
Abstract
KLF11 is a Krüppel-like factor (KLF) family member, which plays a central role in cardiac hypertrophy and cerebrovascular protection during ischemic insults. However, the roles of KLF11 in hypoxia/reoxygenation (H/R) injury of rat cardiomyocytes H9c2 have not been elucidated. The aim of this study was to evaluate the effects of KLF11 on H/R injury and investigate the molecular mechanisms involved. Here, we found that KLF11 was increased following H/R and reached the highest level with 24 h hypoxia followed by 12 h reoxygenation. Moreover, we found that inhibition of KLF11 by small RNA suppressed cell apoptosis, the activity of caspase3, the expression of cleaved-caspase3 and cytochrome C in the cytoplasm and the damage of mitochondrial membrane induced by H/R in H9c2, suggesting that KLF11 silencing protects against H/R injury. In addition, we observed that knockdown of KLF11 elevated the expression of p-JAK2 and p-STAT3 in H9c2, and AG490, a selective inhibitor of JAK2/STAT3 abrogated the potential roles of KLF11 in cell apoptosis and mitochondrial damage. In aggregates, our results showed that depletion of KLF11 protected H9c2 against H/R injury through activating the JAK2/STAT3 signaling pathway, suggesting that KLF11 may be provide therapeutic targets for H/R or other heart diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases remains the leading cause of death in middle-aged people worldwide [1]. Cardiac dysfunction induced by myocardial infarction is one of the most challenging clinical questions. Myocardial ischemia–reperfusion (I/R) caused acute myocardial infarction; Moreover, an abrupt rise in reactive oxygen species (ROS) induced by mitochondria during reperfusion [2] causes further cardiomyocyte apoptosis and mitochondrial damage [3, 4]. Therefore, targeted inhibition of cell apoptosis and mitochondrial damage may be effective in the prevention of I/R injury.
A variety of signaling pathways are involved in the regulation of I/R. Janus kinase (JAKs) proteins are a kind of cytosolic tyrosine kinase associated with the intracellular domain of membrane-bound receptors, which could transduce signals from ligands to the nucleus, activating downstream signaling pathways [5]. There are four family members: JAK1, JAK2, JAK3 and tyrosine kinase (TYKS), which show different receptor affinities. However, all of them transduce signals via the recruitment of signal transducers and activators of transcription (STAT) [6]. It is believed that the JAK/STAT pathway has an important role in diverse cardiac pathophysiologic processes, including hypertrophy, apoptosis and I/R injury [7]. SOCS1 gene transfer accelerates heart failure by inhibiting the JAK/STAT pathway [8]. Postconditioning reduces infarct size and myocyte apoptosis by activating the JAK/STAT signaling pathway [9]. Notably, the majority of STAT activity in the heart is confined to STAT3, which is a key regulator of cell to cell communication in the heart [10].
Krüppel-like transcription factors (KLFs) are zinc finger-containing transcription factors that were implicated in physiological and pathological process, including proliferation, differentiation and cell death [11, 12]. KLF11 (TIEG2) is a member of the KLFs family, and plays a critical role in cerebrovascular protection during ischemic insults [13]. KLF11 is an important regulator in hepatic lipid metabolism [14]. In addition, mutations of KLF11 induced MODY VII diabetes [15]. Moreover, it has been reported that KLF11 suppressed cardiac hypertrophy and fibrosis [16]. However, the roles of KLF11 in cellular apoptosis and mitochondrial damage induced by H/R are unknown.
In the present study, we found higher levels of KLF11 following H/R than in the control group in H9c2 cells. The roles of KLF11 in H/R injury and the underlying mechanisms were determined. Knockdown of KLF11 suppressed cell apoptosis and mitochondrial damage by activating the JAK2/STAT3 pathway. Our results illustrate the roles of KLF11 in regulating cell apoptosis, mitochondrial membrane potential and the expression of Cyto C of H9c2 following H/R. This study provided a new strategy and targets for I/R in the heart.
Methods
Cell culture and establishment of H/R models
The rat embryonic cardiomyoblast cell line H9c2 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were maintained in DMEM (Invitrogen, CA, USA) supplemented with 10% (v/v), H-glutamine (5 mM, Gibco, CA, USA) or L-glutamine (1 mM), fetal bovine serum (FBS, Gibco, CA, USA), 100 U/ml penicillin (Sigma-Aldrich, Inc., Saint Louis, MO, USA) and 100 mg/ml streptomycin (Sigma-Aldrich), at 37 °C (5% CO2 and 95% air). When cells reached about 80% confluence in culture, cells were treated with medium containing 0.5% FBS for 12 h before every experiment. To induce H/R injury, H9c2 cells were cultured in serum-free and L-glucose DMEM for 24 h in a hypoxia chamber (37 °C, 1% O2/94% N2), which was followed by various periods of reoxygenation (3–18 h) during which H9c2 cells were cultured in serum-free DMEM in a normal chamber (37 °C, 5% CO2/95% air). Cells were divided into following groups: CON group (untreated cells); H/R group [Cells were exposed to hypoxia/reoxygenation (24/6 h)]; NC group (Cells were transfected with NC for 48 h); KLF11 siRNA group (Cells were transfected with KLF11 siRNA for 48 h); NC + H/R group (Cells were transfected with negative control of siRNA following by exposed to H/R); KLF11 siRNA + H/R group (Cells were transfected with KLF11 siRNA followed by exposed to H/R); AG490 + KLF11 siRNA + H/R group (Cells were pretreated with AG490 for 30 min, transfected with KLF11 siRNA followed by exposed to H/R). AG490 + H/R group (cells were pretreated with 30 min followed by exposed to H/R).
Transfection
H9c2 cells were plated into six-well plates at about 50% confluence before transfection. The negative control (NC for siRNA) and KLF11 siRNA were bought from ABI (Abilene, Texas, USA). Cells were transfected with NC, KLF11 siRNA (50 nM) using lipofectamine 2000 (Invitrogen, Camarillo, CA, USA) for 5 h, then added to DMEM containing FBS for 48 h, according to the manufacturer’s manual. The transfection efficiency of the cells was determined by RT-PCR.
Measurement of LDH activity
The LDH-Cytotoxicity assay kit (BioVision, Palo Alto, CA, USA) was used to determine the LDH activity according to the manufacturer’s instruction. LDH activity can be easily quantified by a spectrophotometer or plate reader at OD 450 nm.
Apoptosis detection
Apoptosis was determined by the Annexin V-FITC Apoptosis Dectection Kit (BD Biosciences, San Diego, CA) following the manufacturer’s protocol. Briefly, the cells were washed thrice with chilled PBS buffer, then trypsinizedwith trypsin, centrifuged with 21×g for 5 min, resuspended in 200 µL binding buffer, and stained with 5 µL Annexin V-FITC and 10 µL propidium iodide (PI) for 15 min at room temperature in the dark. A FACSCcan Flow Cytometer (BD, Franklin Lakes, NJ) was used to assay the fluorescent signals. To detect JAK2/STAT3 whether involved in the cardioprotective effect of KLF11 inhibition, H9c2 were cells were incubated in serum free medium for 12 h then exposed to H/R with or without AG490 (Sigma Chemicals, St Louis, MO, USA; 50 μM).
Caspase3 activity detection
The Caspase-3/CPP32 fluorometric assay kit (BioVision, Palo Alto, CA, USA) was used to measure the activity of caspase3 according to the manufacturer’s protocol. Cells were grown in six-well plates. After the appropriate treatment, cells were treated with 50 µL cell lysis buffer on ice for 10 min, and then the lysis solution was collected and centrifuged at 4800×g for 15 min. The supernatant was transferred in to a new microtube. Then, 50 µL of 2× reaction buffer and 5 µL of 1 mM DEVD-AFC sbustrate (50 µM, final) was added and the solution was incubated at 37 °C for 2 h. After incubation, 100 µL of each sample was transferred to 96-well plates. Samples were read in a fluorometer equipped with a 400 nm excitation filter and 505 nm emission filter. The protein concentration was assayed using the BCA Protein Assay Kit (Beyotime, China) to normalize the expression of c-caspase3.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential was assayed by staining with the potential-sensitive fluorescent dye JC-1 (Sigma-Aldrich, Inc., Saint Louis, MO, USA) and determined by flow cytometry. Briefly, H9c2 cells were incubated with JC-1 (5 μM) for 15 min at 37 °C, The stained cells were washed with PBS buffer, and analyzed by flow cytometry (Becton Dichinson FACScan, BD, Franklin Lakes, NJ). JC-1 monomers emit at 527 nm and “J-aggregates” emit at 590 nm.
RNA extraction and RT-PCR
Total RNA was isolated using the Trizol regent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. The concentration of RNA was assayed by a microplate reader, and then 5 μg of RNA was reverse-transcribed to cDNA with specific primers. Quantitative real-time PCR analysis was performed on an ABI Step One Plus sequence detection system (Life Technologies) using the conditions and primer concentrations suggested by the SYBR Green PCR master mix (Life Technologies) protocol. GAPDH was used as an internal control. Each experiment was carried out in triplicate. Differences in gene expression, reported as fold changes, were calculated using the 2−ΔΔCt method.
The following primers were used:
Gene | Sequence (5′-3′) |
|---|---|
KLF11 | CTCCTGCAGGGCCGTGATGAC |
GGGGAACAGGCCACCAGACTTG | |
GAPDH | TCCCTCAAGATTGTCAGCAA |
AGATCCACAACGGATACATT |
Subcellular fraction
To detect the Cyto C leaked from mitochondrial, a cytosolic and mitochondrial fraction kits (Beyotime, China) was used to separate the protein into cytosolic and mitochondrial fraction. Briefly, Cells were harvested and incubated with mitochondrial isolation regents at 4 °C. The lysates were centrifuged at 600×g for 15 min. The supernatants were collected and centrifuged at 10,000×g for 30 min. The mitochondrial fraction was contained in the pallets, while the cytosolic fraction was in supernatant.
Western blotting
After the proper treatment, total proteins were extracted and quantified. 30 µg proteins from each sample was separated by SDS-PAGE gel electrophoresis and transferred to a polyvinylidene fluoride (PVDF) membrane. All membranes were blocked with 5% milk or BSA and incubated with anti-KLF11 antibody, anti-Cyto C antibody, anti-COX IV antibody, anti-p-JAK2 antibody, anti-t-JAK2 antibody, anti-p-STAT3 antibody and anti-t-STAT3 antibody (Abcam, Cambridge, MA, USA), anti-c-caspase3 antibody (CST, Danvers, MA, USA) and anti-β-actin antibody (Santa Cruz, CA, USA) overnight at 4 °C then with horseradish peroxidase-conjugated secondary antibody (ZSGB, China) for 1 h at room temperature. Proteins were assayed with an enhanced chemiluminescence kit (Thermo Scientific, Waltham, MA, USA). OD measurements were then normalized to β-actin readings.
Statistical analysis
Statistical analysis was performed using the SPSS 11.0 software package. Quantitative data from at least three experiments were compared. Differences of more than two groups were analyzed by one way ANOVA, followed by Newman-Keuls test. A value of P < 0.05 was considered statistically significant.
Results
The expression level of KLF11 was increased following H/R
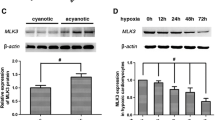
After H9c2 cells were exposed to various times of reoxygenation (3, 6, 12 and 18 h) followed by 24 h hypoxia, the expression of KLF11 was assessed using RT-PCR and Western blot. We found that H/R injury significantly increased KLF11 expression both in mRNA (Fig. 1a) and protein (Fig. 1b) levels when compared with the controal group. Following exposure to 24 h hypoxia and 3 h reoxygenation, the mRNA levels of KLF11 increased 3.21-fold compared with that of the controlgroup (Fig. 1a), while 6, 12 and 18 h of reoxygenation led the mRNA levels of KLF11 to increase up to 3.92-, 7.19- and 5.06-fold respectively (Fig. 1a). In addition, cells were exposed to 24 h hypoxia and 3 h reoxygenation, and the protein expression of KLF11 increased 1.53-fold, but there is no significant difference compared with 6 h reoxygenation. Exposure to 12 and 18 h reoxygenation led to the protein levels of KLF11 increasing up to 2.87- and 2.16-fold, respectively (Fig. 1b). In particular, when H9c2 cells were subjected to 24 h hypoxia and 12 h reoxygenation, the KLF11 expression level reached the highest level. Based on these results, treatment with 24 h hypoxia followed by 12 h reoxygenation was selected for experiments to examine the potential role of KLF11 in H/R injury.

The expression of KLF11 was increased following H/R. a The mRNA levels of KLF11 after 24 h hypoxia and various times of reoxygenation. b The protein expression of KLF11 after 24 h hypoxia and various times of reoxygenation. CON control, *P < 0.05, **P < 0.01 vs. CON group
Knockdown of KLF11 inhibited cell apoptosis induced by H/R
LDH is a stable enzyme that is rapidly released into the medium upon injury of cell plasma membrane. To determine the effects of KLF11 on H/R injury in H9c2, we detected LDH activity, apoptosis rate, caspase3 activity and the expression of cleaved-caspase3 (c-caspase3). KLF11 silencing markedly reduced KLF11 expression in H9c2 cells (Fig. 2a). Control = 1.12 activity compared to H/R treatment = 4.15 activity, suggesting that H/R induced cell damage. Compared with the NC + H/R = 4.32 activity, the depletion of KLF11 (1.53 activity) obviously inhibited LDH activity caused by H/R treatment (Fig. 2b). In the H/R group, the expression levels of c-caspase3 obviously increased in comparison to that of the CON group; however, the knockdown of KLF11 reduced c-caspase3 expression compared with the NC + H/R group (Fig. 2c). Moreover, H/R caused cell apoptosis increased up to (43.40 ± 2.35%) compared with the CON group (10.65 ± 0.29%), as well as in the NC + H/R group (40.94 ± 3.62%); however, the depletion of KLF11 reduced cell apoptosis (15.63 ± 3.08%) (Fig. 2d). In addition, we observed that caspase3 activity was increased following H/R; however, the increased activity of caspase3 could be reduced by the depletion of KLF11 (Fig. 2e). These data illustrate that the knockdown of KLF11 suppressed cell apoptosis induced by H/R in H9c2 cells.

Depletion of KLF11 inhibited cell apoptosis and mitochondrial damage induced by H/R in H9c2. a The expression of KLF11 in H9c2 cells transfected with either NC or KLF11 siRNA plasmid. b The LDH activity was measured using ELISA. c The caspase3 activity was detected using ELISA. d Apoptosis rate was detected by flow cytometry. e Representative blot of c-caspase3 levels was assayed using western blot. f Mitochondrial membrane potential was detected using flow cytometry after staining with JC-1. g, h Representative blot of Cyto C levels was assayed using western blot in plasma (g) and mitochondrial (h). CON control, *P < 0.05, **P < 0.01 vs. NC or CON group, # P < 0.05 vs. NC + H/R group
H/R caused mitochondrial dysfunction resulting in altered mitochondrial membrane potential; mitochondrial damage may initiate apoptosis by releasing pro-apoptotic factors, such as Cyto C, from the mitochondrial intermembrane space into the cytoplasm [17]. To further study the roles of KLF11 in H/R, we assessed the mitochondrial membrane potential and Cyto C expression. Our results showed that H/R-induced mitochondrial membrane potential decreased compared with the CON group, and there was no difference between the H/R group and the NC + H/R group; however, downregulation of KLF11 prevented the decrease in mitochondrial membrane potential (Fig. 2f). Moreover, H/R resulted in the augment of Cyto C in the cytoplasm (Fig. 2g) while reduction in mitochondrial (Fig. 2h) compared with CON group. Knockdown of KLF11 markedly attenuated Cyto C in cytoplasm while increased in mitochondrialafter exposure to H/R compared with the NC + H/R group. The results suggested that the depletion of KLF11 reduced mitochondrial damage in H9c2 cells.
Knockdown of KLF11 activated the JAK2/STAT3 pathway
To determine the mechanisms of KLF11 on H/R injury, we investigated the expression of p-JAK2 and p-STAT3 after the inhibition of KLF11. The total level of JAK2 and STAT3 in each group of cells remained unchanged. As shown in Fig. 3, compared with the CON group, the p-JAK2 was increased significantly. In addition, there is no obvious change of p-STAT3 in the H/R group compared with CON group. However, the protein levels of p-JAK2 and p-STAT3 were greatly elevated in the KLF11 siRNA + H/R group compared with the NC + H/R group, and AG490 significantly reduced the expression of p-JAK2 and p-STAT3. These data show that KLF11 activated the JAK2/STAT3 signaling pathway in H9c2 cells.

Depletion of KLF11 activated JAK2/STAT3 pathway. Representative blot of p-JAK2 and p-STAT3 levels were assayed using western blot. CON control, & P < 0.05 vs. CON group, *P < 0.05 vs. NC + H/R group, # P < 0.05 vs. KLF11 siRNA + H/R group
The JAK2/STAT inhibitor AG490 reversed the effect of KLF11 on cell apoptosis and mitochondrial damage
The JAK2/STAT3 pathway has been shown to play an important role in multiple processes within the heart, including hypertrophy and ischemia–reperfusion (I/R) injury [18]. To investigate whether the JAK2/STAT3 pathway is involved in the regulation of KLF11 in H/R injury, cells were pretreated with AG490, which is a selective inhibitor; therefore, we evaluated cell apoptosis, mitochondrial membrane potential and the expression of Cyto C in cytosolic and mitochondrial fraction. There is no effect of AG490 on the expression of KLF11 (Supplementary data 1). Compared with NC + H/R group (39.82 ± 4.29%), knockdown of KLF11 (21.73 ± 1.06%) caused the reduction of apoptosis rate. However, AG490 (36.58 ± 2.38%) abrogated the inhibitory effect of KLF11 on apoptosis (Fig. 4a). Moreover, the apoptosis rate was increased in AG490 + H/R (49.37 ± 4.19%) group compared with H/R group (37.81 ± 3.27%), and depletion of KLF11 suppressed the injury action of AG490 exposed to H/R (Fig. 4a), suggesting a dependence on JAK2/STAT3 for KLF11 siRNA-mediated cardioprotection. The activity of caspase3 (Fig. 4b) was assayed, as well as c-caspase3 expression (Fig. 4c); as expected, AG490 abolished the roles of KLF11 in apoptosis induced by H/R. In addition, we observed the activity of caspase3 was obviously enhanced in AG490 + H/R group compared with H/R group, but depletion of KLF11 resulted in the reduction of caspase3 acitviy (Fig. 4b). Moreover, AG490 pretreatment increased the mitochondrial membrane potential compared with the KLF11 siRNA + H/R group (Fig. 4d). There is no difference of mitochondrial membrane potential between AG490 + H/R group and H/R group, however, KLF11 silenced caused the augment of mitochondrial membrane potential compared with AG490 + H/R group. The expression of Cyto C in the cytoplasm was reduced (Fig. 4e) while conversely elevated in mitochondrial fraction in the KLF11 siRNA + H/R group, but AG490 reversed the effects of KLF11 on Cyto C expression (Fig. 4e, f). These results indicated that the protective effects of KLF11 on cell apoptosis and mitochondrial damage were abrogated by AG490. The depletion of KLF11 protected H/R-induced cell apoptosis and mitochondrial damage in H9c2 cells via activating the JAK2/STAT3 signaling pathway.

Downregulation of KLF11 protected against H/R induced apoptosis and mitochondrial damage via JAK2/STAT3 signaling pathway. a Cell apoptosis was evaluated using flow cytometry. b The activity of caspase3 was detected using ELISA. c Representative blot of c-caspase3 levels was assayed using western blot. d Mitochondrial membrane potential was detected using flow cytometry after staining JC-1. e, f Representative blot of Cyto C levels was assayed using western blot in plasma (e) and mitochondrial (f). *P < 0.05, **P < 0.01 vs. CON group, # P < 0.05 vs. NC + H/R group, $ P < 0.05 vs. H/R group, & P < 0.05 vs. KLF11 siRNA + H/R group, ^ P < 0.05 vs. AG490 + H/R group
Discussion
Myocardial ischemia and reperfusion injury triggers various mechanisms aimed at attenuating the cellular damage induced by apoptosis, and inflammatory mediators [19]. KLF11 is involved in the regulation of ischemic disease; for instance, KLF11 confers cerebrovascular protection in ischemic stroke [13]. In the present study, we investigated the roles of KLF11 in cell apoptosis and mitochondrial dysfunction following H/R in H9c2. The results demonstrated that the depletion of KLF11 efficiently prevented H/R-induced cell apoptosis and mitochondrial dysfunction. Specially, we focused on the potential role of the JAK2/STAT3 pathway in KLF11 siRNA-mediated cardioprotection, and found that the knockdown of KLF11 activated the JAK2/STAT3 pathway. In addition, A490, a selective inhibitor of JAK2/STAT3, abolished the cardioprotective effect of KLF11 siRNA.
Several reports have demonstrated that KLF11 plays a role in response to injuries. For example, KLF11 protects from ischemia stroke by mediating PPARγ [13]. Moreover, KLF11 suppressed endothelial cell activation via the nuclear factor-κB signaling pathway [20]. In conclusion, these reports show the importance of the role of KLF11 in different cell types, tissues, and context. In the current study we detected the expression of KLF11 induced by H/R in H9c2 cells. The results showed that the mRNA and protein levels of KLF11 were clearly raised following H/R. We next ascertained the roles of KLF11 in the regulation of cell apoptosis, the activity and expression of caspase3 and mitochondrial damage following H/R. We found that the depletion of KLF11 effectively suppressed cell apoptosis, which is consistent with a previous report that the overexpression KLF11 activated caspase3 and induced apoptosis in OLI-neu [21], oligodendroglial [22] and mesenchymal cells [23]. It has been reported that KLF11 is involved in the regulation of mitochondrial function [24]. Similarly, we observed that the knockdown of KLF11 improved mitochondrial function. Cyto C is a heme protein which is essential for aerobic respiration. The release of Cyto C from mitochondria is an important signal in mitochondrial dysfunction and apoptosis initiation [25]. We observed that the knockdown of KLF11 inhibits Cyto C expression, suggesting that the depletion of KLF11 protects H9c2 cells from H/R via inhibiting cell apoptosis and mitochondrial damage.
In particular, we focused on the potential role of the JAK2/STAT3 pathway in KLF11 mediated cell apoptosis and mitochondrial damage. This is because JAK-STAT signaling plays a central role in cardio-protection against ischemia and reperfusion [26]. The inhibition of JAK2 signaling ameliorates damage due to ischemia and reperfusion [27]. In the present study, we observed that H/R induced the p-JAK2 increased slightly, and AG490 exacerbated the cell apoptosis induced by H/R. These findings are consistent with other studies that also reported a similar results [28, 29]. Moreover, among JAK2V617F-negative ET patients, KLF10 and KLF5 were markedly increased compared with JAK2V617F-positive ET patients [30], and KLF5 protects against murine colitis and activates JAK-STAT signaling [31], increasing the possibility that KLF11 is associated with JAK2/STAT3 signaling. In H9c2 cells, we observed that the depletion of KLF11 elevated the expression of p-JAK2 and p-STAT3. To further investigate whether JAK2/STAT3 is involved in the regulation of KLF11 in H/R injury, we inhibited JAK2/STAT3 using AG490 and silenced KLF11 in H9c2, and found that AG490 partly abolishes the potential function of KLF11 in cell apoptosis and mitochondrial damage. Therefore, JAK2/STAT3 signaling plays a central role in the regulation of cell apoptosis by KLF11.
In summary, we used a small RNA silencing KLF11 to demonstrate an essential role for KLF11 in cell apoptosis and mitochondrial damage induced by H/R. Following H/R, the knockdown of KLF11 inhibited cell apoptosis, the activity of caspase3, the expression of c-caspase3 and mitochondrial damage. Moreover, the KLF11 is associated with the activation of JAK2/STAT3 signaling. Indeed, KLF11 may represent a desirable approach to treat heart failure. A future challenge will be identifying the responsible chemokines and better defining the interaction between KLF11 and JAK2/STAT3 signaling.
Conclusions
Depletion of KLF11 inhibited apoptosis and mitochondrial damage inducecd by H/R in H9c2 through JAK2/STAT3 pathway. KLF11 might be a novel approach of the treatment of I/R or other cardiovascular disease.
References
Gersh BJ, Sliwa K, Mayosi BM, Yusuf S (2010) Novel therapeutic concepts: the epidemic of cardiovascular disease in the developing world: global implications. Eur Heart J 31(6):642–648
Zweier JL, Talukder MH (2006) The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res 70(2):181–190
Frank A, Bonney M, Bonney S, Weitzel L, Koeppen M, Eckle T (2012) Myocardial ischemia reperfusion injury from basic science to clinical bedside. In: Seminars in cardiothoracic and vascular anesthesia. SAGE Publications, pp 123–132
Hausenloy DJ, Yellon DM (2013) Myocardial ischemia–reperfusion injury: a neglected therapeutic target. J Clin investig 123(1):92–100
Stark GR, Darnell JE (2012) The JAK-STAT pathway at twenty. Immunity 36(4):503–514
Levy DE, Darnell J (2002) Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 3(9):651–662
Barry SP, Townsend PA, Latchman DS, Stephanou A (2007) Role of the JAK–STAT pathway in myocardial injury. Trends Mol Med 13(2):82–89
Cittadini A, Monti MG, Iaccarino G, Castiello MC, Baldi A, Bossone E, Longobardi S, Marra AM, Petrillo V, Saldamarco L (2012) SOCS1 gene transfer accelerates the transition to heart failure through the inhibition of the gp130/JAK/STAT pathway. Cardiovasc Res 96(3):381–390
You L, Li L, Xu Q, Ren J, Zhang F (2011) Postconditioning reduces infarct size and cardiac myocyte apoptosis via the opioid receptor and JAK-STAT signaling pathway. Mol Biol Rep 38(1):437–443
Haghikia A, Ricke-Hoch M, Stapel B, Gorst I, Hilfiker-Kleiner D (2014) STAT3, a key regulator of cell-to-cell communication in the heart. Cardiovasc Res 102(2):281–289
McConnell BB, Yang VW (2010) Mammalian Krüppel-like factors in health and diseases. Physiol Rev 90(4):1337–1381
Pearson R, Fleetwood J, Eaton S, Crossley M, Bao S (2008) Krüppel-like transcription factors: a functional family. Int J Biochem Cell Biol 40(10):1996–2001
Yin K-J, Fan Y, Hamblin M, Zhang J, Zhu T, Li S, Hawse JR, Subramaniam M, Song C-Z, Urrutia R (2013) KLF11 mediates PPARγ cerebrovascular protection in ischaemic stroke. Brain 136(4):1274–1287
Zhang H, Chen Q, Yang M, Zhu B, Cui Y, Xue Y, Gong N, Cui A, Wang M, Shen L (2013) Mouse KLF11 regulates hepatic lipid metabolism. J Hepatol 58(4):763–770
Mathison A, Escande C, Calvo E, Seo S, White T, Salmonson A, Faubion WA Jr, Buttar N, Iovanna J, Lomberk G (2015) Phenotypic characterization of mice carrying homozygous deletion of KLF11, a gene in which mutations cause human neonatal and MODY VII diabetes. Endocrinology 156(10):3581–3595
Zheng Y, Kong Y, Li F (2014) Krüppel-like transcription factor 11 (KLF11) overexpression inhibits cardiac hypertrophy and fibrosis in mice. Biochem Biophys Res Commun 443(2):683–688
Yue R, Hu H, Yiu KH, Luo T, Zhou Z, Xu L, Zhang S, Li K, Yu Z (2012) Lycopene protects against hypoxia/reoxygenation-induced apoptosis by preventing mitochondrial dysfunction in primary neonatal mouse cardiomyocytes. PloS one 7(11):e50778
Haghikia A, Stapel B, Hoch M, Hilfiker-Kleiner D (2011) STAT3 and cardiac remodeling. Heart Fail Rev 16(1):35–47
Thind GS, Agrawal PR, Hirsh B, Saravolatz L, Chen-Scarabelli C, Narula J, Scarabelli TM (2015) Mechanisms of myocardial ischemia–reperfusion injury and the cytoprotective role of minocycline: scope and limitations. Future Cardiol 11(1):61–76
Fan Y, Guo Y, Zhang J, Subramaniam M, Song C-Z, Urrutia R, Chen YE (2012) Krüppel-like factor-11, a transcription factor involved in diabetes mellitus, suppresses endothelial cell activation via the nuclear factor-κB signaling pathway. Arterioscler Thromb Vasc Biol 32(12):2981–2988
Gohla G, Krieglstein K, Spittau B (2008) Tieg3/Klf11 induces apoptosis in OLI-neu cells and enhances the TGF-βeu cells and enhances the TGF-β signaling pathway by transcriptional repression of Smad7. J Cell Biochem 104(3):850–861
Wang Z, Spittau B, Behrendt M, Peters B, Krieglstein K (2007) Human TIEG2/KLF11 induces oligodendroglial cell death by downregulation of Bcl-XL expression. J Neural transm 114(7):867–875
Mathison A, Grzenda A, Lomberk G, Velez G, Buttar N, Tietz P, Hendrickson H, Liebl A, Xiong YY, Gores G (2013) Role for Kruppel-like transcription factor 11 in mesenchymal cell function and fibrosis. PloS one 8:e75311
Loft A, Forss I, Siersbæk MS, Schmidt SF, Larsen A-SB, Madsen JGS, Pisani DF, Nielsen R, Aagaard MM, Mathison A (2014) Browning of human adipocytes requires KLF11 and reprogramming of PPARγ superenhancers. Genes Dev 29(1):7–22
Babbitt SE, Sutherland MC, San Francisco B, Mendez DL, Kranz RG (2015) Mitochondrial cytochrome c biogenesis: no longer an enigma. Trends Biochem Sci 40(8):446–455
Bolli R, Dawn B, Xuan Y-T (2003) Role of the JAK–STAT pathway in protection against myocardial ischemia/reperfusion injury. Trends Cardiovasc Med 13(2):72–79
Freitas MCS, Uchida Y, Zhao D, Ke B, Busuttil RW, Kupiec-Weglinski JW (2010) Blockade of Janus kinase-2 signaling ameliorates mouse liver damage due to ischemia and reperfusion. Liver. Transplantation 16(5):600–610
Wang Y, Wang D, Zhang L, Ye F, Li M, Wen K (2016) Role of JAK-STAT pathway in reducing cardiomyocytes hypoxia/reoxygenation injury induced by S1P postconditioning. Eur J Pharmacol 784:129–136
Li L, Li M, Li Y, Sun W, Wang Y, Bai S et al (2016) Exogenous H2S contributes to recovery of ischemic post-conditioning-induced cardioprotection by decrease of ROS level via down-regulation of NF-κB and JAK2-STAT3 pathways in the aging cardiomyocytes. Cell Biosci 6(1):1
Puigdecanet E, Espinet B, Lozano J, Sumoy L, Bellosillo B, Arenillas L, Alvarez-Larran A, Sole F, Serrano S, Besses C (2008) Gene expression profiling distinguishes JAK2V617F-negative from JAK2V617F-positive patients in essential thrombocythemia. Leukemia 22(7):1368–1376
Tetreault M-P, Alrabaa R, McGeehan M, Katz JP (2012) Krüppel-like factor 5 protects against murine colitis and activates JAK-STAT signaling in vivo. PloS one 7(5):e38338
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, Y., Shi, X., Li, J. et al. Knockdown of KLF11 attenuates hypoxia/reoxygenation injury via JAK2/STAT3 signaling in H9c2. Apoptosis 22, 510–518 (2017). https://doi.org/10.1007/s10495-016-1327-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-016-1327-1