patients were collected in the present study. In acyanotic group, 15 patients were diagnosed with tetralogy of Fallot (TOF) and the one remaining was ventricular septal defect (VSD) combined with pulmonary atresia (PA). All patients in cyanotic group underwent operations for VSD combined with right ventricular outflow tract obstruction (RVOS). The median blood oxygen saturation in acyanotic group was 97.3% which was greater than that in cyanotic group (78.9%). In addition, there was significant difference between two groups in preoperative hemoglobin concentration [197 (121–308) g/L vs. 131 (97–210) g/L, p < 0.05] and hematocrit [55.7 (41.9–68.4) % vs. 42.9 (34.2–51.0) %, p < 0.05]. Patients in two groups were well matched for age, gender, and weight.
Expression of MLK3 under hypoxic conditions
Previously, we have demonstrated that miR-138 was upregulated in hypoxic H9C2 cells. As the target of miR-138, the expression of MLK3 under hypoxic conditions was further studied in the present research. Firstly, RNA was extracted from myocardial samples of patients to test the expression of MLK3 mRNA. We found that its expression had no significant difference between cyanotic and acyanotic patients (Fig. 1a). The same result was observed in hypoxic cell model in vitro. H9C2 cells were cultured in hypoxic conditions for 0, 12, 24, 48, and 72 h, respectively. The difference of MLK3 mRNA expression did not appear with prolonged time of hypoxia (Fig. 1b). Subsequently, the expression of MLK3 protein was examined. In patients with cyanotic CHD, its protein expression was only 71% of that in patients with acyanotic CHD (Fig. 1c, #p < 0.05). In hypoxic H9C2 cells, the expression was found to be gradually decreased in a time-dependent manner (#p < 0.05). As shown in Fig. 1e, immunohistochemical analysis revealed that MLK3 was stably expressed in cytoplasm of cardiac cells, and cyanotic group had obviously lower expression compared with acyanotic group. All these data suggested that the expression of MLK3 was regulated by hypoxia in a posttranscriptional way, which was consistent with the regulatory mechanism of microRNA.

Expression of MLK3 in hypoxic cardiomyocytes. a qRT-PCR revealed that expression of MLK3 mRNA had no significant difference between cyanotic (n = 16) and acyanotic (n = 19) patients. b After H9C2 cells were cultured in hypoxic conditions for duration of 0, 12, 24, 48, and 72 h, respectively, there was still no significant difference about expression of MLK3 mRNA among various groups (n = 9). cWestern blot revealed that expression of MLK3 protein was significantly decreased in myocardial samples of patients with cyanotic CHD (#p < 0.05). d In hypoxic H9C2 cells, the expression of MLK3 protein was gradually downregulated in a time-dependent manner (#p < 0.05, n = 15). e Immunohistochemical analysis revealed that expression of MLK3 protein was obviously lower in patients with cyanotic CHD. Values were mean ± SEM
Hypoxia-induced downregulated MLK3 promoted cell survival
In order to analyze the potential role of MLK3 in hypoxic H9C2 cells, shRNA transfection was used to induce stable silencing of MLK3. Cell growth curve was drawn by using MTT assay at the time point of hypoxic 0, 12, 24, 48, and 72 h. Among three groups, MLK3-shRNA group had the fastest cell growth rate. At 72 h, difference reached the peak, and the amount of cells in MLK3-shRNA group was almost 1.4-fold of that in scramble-shRNA group (Fig. 2a, #p < 0.05). Cell death was examined by using LDH assay. After incubation in hypoxic conditions for 72 h, we found that once MLK3 expression was down-regulated by shRNA, the cell death rate was significantly decreased from 19.9 to 8.2% (Fig. 2b, #p < 0.05). Furthermore, TUNEL assay was used to evaluate the effect of MLK3 on apoptosis. As shown in Fig. 2d, chronic hypoxia led to the increase of TUNEL-positive H9C2 cells. This pro-apoptotic effect could be efficiently eased up by silencing of MLK3. The number of TUNEL-positive cells in MLK3-shRNA group was found to be only 38% of that in scramble-shRNA group (Fig. 2c, #p < 0.05). Taken together, all these findings strongly supported that hypoxia-induced downregulation of MLK3 is beneficial for cell survival.

Downregulated MLK3 was beneficial for cell viability in hypoxic conditions. a Cell survival curve was drawn by MTT assay, which revealed MLK3-shRNA group had the fastest growth rate (#p < 0.05, MLK3-shRNA group vs. scramble-shRNA group, n = 15). b Cell death was examined by LDH assay, which revealed death rate of H9C2 cells was lower in MLK3-shRNA compared with scramble-shRNA group (#p < 0.05, n = 15). c Cell apoptosis was evaluated by TUNEL assay, which revealed less apoptotic H9C2 cells could be observed in MLK3-shRNA group (#p < 0.05 vs. scramble-shRNA group, n = 9). d TUNEL-positive cells were labeled by Cy3 in red fluorescence; total cells were stained by Hoechst in blue fluorescence; percentage of apoptotic cells were calculated in merged picture. Each value was counted in six random fields. e A complementary flow cytometry-based assay was employed to assess apoptotic cell, showing that silencing of MLK3 could significantly reduce the proportion of apoptotic cells (#p < 0.05, MLK3-shRNA group vs. scramble-shRNA group, n = 9). f Flow cytometry via Annexin V-PE/7-AAD staining (a normoxia group, b hypoxia_control group, c hypoxia_MLK3-shRNA group, d hypoxia_scramble-shRNA group). *p < 0.05 vs. normoxia group. Values were mean ± SEM
Effect of MLK3 on expression of apoptosis-related proteins
To further verify the influence of MLK3 on apoptosis, we detected the expression of caspases as well as Bcl-2 family proteins by using Western blotting. Caspase-3 is a key monitor for cell apoptosis, and PARP is the direct substrate of caspase-3. When exposed to hypoxia for 72 h, the expression of cleaved caspase-3 and cleaved PARP was significantly upregulated, which could be attenuated in case of silencing of MLK3. Compared with corresponding scramble-shRNA group, the expression of cleaved caspase-3 in MLK3-shRNA group was reduced by 49% (Fig. 3b, #p < 0.05) while the expression of cleaved PARP had half decrease (Fig. 3c, #p < 0.05). Bcl-2 family proteins are thought to play a crucial role in regulation of cell apoptosis, so they are also chosen as the study objects. We found that transfection of MLK3 shRNA could significantly decrease the expression of Bad (Fig. 3d, #p < 0.05 vs. scramble-shRNA group) and Bax (Fig. 3f, #p < 0.05 vs. scramble-shRNA group). The expression of Bcl-2 in MLK3-shRNA group was obviously upregulated compared with scramble-shRNA group (Fig. 3e, #p < 0.05). The same trend could be observed in the ratio of Bcl-2/Bax. In the MLK3-shRNA group, this ratio was 3.5-fold of that in the scramble-shRNA group (Fig. 3g, #p < 0.05). Flow cytometry results revealed that the amount of apoptotic cells in all hypoxic groups greatly increased after exposure for 72 h. When MLK3 was silenced by shRNA in hypoxic H9C2 cells, the pro-apoptotic actions were significantly weakened compared with scramble-shRNA group (Fig. 2e, f, #p < 0.05). All above results indicated that MLK3 has an efficient pro-apoptotic role in hypoxic H9C2 cells.

Effect of MLK3 on expression of apoptosis-related proteins. a Western blot results showed relative expression of proteins in both normoxic and hypoxic conditions at 72 h. Silencing of MLK3 significantly reduced the expression of cleaved caspase-3 (b), cleaved PARP (c), Bad (d), and Bax (f), together with increased expression of Bcl-2 (e), and ratio of Bcl-2/Bax (g). #p < 0.05, MLK3-shRNA group vs. scramble-shRNA group (n = 5). *p < 0.05 vs. normoxia group. β-actin was used as an internal control. Values were means ± SEM
Effect of MLK3 on JNK/c-jun pathway in hypoxic conditions
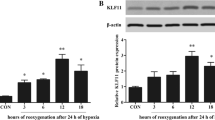
MLK3 is supposed to exert its role by phosphorylating the downstream proteins JNK and c-jun. We speculate it is probably the mechanism that MLK3 functions in hypoxic cardiomyocytes. So we further examined the activation of JNK and c-jun. Western blotting results revealed that the expression of phosphorylated JNK and c-jun was significantly increased after exposure to hypoxia after 72 h. Once MLK3 was silenced by shRNA, this activation could be efficiently blocked. In MLK3-shRNA group, the ratio of phospho-JNK/total JNK was 46% of that in scramble-shRNA group (Fig. 4a, #p < 0.05). The ratio of phospho-c-jun/total c-jun was also decreased by 56% compared with its scramble-shRNA group (Fig. 4b, #p < 0.05). Therefore, this finding reflects that it is via activation of JNK/c-jun pathway that MLK3 involves in the myocardial adaptation to chronic hypoxia.

Silencing of MLK3 inhibited activation of JNK/c-jun pathway, and attenuated H/R injury in hypoxic cardiomyocytes. a Western blot results showed relative expression of proteins in both normoxic and hypoxic conditions at 72 h. MLK3 shRNA significantly decreased ratio of phospho-JNK/total JNK (b) and ratio of phospho-c-jun/total c-jun (c) (#p < 0.05, MLK3-shRNA group vs. scramble-shRNA group, n = 5). After 72 h for hypoxia, H9C2 cells were subjected to normoxia for 30 min, 1, 2, and 4 h, respectively. Time at the initiation of reoxygenation was set as the base line. MLK3-shRNA group had higher cell growth rate (d) and lower death rate (e) compared with scramble-shRNA group at every time point (n = 15). *p < 0.05 vs. normoxia group. Values were means ± SEM
Downregulation of MLK3 alleviated H/R injury
Given that myocardial adaptation to chronic hypoxia may attenuate H/R injury, we next determined whether hypoxia-induced down-regulated MLK3 had this protective role. Time at the initiation of reoxygenation was set as the base line. According to MTT results, we found that reoxygenation made cell viability decreased sharply. At every time point, cell survival rate in MLK3-shRNA group was significantly higher than scramble-shRNA group (Fig. 4d). Opposite trend could be observed in LDH results (Fig. 4e). This demonstrated that silencing of MLK3 could protect cardiomyocytes from reoxygenation injury after exposure to chronic hypoxia.
Discussion
Chronic hypoxia is the common pathophysiological characteristic in patients with CHD and those who live in high altitude region. Mitochondrion is most sensitive cell organelle to oxygen. Hypoxia could cause the dysfunction of its oxidation leading to the lack of energy. And then cytochrome c and some proteins may be released from the damaged membrane, thus making an end with apoptosis and pathological remodeling [5]. On the other hand, compensatory adaptative responses are spontaneously evoked. Some potassium channels sensitive to ATP will be activated in hypoxic conditions, combined with changes of ROS, NO, and some protein kinases. The increased oxygen release would balance energy supply and demand, thereby creating a state of cell homeostasis and hypoxic tolerance [13]. In previous literature, several signaling pathways were demonstrated to be involved in chronically hypoxic adaptation. Chronic hypoxia induced increased expression of migration inhibitory factor (MIF) and activation of 5′-adenosine monophosphate activated protein kinase (AMPK) in metabolic process [10]. Suppressor of cytokine signaling 3 (SOCS3) was proven to be a rapid hypoxia-inducible gene and acted to regulate cell signaling crosstalk between NF-κB and STAT3 in a negative feedback loop during chronic hypoxia [4]. Activating transcription factor 6α (ATF6α) was increased in hypoxic cardiomyocytes, which could alleviate endoplasmic reticulum (ER) stress via facilitating ER proteins folding [9] and attenuate myocardial I/R injury through regulation of Akt signaling pathway [8]. MicroRNA-138 [7] and microRNA-146b [14] were two key factors that protected cardiomyocytes from hypoxia-induced apoptosis. Erythropoietin (EPO) and its receptor were significantly upregulated in patients with cyanotic CHD, and EPO treatment could enhance mitochondrial biogenesis in cardiomyocytes exposed to chronic hypoxia, partly through Akt/eNOS signaling [20]. Therefore, studying the molecular regulatory mechanisms will provide a new target for clinical myocardial protection.
MLK3 was previously demonstrated to be one of the targets of miR-138 and was supposed to potentially participate in chronically hypoxic adaptation [7]. So in the present study, we intend to further analyze the performance of MLK3 under hypoxic conditions. Firstly, we found that expression of MLK3 protein was decreased as expected in hypoxic H9C2 cells whereas mRNA expression had no significant changes. As we all know, microRNAs exert its function by controlling the translation of mRNAs instead of directly affecting expression level of mRNAs [3]. Therefore, our results confirmed that MLK3 was regulated by miR-138 in a posttranscriptional manner. MLK3 is the best characterized of MAPK family in terms of its biochemical functions. The role of MLK3 in cell death and apoptosis is well established and has been extensively reviewed. Activation of MLK3 and its downstream cascades were considered to be crucial for amyloid-β peptide mediated cell apoptosis [22]. The hepatitis B virus X protein contributed to apoptosis of renal tubular cells, and recent studies indicated that this process depended on the activation of MLK3 signaling module [6]. MLK3 inhibitors could efficiently prevent neuronal cell death and neurodegenerative disease progression [18]. In our research, silencing of MLK3 was achieved by transfection of shRNA into hypoxic H9C2 cells to verify its role in myocardial adaptation. It was observed that MLK3 shRNA could significantly attenuate cell apoptosis, implying that hypoxia-induced downregulation of MLK3 was a protective response to chronic hypoxia and was beneficial for cell survival. Although we intend to evaluate the protective role of down-regulated MLK3 in cardiomyocytes, using shRNA for downregulation of already downregulated MLK3 is definitely notable limitation in the present study. Over-expression interference to explore more comprehensive functions of MLK3 is urgently needed in future research.
MLK3 functions as MAP3Ks to activate specific MAP2ks, and then in turn phosphorylate specific MAPKs, of which JNK is the well-described one. Once activated, JNK will be translocated from cytoplasm to nucleus and enhance the activity of transcription factor c-jun, thus promoting the expression of apoptosis-related proteins [1]. In addition, the absence of JNK induces the release of cytochrome c from mitochondrion to cytoplasm and interacts with caspase-3 to accelerate cell apoptosis [21]. In order to elucidate the mechanism that MLK3 was involved in myocardial adaptation, we subsequently examined the activation of its downstream proteins, JNK and c-jun. We demonstrated that silencing of MLK3 could significantly reduce the ratio of phosphorylated JNK and c-jun under chronically hypoxic conditions. Although the upstream protein MLK3 was decreased in hypoxic cardiomyocytes, JNK and c-jun was still found to be activated probably because of the adaptive regulatory mechanism of other signaling pathways. Actually, JNK signaling has already been found to be associated with numerous cellular responses in heart. The activation of JNK signaling could cause characterized changes of myocardial hypertrophy, leading to serious disorder of cardiac function [2]. Specific JNK inhibitors are capable of reversing cardiac remodeling, reducing the expression of proteins related to hypertrophy [16], improving myocardial systolic ability, and alleviating cardiac injury caused by ischemia [11]. In clinical, myocardium of patients with cyanotic CHD will experience H/R injury once heart malformations are surgically corrected. JNK signaling is supposed to be a key regulator in the process of H/R injury. Since MLK3 is the upstream molecule of JNK, we speculate cell performance will certainly influenced by MLK3. So in the present study, we further established the H/R model in vitro and demonstrated that MLK3 shRNA could significantly promote cell survival and decrease cell death according to MTT and LDH, suggesting downregulation of MLK3 could efficiently attenuate H/R injury. Accordingly, we suppose that suppressed activation of MLK/JNK/c-jun pathway is probably a protective mechanism in hypoxic H9C2 cells. This finding will give us a new insight and strategy for improving clinical treatments.
In summary, the present study firstly demonstrated that MLK3 was lowly expressed in patients with cyanotic CHD. Chronic hypoxia induced the downregulation of MLK3 in a posttranscriptional way. Decreased expression of MLK3 was beneficial for cell survival and protected H9C2 cells from hypoxia-induced apoptosis. Our results suggested the involvement of MLK3/JNK/c-jun signaling pathway in adaptation to chronic hypoxia, thus providing a potential target for clinical cardioprotection.
Change history
05 October 2017
Volume 73 issue 3 was published with an incorrect cover date. Correct is August 2017. The Publisher apologizes for this mistake and all related inconveniences caused by this.
References
Abdelli S, Abderrahmani A, Hering BJ, Beckmann JS, Bonny C (2007) The c-Jun N-terminal kinase JNK participates in cytokine- and isolation stress-induced rat pancreatic islet apoptosis. Diabetologia 50:1660–1669
Cao S, Zeng Z, Wang X, Bin J, Xu D, Liao Y (2013) Pravastatin slows the progression of heart failure by inhibiting the c-Jun N-terminal kinase-mediated intrinsic apoptotic signaling pathway. Mol Med Rep 8:1163–1168. doi:10.3892/mmr.2013.1622
Esteller M (2011) Non-coding RNAs in human disease. Nat Rev Genet 12:861–874
Gu Q, Kong Y, Yu ZB, Bai L, Xiao YB (2011) Hypoxia-induced SOCS3 is limiting STAT3 phosphorylation and NF-kappaB activation in congenital heart disease. Biochimie 93:909–920
Han Q, Yeung SC, Ip MS, Mak JC (2014) Cellular mechanisms in intermittent hypoxia-induced cardiac damage in vivo. J Physiol Biochem 70:201–213
He P, Zhang B, Liu D, Bian X, Li D, Wang Y, Sun G, Zhou G (2016) Hepatitis B virus X protein modulates apoptosis in NRK-52E cells and activates Fas/FasL through the MLK3-MKK7-JNK3 signaling pathway. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 39:1433–1443
He S, Liu P, Jian Z, Li J, Zhu Y, Feng Z, Xiao Y (2013) miR-138 protects cardiomyocytes from hypoxia-induced apoptosis via MLK3/JNK/c-jun pathway. Biochem Biophys Res Commun 441:763–769
Jia W, Jian Z, Li J, Luo L, Zhao L, Zhou Y, Tang F, Xiao Y (2016) Upregulated ATF6 contributes to chronic intermittent hypoxia-afforded protection against myocardial ischemia/reperfusion injury. Int J Mol Med 37:1199–1208
Jian Z, Li JB, Ma RY, Chen L, Wang XF, Xiao YB (2012) Pivotal role of activating transcription factor 6alpha in myocardial adaptation to chronic hypoxia. Int J Biochem Cell Biol 44:972–979
Jian Z, Li JB, Ma RY, Chen L, Zhong QJ, Wang XF, Wang W, Hong Y, Xiao YB (2009) Increase of macrophage migration inhibitory factor (MIF) expression in cardiomyocytes during chronic hypoxia. Clinica chimica acta; international journal of clinical chemistry 405:132–138
Kaiser RA, Liang Q, Bueno O, Huang Y, Lackey T, Klevitsky R, Hewett TE, Molkentin JD (2005) Genetic inhibition or activation of JNK1/2 protects the myocardium from ischemia-reperfusion-induced cell death in vivo. J Biol Chem 280:32602–32608
Kim SO, Irwin P, Katz S, Pelech SL (1998) Expression of mitogen-activated protein kinase pathways during postnatal development of rat heart. J Cell Biochem 71:286–301
Kolar F, Ostadal B (2004) Molecular mechanisms of cardiac protection by adaptation to chronic hypoxia. Physiological research / Academia Scientiarum Bohemoslovaca 53(Suppl 1):S3–13
Li JW, He SY, Feng ZZ, Zhao L, Jia WK, Liu P, Zhu Y, Jian Z, Xiao YB (2015) MicroRNA-146b inhibition augments hypoxia-induced cardiomyocyte apoptosis. Mol Med Rep 12:6903–6910
Li X, Liu Y, Ma H, Guan Y, Cao Y, Tian Y, Zhang Y (2016) Enhancement of glucose metabolism via PGC-1alpha participates in the cardioprotection of chronic intermittent hypobaric hypoxia. Front Physiol 7:219
Liang Q, Molkentin JD (2003) Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. J Mol Cell Cardiol 35:1385–1394
Ma HJ, Li Q, Ma HJ, Guan Y, Shi M, Yang J, Li DP, Zhang Y (2014) Chronic intermittent hypobaric hypoxia ameliorates ischemia/reperfusion-induced calcium overload in heart via Na/Ca2+ exchanger in developing rats. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 34:313–324
Mishra P, Senthivinayagam S, Rangasamy V, Sondarva G, Rana B (2010) Mixed lineage kinase-3/JNK1 axis promotes migration of human gastric cancer cells following gastrin stimulation. Mol Endocrinol 24:598–607
Ola A, Kerkela R, Tokola H, Pikkarainen S, Skoumal R, Vuolteenaho O, Ruskoaho H (2010) The mixed-lineage kinase 1-3 signalling pathway regulates stress response in cardiac myocytes via GATA-4 and AP-1 transcription factors. Br J Pharmacol 159:717–725
Qin C, Zhou S, Xiao Y, Chen L (2014) Erythropoietin enhances mitochondrial biogenesis in cardiomyocytes exposed to chronic hypoxia through Akt/eNOS signalling pathway. Cell Biol Int 38:335–342
Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288:870–874
Zhou F, Xu Y, Hou XY (2014) MLK3-MKK3/6-P38MAPK cascades following N-methyl-D-aspartate receptor activation contributes to amyloid-beta peptide-induced apoptosis in SH-SY5Y cells. Journal of neuroscience research.
Acknowledgements
The present work was supported by the project of youth scientific and technological innovation in Chengdu Military General Hospital (No. 41732C11K) and technological project of Sichuan health and family planning commission (No. 16PJ022).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
All procedures were approved by hospital’s Human Ethical Committee and were performed in accordance with the Declaration of Helsinki. Informed consent was obtained from all individual participants included in the study.
Additional information
An erratum to this article is available at https://doi.org/10.1007/s13105-017-0593-x.
Electronic supplementary material

Supplementary Fig 1
Transfection agents information and efficiency detection. (A) The transfection carrier atlas. (B) The constructed shRNA was encoded with green fluorescence protein (GFP). Almost all transfected cardiomyocytes were emitting green light by using a fluorescence microscope. Expression of MLK3 mRNA in MLK3-shRNA group was only 11% of that in scramble-shRNA group (C) (#p < 0.05, n = 9), while protein expression was decreased to 36% once MLK3 shRNA was transfected (D) (#p < 0.05, n = 3). Values were means ± SEM. (JPEG 615 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
He, S., Liu, S., Wu, X. et al. Protective role of downregulated MLK3 in myocardial adaptation to chronic hypoxia. J Physiol Biochem 73, 371–380 (2016). https://doi.org/10.1007/s13105-017-0561-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-017-0561-5