
Abstract
Tyramine is naturally occurring in food and induces pressor responses. Low-tyramine diets are recommended for patients treated with MAO inhibitors to avoid the fatal hypertensive crisis sadly known as “cheese effect”. Hence, tyramine intake is suspected to have toxicological consequences in humans, while its administration to type 1 diabetic rodents has been reported to improve glucose tolerance. We investigated in mice whether prolonged tyramine ingestion could alter glucose homeostasis, insulin sensitivity, adipose tissue physiology or cardiovascular functions. Tyramine was added at 0.04 or 0.14 % in the drinking water since this was estimated to increase by 10- to 40-fold the spontaneous tyramine intake of control mice fed a standard diet. Ten to 12 weeks of such tyramine supplementation did not influence body weight gain, adiposity or food consumption. Both doses (reaching approx. 300 and 1100 μmol tyramine/kg bw/day) decreased nonfasting blood glucose but did not modify glucose tolerance or fasting levels of glucose, insulin or circulating lipids. Blood pressure was not increased in tyramine-drinking mice, while only the higher tested dose moderately increased heart rate without change in its variability. Markers of cardiac tissue injury or oxidative stress remained unaltered, except an increased hydrogen peroxide production in heart preparations. In isolated adipocytes, tyramine inhibited lipolysis similarly in treated and control groups, as did insulin. The lack of serious adverse cardiovascular effects of prolonged tyramine supplementation in normoglycemic mice together with the somewhat insulin-like effects found on adipose cells should lead to reconsider favourably the risk/benefit ratio of the intake of this dietary amine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tyramine belongs to a wide family of amines having interchangeable names: sympathomimetic amines, trace amines or dietary amines. It is abundant in tyramine-rich fermented foods, such as aged cheese, wine, beer and sausages. Tyramine is known for being a “false transmitter” in mammals, while it has definitely the status of neurotransmitter in insects. In fact, tyramine is widely and sadly known to be involved in the “cheese effect”, which is a fatal hypertensive crisis induced by dietary amines in depressive patients treated with monoamine oxidase inhibitors (MAOIs). It has been demonstrated from a long while that ingested or injected tyramine displaces noradrenaline from adrenergic neurones, resulting in an increase in systolic blood pressure (SBP), which is dramatically exaggerated in the presence of irreversible MAOIs that inhibit the catabolism of both biogenic and dietary amines.
On a nutritional point of view, the implication of dietary tyramine in the adverse cardiovascular effects of MAOIs is completed by an elusive link between amine-rich food and migraine [27]. All this panel of deleterious actions led to consider tyramine dietary intake as a serious matter of toxicological safety. While the restriction of tyramine oral intake has prevented fatal SBP elevations during the therapeutic use of non-selective MAOIs in depressed patients, the consumption of foods in which tyramine is naturally present at high concentration is banned in a large number of consumers with diverse clinical disorders on the basis of more or less convincing documentation. In this context, the threshold of 6 mg tyramine per serving has been proposed as clinically significant content and for categorisation into low- and high-tyramine foods [56]. Actually, biogenic amines (tyramine, histamine, polyamines) emerge in foods as a consequence of poor hygiene, temperature or storage time abuse, and their (re)analysis in foods is currently used as a biosensor of freshness for quality control [33]. However, dietary tyramine is not the culprit of all previously reported intoxications, and the use of prebiotics and probiotics claimed to promote gut colonisation by “friendly” bacteria tends to increase the proportion of lactic-acid-producing bacteria (e.g. several Lactobacillus strains), which constitute a potential source of tyramine [8, 46] similar to that reported in more classical fermented foods and beverages [15]. Consequently, many recommendations for low-tyramine diets may appear at the present time as dietary restrictions that are almost unnecessary. Moreover, numerous novel antidepressive agents have replaced the historical irreversible MAOIs for the treatment of depression [33], and no relationship has been clearly evidenced between dietary amines and food intolerance reactions [27].
On a pharmacological point of view, tyramine is an indirectly acting sympathomimetic amine that releases noradrenaline and also an agonist for trace amine-associated receptors (TAARs): at least nine types in human and 16 in mouse [31] and a substrate of monoamine oxidases (MAO). The short-term effects of tyramine on the cardiovascular system mainly consist in an increase of blood pressure that results from complex mechanisms: release of noradrenaline and increased cardiac output (positive inotropy) [44], vasoconstriction in selected vascular beds (mediated by TAAR activation), and compensation by a baroreceptor-mediated reflex vasodilation [16, 28] and by nitric oxide release; all these components are reviewed in [7]. By contrast to its rapid pressor effects [22], the long-term actions of tyramine prolonged exposure are poorly known, at least to our knowledge. In the cardiovascular system, it is known that the degradation of noradrenaline or serotonin by MAO generates reactive oxygen species (ROS) which, in turn, lead to an increase in oxidative stress, mitogenic signalling in smooth muscle cells [14], cardiomyocyte hypertrophy [5] and apoptosis [4]. Thus, in heart, a chronic and sustained increase in oxidative stress generated by MAO activation seems to translate into enhanced sensitivity to apoptosis or necrosis. Accordingly, mice overexpressing MAO-A present deleterious effects in the heart, such as oxidative DNA damage, fibrosis, increased inflammatory response, necrosis, ventricular remodelling and heart failure [52].
All these issues incited us to study the influence of sustained tyramine ingestion, with a special attention focused on cardiovascular system, since tyramine is a pressor agent, able to induce cardiac arrhythmias [50], and a MAO substrate. The choice of the dose(s) of ingested tyramine needed justification before performing our supplementation studies. The clinical use of the pressor response to oral tyramine is a widely accepted method for quantifying the potential hypertensive complication triggered by any novel drug supposed to inhibit monoamine oxidases [18]. This test is based on the determination of the tyramine dose producing a >30 mm Hg increase in SBP in subjects previously treated by novel drug under development vs placebo [17]. It has been intensively used to compare various types of drugs [9, 16, 26] to classical MAOIs, since the latter typically reduce the tyramine dose required to raise blood pressure. However, this pressor test is poorly indicative regarding the oral tyramine dose to be used for prolonged experiments. On the one hand, the route of administration is crucial for the response to tyramine: When intravenously (i.v.) injected, only about 10 mg/subject is sufficient to trigger an increase of SBP in healthy volunteers [9], while the typical oral dose required to produce a 30 mm Hg rise in SBP is approximately 6–7 mg tyramine/kg bw, i.e. about 500 mg/subject (reviewed in [7]). On the other hand, tyramine appears to act differently whether it is orally administered alone or mixed with food [51], and the presence of low and high responders has been suggested. For unmedicated adults, an oral dose of 75 mg/subject can increase SBP only in less than 2 % of tested individuals [16], while it has been reported that dietary tyramine generally needs to exceed 200–800 mg/subject to induce a modest rise in blood pressure when ingested during a meal [27]. It can be estimated that such high intake is “supra-nutritional” since, without any food supplement, it can be reached only after the consumption of either kilogram(s) of aged cheeses (having a maximal tyramine content of 300 mg/kg) [7], hundred litres of red wines (averaging 5 mg/L) [2], or their combination. Thus, to determine the influence of sustained tyramine ingestion in a rodent model, we first estimated the spontaneous daily consumption of this dietary amine in control conditions before making a choice of the dose(s) valuable for tyramine supplementation. Such an approximation was feasible knowing the tyramine content we previously determined in pelleted standard diet for rodents: 45 mg/kg [54]. Assuming that the daily food intake is quantitatively equivalent to the water intake in a mouse weighing 25–30 g (approx. 3.5 g) and that the pellets contain 0.004 % tyramine, we performed successively two sets of experiments for providing tyramine in the drinking water at 0.04 and at 0.14 % to increase the estimated spontaneous dietary intake by 10- and >30-fold.
Indeed, our aim was to evaluate the risk/benefit ratio of tyramine supplementation not only by investigating its consequences on cardiovascular parameters but also by determining its putative actions on adipose tissue and metabolic functions as well as on energy balance. Yet, tyramine is oxidised by MAO and by semicarbazide-sensitive amine oxidase (SSAO) as well. Since the latter is highly expressed in white adipose tissue (WAT), one can expect that tyramine is not targeting solely nervous and cardiovascular systems. Whatever the amine oxidase or the anatomical site of oxidation, tyramine generates hydrogen peroxide once oxidised. While this member of ROS family is deleterious in cardiomyocytes, it has been proved to act as an insulin-mimicking agent in fat cells: involved in tyramine-induced activation of glucose transport in rat adipocytes [32, 55], and in adipogenesis stimulation in murine [11, 47] and human [6] preadipocytes. Such insulin mimicry also functions in vivo since tyramine improves glucose tolerance, when injected in combination with vanadium [54], when continuously delivered by osmotic minipumps [53], or when orally administered to type 1 diabetic rats [30].
The following results will show therefore how elevated tyramine intake in normoglycemic, normotensive mice could influence glucose handling and adipose tissue functions without promoting adverse cardiovascular effects.
Materials and methods
Animals and treatments
C57BL/6 male mice (Charles River, l’Arbresle, France) were housed at four animals per plastic cage with free access to water and standard rodent chow (Global rodent diet, Harlan, France) in a temperature-controlled room, with a 12-h light–dark cycle in the GENOTOUL animal housing facility meeting all the European standards. All animal procedures were approved by the University of Toulouse and INSERM Institutional Animal Care Committee.
In a first set of experiments, mice were separated in two groups of 12 with equivalent body weight (bw) at the age of 9 weeks. Twelve mice received tyramine hydrochloride (Sigma-Aldrich, Saint Quentin Fallavier, France) in the drinking water during 10 weeks (TYR-drinking mice) under the form of a 0.04 % solution that was changed weekly. Other 12 mice were without treatment (control). Non-fasting blood glucose levels were determined weekly (with glucose monitor from Roche Diagnostic between 15:00 and 15:45), as food and water consumption, while cardiovascular parameters were checked on three occurrences and glucose tolerance test was performed at week 9. Body composition was assessed by nuclear magnetic resonance (Echo MRI 100TM3 device, Echo Medical Systems, Houston, TX) a few days before sacrifice.
In a second set of experiments, tyramine was orally administered for a longer period and at a higher dose, to a group of eight mice (11-week old, 28 g) under the form of a 0.14 % solution in drinking water during 12 weeks. In parallel, age- and weight-matched controls received water and chow ad libitum. Both groups were subjected to the same non-invasive explorations as described above. Such tyramine supplementation led to a daily intake that could be considered as a >30-fold larger than the spontaneous dietary tyramine intake in control mice fed a pelleted standard diet.
Blood pressure and electrocardiogram analyses
Systolic blood pressure (SBP) was measured in conscious mice by a non-invasive plethysmographic tail-cuff method, using a PowerLab 800S and Chart Software (Phymep, Paris, France). Experiments were conducted in quietness by the same investigators and comprised two sessions of animal training and acclimatisation, followed by three sessions in which the pressure of each mouse was measured about ten times over a 10-min period.
Surface electrocardiograms (ECG) were recorded during 10 min on three animals simultaneously using Animal BioAmps, a PowerLab 8/30 and analysed with ChartPro software (AD-Instrument, UK). Mice were anaesthetised with isoflurane (induction 1.5 %, maintenance 1 % vol) under oxygen (0.6 L/min). Time-domain analysis of heart rate variability was performed to assess the square root of the mean of the squares of the successive differences between adjacent RR intervals (RMSSD) and pNN 6 % (percentage of normal RR intervals differing by >6 ms). Frequency-domain analysis was carried out using a fast Fourier transform size of frequency ranges of 0.15–1.5 Hz for the low frequencies (LF) and 1.5–5 Hz for the high frequencies (HF), according to [48].
Post-mortem determinations of cardiac morphology
Hydrogen peroxide production
Hydrogen peroxide production in the auricle was determined with Amplex Red (AR)-based fluorometric method. Briefly, auricles were removed from the heart and immediately incubated in 400 μl of phosphate buffer containing 100 μM AR and 1 IU/ml horseradish peroxidase. Resorufin generated after AR oxidation was measured after 30-min incubation as previously reported [10]. Hydrogen peroxide production was expressed per milligrams of protein.
Aconitase activity
Aconitase activity was spectrophotometrically measured using a commercial enzymatic kit (OxisResearch), and decrease of its activity was considered as a marker of short- or long-term oxidative stress [36].
Real-time RT-PCR
Extraction of RNA from cardiac ventricles was performed using column affinity purification (Qiagen, Courtaboeuf, France). Complementary DNAs (cDNAs) were synthesised using the superscript II RT-PCR system (Invitrogen) with random hexamers. Real-time PCR was performed in an ABI prism 7900 using Taqman Low density Arrays (Applied Biosystem). This allowed simultaneous measurement of 45 genes and 3 housekeeping genes using 200 ng of cDNA. Genes were expressed after normalisation to the geometric mean of three housekeeping genes: β-glucuronidase (GUSb), peptidyl-prolyl-isomerase A (PPIA) and hypoxanthine phosphoribosyltransferase (HPRT).
Histological analysis
Ventricles were incubated in Carnoy’s fixative solution (ethanol 60 %, chloroform 30 %, acetic acid 10 %), embedded in paraffin and transversally sectioned. Five-micrometer tissue sections were stained with hemalun-eosin. Analyses were performed as already reported [35].
Adipose tissue functional explorations
Lipolytic activity was assessed on freshly isolated adipocytes as previously described [23]. Lipogenic activity was also determined on freshly isolated adipocytes by measuring the radioactivity incorporated from [3-3H]-glucose (PerkinElmer Life Science, Boston, MA, USA) into cellular lipids as already described [25].
Statistical analyses
Biochemical data were analysed by ANOVA and post hoc tests, unless otherwise stated, such as, for example, Student’s t test for the production of hydrogen peroxide by heart preparations of control and TYR-drinking mice. Statistical analysis of cardiovascular data was performed after checking values for Gaussian distribution with D’Agostino and Pearson test and then compared with either unpaired t test or Mann–Whitney test. For all collected parameters, no comparison was tested between successive experiments 1 and 2 since each contained its own internal control. Values are given as mean ± standard error of the mean (SEM), and NS denotes a non-statistical significance.
Results
Body mass, adiposity, food and water intake in mice drinking tyramine
The growth curve of mice drinking a 0.04 % tyramine solution was superimposed to that of control group (F(1,198) = 0.3, NS) (Fig. 1a). The respective weight gain at the end of the 10-week period of this “experiment 1” was 5.1 ± 0.3 and 4.8 ± 0.4 g, respectively (n = 12, NS). Tyramine supplementation did not modify food intake since the mean values for food consumption were 3.2 ± 0.1 and 3.3 ± 0.1 g/mouse/day for TYR-drinking and control (NS), respectively. Similarly, the daily water intake was unchanged (3.29 ± 0.06 and 3.24 ± 0.06 g, NS). At the end of experiment, no change in adiposity was detected when fat depots were dissected and weighed and their lumped mass expressed as relative to body mass (adiposomatic index, Table 1). This lack of difference between TYR-drinking and control group was also found when body composition was measured during (Fig. 1b) or at the end of tyramine supplementation (Table 1).

Lack of influence of tyramine supplementation on mouse growth. a Body mass evolution in control (black squares) and mice drinking tyramine at 0.04 % (open triangles). b Fat mass as assessed by measurement of body composition by nuclear magnetic resonance. Mean ± SEM of 12 mice per group. No statistical difference between tyramine-drinking mice and their respective control
Similarly, in mice drinking a 0.14 % tyramine solution during 12 weeks, the growth curve was superimposed to respective control (experiment 2, not shown). Tyramine supplementation did not modify final body mass (31.7 ± 1.7 and 31.1 ± 0.8 g for TYR-drinking and control), daily food intake (4.2 ± 0.1 and 4.0 ± 0.1 g/mouse) or water consumption (4.23 ± 0.16 and 4.05 ± 0.23 g/mouse), and no change in adiposity was detected when fat depots were dissected and weighed or when body composition was assessed in a non-invasive manner (Table 1).
Taking into account body mass and water intake, the daily doses of tyramine ingested via the drinking solutions were calculated to bring approximately 300 and 1100 μmol of tyramine per kilogram per day for experiments 1 and 2, respectively, i.e. 10- to 37-fold more than the estimated spontaneous intake in control mice fed the standard chow.
Effect of tyramine supplementation on glucose handling and circulating lipids
While non-fasting blood glucose was not different between the groups at the start of treatment, a significant reduction was observed in the 0.04 % TYR-drinking group when taking into account all the repeated determinations performed during the 10-week treatment (F(1,176) = 9.4, p = 0.005) (Fig. 2a). With the higher tyramine dose, a modest but significant reduction of unfasted blood glucose was also found (p = 0.015) (Fig. 2b). The overall mean values of unfasted blood glucose were reduced in TYR-drinking mice by 4.7 % in experiment 1 and by 3.7 % in experiment 2, suggesting a hypoglycaemic effect of sustained tyramine intake.

Influence of tyramine oral treatments on non-fasting glycaemia. Unfasted blood glucose was determined weekly on control (black squares) and mice drinking tyramine at 0.04 % (open triangles) or 0.14 % (open squares), between 15:00 and 15:45 and during a supplementation period that is indicated by the horizontal arrow. Mean ± SEM (error bar lies inside symbol size in many occurrences) of 12 (a 0.04 % tyramine) or 8 (b 0.14 % tyramine) mice
After 9 weeks of treatment, mice were fasted for 5 h and subjected to intraperitoneal glucose tolerance test. The hyperglycaemic excursions were similar in control and TYR-drinking mice in both experiments (NS, not shown).
At sacrifice, after an overnight fasting, circulating glucose levels slightly tended to be lowered by tyramine supplementation with the 0.04 % dose, although the difference was at the limit of statistical significance. Similarly, a tendency to improve insulin sensitivity (HOMA index) was detected in TYR-drinking mice (Table 2). There was not any change of fasting glucose and insulin with the higher dose of tyramine. Circulating lipid levels (triglycerides, free fatty acids and glycerol) remained unmodified by tyramine supplementation, irrespective of the dose (not shown).
Influence of tyramine treatment on lipolytic and lipogenic activities of adipose cells
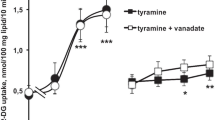
The responsiveness of isolated adipocytes to the inhibitory control of lipolysis exerted by insulin was used to assess ex vivo the insulin sensitivity in the different experimental groups. The insulin-dependent inhibition of lipolysis was unaltered by any of the tyramine treatments and was very reproducible among the two sets of experiments (Fig. 3). Similarly, the curves of the dose-dependent stimulation of lipolysis by isoprenaline were superimposed in adipocytes from TYR-drinking mice and respective control, with maximum reached with 1 μM of the beta-adrenergic agonist (insets of Fig. 3), therefore ruling out the emergence of a desensitisation to adrenergic stimuli in TYR-drinking mice. Figure 3 also shows that the addition of tyramine at doses between 0.1 and 1 mM mimicked the antilipolytic insulin action since it dose-dependently counteracted isoprenaline stimulation. The antilipolytic effect of tyramine was unaltered in both groups of TYR-drinking mice.

Lipolytic and antilipolytic responses in mouse adipocytes after tyramine chronic treatments. Comparison between mice drinking tyramine at 0.04 % (a) or 0.14 % (b) and their respective control. Dose-dependent antilipolytic effects of insulin and tyramine (left panels). Adipocytes were incubated with 10 nM isoprenaline and with insulin (circles) or tyramine (squares) at the indicated concentrations. Data are expressed as inhibition percentage of the lipolytic response to 10 nM isoprenaline alone taken as 0 % reference with return to basal lipolysis set at 100 %. Insets show the dose–response curves for isoprenaline when expressed as per cent of maximal response. Mean ± SEM of 6 (a) or 8 (b) adipocyte preparations. No significant difference was found between TYR-drinking (open symbols) and respective control (closed symbols)
Insulin responsiveness was also unaltered in adipocytes from TYR-drinking mice when measuring its capacity to activate de novo lipogenesis. The stimulation of the incorporation of [3H]-glucose into lipids by 100-nM insulin was equivalent to an increase over basal of 3.29 ± 0.76 vs 3.44 ± 0.73 in 0.04 % TYR-drinking and their control for experiment 1 (n = 4, NS), and of 3.48 ± 0.82 vs 3.93 ± 0.50 in experiment 2 (n = 5, NS). Lastly, the effect of the insulin-like combination of 0.1 mM benzylamine plus vanadate, already reported to be a SSAO-dependent response [24], was unaltered by tyramine supplementation, reaching 73 to 85 % of insulin maximal lipogenic action in experiments 1 and 2 (not shown) and indicating that SSAO activity was not altered.
Influence of tyramine treatment on cardiovascular parameters
As assessed by a non-invasive plethysmographic tail-cuff method, the blood pressure of conscious mice, especially the SBP, was not significantly modified by tyramine administration, irrespective of the ingested dose (Fig. 4a). Analysis of electrocardiograms under light anaesthesia did not reveal any difference between TYR-drinking and control for experiment 1 but revealed a modest, though significant, increase in heart rate after prolonged exposure to 0.14 % tyramine (Fig. 4b). However, atrioventricular and ventricular conduction, reflected by PR interval and QRS length, were unaffected (respectively ranging around 40 and 9 ms in all the groups, Fig. 4c, d), while there was no change in QT interval (not shown). Visual examination of ECG indicated the presence of a sinus rhythm and an absence of ectopic beats. Further analysis detected only a reduction in RR interval in the mice drinking 0.14 % tyramine (Fig. 4e).

Cardiovascular physiological parameters measured in vivo. a Systolic blood pressure. b Heart rate under anaesthesia. c Duration of the QRS complex. d Duration of the PR interval. e Beat-to-beat RR interval. Each column is mean ± SEM of 12 (expt 1) or 8 mice (expt 2). Difference between 0.14 % TYR-drinking mice (closed columns) and respective control (open columns) at **p < 0.01
Time-domain analysis of heart rate variability (HRV) did not show difference between treated and control groups, as reflected by unchanged standard deviation of normal RR intervals (SDNN), including in experiment 2 (Table 3). Similarly, root mean square of successive differences (RMSSD) and pNN6 values, reflecting respectively short-term variability and parasympathetic tone, were not modified in TYR-drinking mice, ruling out dysregulation of para- or ortho-sympathetic control.
Frequency–domain HRV analysis also revealed a lack of difference in HF power, reflecting parasympathetic activity, or in LF power, reflecting sympathetic activity. Furthermore, the LF/HF ratio, indicative of the sympathovagal balance, was undistinguishable between TYR-drinking and respective control in both sets of experiments. Only the component of very low frequency was altered in the mice ingesting the higher amounts of tyramine (Table 3).
Altogether, these results point out the lack of clear-cut pressor or sympathomimetic effect of sustained oral tyramine treatment, at least at the two doses tested. Only a sustained administration of tyramine estimated to be about 40-fold higher than the spontaneously ingestion found in standard condition in rodents fed a chow diet induced a moderate tachycardia, traduced by a reduced beat-to-beat interval that was not accompanied by major alterations in heart rate variability or blood pressure as it is the case for various cardiovascular diseases.
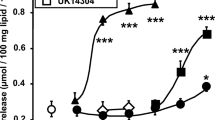
However, the appearance of such modest tachycardia after high tyramine ingestion prompted us to further explore cardiac function and morphology by another in vivo approach and by ex vivo analyses. When measuring HR in awake animals by plethysmography, we confirmed the presence of a limited rhythm acceleration: 678 ± 8 bpm in 0.14 % TYR-drinking vs 652 ± 9 bpm in control (n = 8; p < 0.05). The results of ex vivo explorations are shown in Fig. 5. There was no difference in the cardiac mass (Fig. 5a) or in the expression of various genes classically involved in cardiac remodelling (βMHC, ANP, BNP, IL6) (Fig. 5b) after the treatment with the high dose of tyramine. The ex vivo production of hydrogen peroxide by freshly dissected pieces of heart was greater in the mice subjected to tyramine (Fig. 5c). However, there was no difference in aconitase activity, a marker of oxidative stress, in the heart of these mice when compared to respective control (Fig. 5d). Likewise, there was no difference in the expression of genes related with oxidative stress (Fig. 5e). Histological examination of the hearts did not show evidence of structural abnormalities in the mice treated with 0.14 % tyramine. There was no sign of fibrosis, no inflammatory infiltrates and no degenerative necrosis of cardiomyocytes (not shown). All these observations were consistent with a lack of intensive cardiac remodelling upon oral tyramine exposure.

Influence of tyramine supplementation on ex vivo heart parameters. Heart determinations were performed in 0.14 % tyramine-drinking mice and their control. a Heart mass. b Expression of genes related to cardiac remodelling and inflammation assessed by quantitative RT-PCR. βMHC myosin heavy chain 7 beta, Bnp natriuretic peptide B, Anp natriuretic peptide A, IL6 interleukin 6. c Basal hydrogen peroxide production in heart. The production of hydrogen peroxide was measured in intact fresh heart for 30 min. d Aconitase activity in heart preparations. e Expression of genes related to oxidative stress. Cat catalase, Gpx1 glutathione peroxidase 1, Xdh xanthine dehydrogenase, Hmox1 heme oxygenase, Hif1 hypoxia inducible factor 1, NFκB1 nuclear factor of kappa polypeptide gene enhancer in B cells, Nos3 nitric oxide synthase 3. Mean ± SEM of eight mice per group. Different from control (black column) at *p < 0.05 by Student’s t test
Discussion
Initially defined as false transmitter, tyramine is a substrate for MAOs and a ligand for trace amine-associated receptors [31], the roles of which remain to be elucidated. Even less known is that tyramine acts as a precursor for octopamine and is an active messenger in insects [29]. However, the major interest for this notorious member of trace amine family is of nutritional and public health importance. A list of restricted foods (cheeses, sausages, sauerkraut, bananas, wines and beers) has been initially established and updated for patients treated with MAOIs to avoid hypertensive crisis [20, 33, 40, 56]. Then, this list, made on the basis of high-tyramine content, has been extended to many other food items and to other questionable drug-related events, resulting in the propagation of controversial dietary restrictions [27]. Meanwhile, clinical trials have shown that tyramine restrictions for novel MAO-B inhibitors (rasagiline) appeared much less necessary [12, 16, 19] than for the historical MAO-A irreversible inhibitors [43]. The aim of our study was not to re-establish a reliable information to patients or consumers but to further explore the real functional and nutrigenomic influences of high tyramine intake in a laboratory model. Taken as a whole, our observations brought evidence that high-tyramine ingestion was not as deleterious as it could be expected since it provoked only modest modifications of a few cardiovascular parameters and did not alter insulin or adrenergic responsiveness in mouse. These results are in line with a meta-analysis showing that the purported intolerance to dietary biogenic amines in susceptible patients has been overestimated [27].
A review of the tyramine found in foods or organisms is not performed here since it is available elsewhere [2, 19, 40, 42]. We just make a comment about the fact that attention must be paid not only to the content of tyramine in food items but also to its bioavailability once ingested, considering that food processing or food contaminants might change the fate of dietary amines. In this view, oleamide, a slip agent that can be found in plasticware and packaging, behaves as a strong inhibitor of MAOs [34]. Although well characterised in many food items, the amount of tyramine is not defined in rodent chow, with this exception of a previous determination we performed by HPLC that gave a tyramine content equivalent to 45 μg/g of standard pellets [54]. It was then estimated that the spontaneous dietary intake of tyramine averaged 30–35 μmol/kg bw/day in control mouse. Subsequently, the tyramine we orally administered to mice was first fixed to reach a tenfold increase above this spontaneous dietary intake. The daily consumption of drinking water containing 0.04 % tyramine allowed attaining this goal since it resulted in a daily intake of 53 mg/kg bw, which was largely lower than the acute oral toxicity, reported to be higher than 2000 mg/kg bw in rats [49]. Considering the tendency to improve glucose handling (reduced blood glucose in fed state, trend to lower fasting plasma glucose and to increase insulin sensitivity) found in these conditions in the absence of cardiovascular alterations, we decided to increase the tyramine intake in a second set of investigations by approaching the no-observed-adverse-effect level, established to be 180 mg/kg bw/day [49]. The higher tested dose (free access to 0.14 % tyramine solution) resulted in a daily intake of 185 mg/kg bw, which corresponded to an almost fortyfold increase over spontaneous intake. This second experiment allowed the detection of a few changes in cardiovascular parameters without any sign of cardiac injury but did not improve the beneficial effects on glucose handling. At a first glance, it can be summarised that elevated dietary tyramine is well tolerated by the organism. Nevertheless, the observed influences on adipose/insulin sensitivity and cardiovascular aspects will be discussed below in details.
When intentionally administered in humans, tyramine is a reliable way to activate sympathetic nerves: those ending in endocrine pancreas (decreasing insulin secretion) [22], gut (increasing ghrelin release) [39] or eyes (provoking mydriasis). Tyramine also elicits local release of endogenous norepinephrine in adipose tissue and stimulates lipolysis when administered via subcutaneous microdialysis probes but not when directly tested in isolated adipocytes [1]. This last clinical observation is consistent with our demonstrations stating that tyramine inhibited isoprenaline-induced lipolysis in mouse adipocytes as did insulin. The mechanism of such antilipolytic action remains to determine. The hypothesis that tyramine may hamper isoprenaline-induced lipolysis by acting as a β-adrenergic antagonist/partial agonist or via activation of TAAR subtype(s) negatively coupled to adenylyl-cyclase could not be ruled out. However, the tyramine antilipolytic effect remained unchanged in TYR-drinking mice, suggesting a lack of substantial desensitisation process after chronic administration and ruling out the involvement of down-regulatable receptors in such effects. In addition, the fact that millimolar doses of tyramine were required to limit triglyceride breakdown and to reproduce the insulin antilipolytic action suggested that, as previously reported for the stimulation of glucose transport [32], amine oxidation was involved in the tyramine insulin-like antilipolytic effect, via an hydrogen peroxide-dependent mechanism. Indeed, we have already demonstrated that, in rat, several tyramine effects—somewhat insulin-mimicking—are inhibited by MAO and/or SSAO blockade, such as glucose transport activation in preadipocytes and adipocytes [11, 54, 55], glucose uptake activation in skeletal and cardiac muscles [37]. This contrasts with the more known hypertensive effects that are improved by MAO blockade. We extend here the panel of their extra-neuronal actions of tyramine to the acute inhibition of lipolysis in mouse fat cells.
Whatever the mechanism implicated in the antilipolytic effect found in adipocytes, our in vitro observations are in agreement with the lack of increased circulating free fatty acids in TYR-drinking mice, likely indicating that tyramine did not promote adipose tissue lipolysis and lipid mobilisation when repeatedly ingested. Accordingly, high-tyramine intake is devoid of any slimming effect, as it is the also case for its derivative, N-methyltyramine [45]. Thus, into our opinion, tyramine supplementation could not be proposed as an attempt to substantially increase orthosympathetic activation of WAT, subsequent browning of white/beige adipocytes and weight loss. More importantly, tyramine exposure did not hamper the in vitro insulin action on adipocytes (lipolysis inhibition and lipogenesis activation) as well as its in vivo action during glucose tolerance tests. When added to the drinking water of normoglycemic mice, at increasing doses, tyramine even tended to lower circulating glucose in the fed state. This agrees with previous observations indicating that tyramine improves glucose handling in diabetic rats [30, 53, 54]. Before extrapolating to the consumer the putative interest of these metabolic actions, a complete study of the tyramine responsiveness of human adipocytes (lipolysis inhibition, glucose uptake activation) remains required to complete the already demonstrated lack of direct lipolytic effect [1].
With the higher tyramine supplementation, we observed changes in cardiac rhythm rather than in SBP. This is consistent with the fact that the rise in SBP following acute tyramine administration most likely reflects a positive inotropism and not a direct vascular “pressor” response [44] since tyramine primarily increases heart rate and reduces it only later, by reflex bradycardia [16, 28]. The lack of SBP elevation can also be explained when comparing the doses of tyramine used for this prolonged exposure to the values used for rapid pressor effect. In rat, the tyramine doses generally used to induce a threshold pressor response vary between 14 and 20 mg/kg p.o. in solution [17, 26] but rise up to 67 mg/kg p.o. in feed preparation, showing that the presence of food delays the pressor effect [17]. It is not so surprising that a dose of 185 mg/kg discretely administered along the day cannot increase SBP in the TYR-drinking mice.
Despite elevated hydrogen peroxide levels found in the heart of mice treated with the high tyramine dosage, we did not observe any oxidative cardiac damage. It has been previously demonstrated that cellular effects of hydrogen peroxide were directly dependent on the concentration [13]. While low concentrations of hydrogen peroxide were well tolerated, higher doses caused apoptosis and very high doses induced necrosis of cardiac cells. Thus, in our study, the levels of hydrogen peroxide were probably not high enough to promote cardiac cell damage. These supposed low levels of ROS at the cardiac levels are in agreement with the already discussed aspects of the intensity of cardiovascular responses to tyramine that depend on the dose and the mode of administration. One can imagine that the main origin of this increased hydrogen peroxide production is the MAO-dependent oxidation of ingested tyramine. We have already reported an anatomical distribution of MAO and SSAO activities in rat [37] and observed that the following order of amine oxidase richness could be established with tyramine as substrate in different organs: liver > WAT > heart >brain > jejunum > aorta > kidney > skeletal muscles. It can be supposed that intestinal tract and liver play a larger role than the heart in metabolising the amine given per os. Consequently, it can be proposed that, in mouse, the heart is involved only in a minor part of the oxidation of ingested tyramine and hence facing to a surmountable handling of the generated H2O2. Moreover, such H2O2 increase was not accompanied by an inactivation of cardiac aconitase, as it is the case for hearts from Western diet-induced obese rats [38].
The RR interval variations measured during resting conditions represent beat-by-beat variations in cardiac autonomic inputs. Parasympathetic activity, a major contributor to the HF component of HRV, was not altered by prolonged tyramine load. The LF component of HRV, which can be considered as a marker of sympathetic modulation, though not specific, was also unaltered by tyramine intake. Tachycardia resulting from typical sympathetic activation is generally accompanied by a marked reduction in total power. This was not clearly the case for both groups of the TYR-drinking mice studied, when compared to their respective control. Only the VLF power, mostly depending on vascular component, was decreased by tyramine. All these spectral components of the HRV suggested that the sympathetic activation induced by tyramine was moderate. The heart rate analysis indicated that tyramine, at a dose 30- to 40-fold larger than baseline dietary intake increased by 10 % the resting HR in mice without diminishing the fluctuations of HR, a condition that accompanies cardiac injury. However, our study was unsuitable to assess whether tyramine might be cardioprotective and might prevent from or recover cardiac injury, as it is the case for other food components such as stilbenes [3] or flavonoids [41].
In conclusion, our study shows that, in mouse, a prolonged increase of tyramine intake lowers unfasted blood glucose at the expense of very few changes in cardiac function. Though they do not bring precision on other organ functions, our observations underscore the utility of banishing high-tyramine diets, except in the case of patients treated with MAOIs to avoid cardiovascular events. Alongside its well-known deleterious pressor effects, especially enhanced by MAOIs, tyramine can exert some metabolic effects that, on the opposite, are inhibited by MAOIs. Such effects (e.g. stimulation of glucose uptake or lipolysis inhibition in adipocytes), better documented in rat than in mouse [37], deserve to be further studied in man. On this basis, we propose that it is of great value to better define whether the intake of tyramine might exert a benefit, especially when glucotoxicity or lipotoxicity needs to be reduced, i.e. in diabetic and obese states [21], which could overpass the risk of hypertensive crisis, which appears limited when MAO is not inhibited.
References
Adams F, Boschmann M, Schaller K, Franke G, Gorzelniak K, Janke J, Klaus S, Luft FC, Heer M, Jordan J (2006) Tyramine in the assessment of regional adrenergic function. Biochem Pharmacol 72:1724–1729
Ancín-Azpilicueta C, González-Marco A, Jiménez-Moreno N (2008) Current knowledge about the presence of amines in wine. Crit Rev Food Sci Nutr 48:257–275
Arafa MH, Mohammad NS, Atteia HH, Abd-Elaziz HR (2014) Protective effect of resveratrol against doxorubicin-induced cardiac toxicity and fibrosis in male experimental rats. J Physiol Biochem 70:701–711
Bianchi P, Kunduzova O, Masini E, Cambon C, Bani D, Raimondi L, Seguelas MH, Nistri S, Colucci W, Leducq N, Parini A (2005) Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation 112:3297–3305
Bianchi P, Pimentel DR, Murphy MP, Colucci WS, Parini A (2005) A new hypertrophic mechanism of serotonin in cardiac myocytes: receptor-independent ROS generation. FASEB J 19:641–643
Bour S, Visentin V, Grès S, Saulnier-Blache JS, Wabitsch M, Carpéné C (2005) Tyramine, benzylamine, and to a lesser extent histamine, partially mimic the adipogenic effect of insulin in a human preadipocyte cell strain. Inflamm Res 54(suppl 1):S60–S61
Broadley KJ (2010) The vascular effects of trace amines and amphetamines. Pharmacol Ther 125:363–375
Broadley KJ, Akhtar Anwar M, Herbert AA, Fehler M, Jones EM, Davies WE, Kidd EJ, Ford WR (2009) Effects of dietary amines on the gut and its vasculature. Br J Nutr 101:1645–1652
Cantarini MV, Painter CJ, Gilmore EM, Bolger C, Watkins CL, Hughes AM (2004) Effect of oral linezolid on the pressor response to intravenous tyramine. Br J Clin Pharmacol 58:470–475
Carpéné C, Bizou M, Tréguer K, Hasnaoui M, Grès S (2015) Glitazones inhibit human monoamine oxidase but their anti-inflammatory actions are not mediated by VAP-1/semicarbazide-sensitive amine oxidase inhibition. J Physiol Biochem 71:487–496
Carpéné C, Daviaud D, Boucher J, Bour S, Visentin V, Gres S, Duffaut C, Fontana E, Testar X, Saulnier-Blache S, Valet P (2006) Short-and long-term insulinlike effects of substrates of monoamine oxidases and semicarbazide-sensitive amine oxidases in cultured adipocytes. Metabolism 55:1397–1405
Chen JJ, Wilkinson JR (2012) The monoamine oxidase type B inhibitor Rasagiline in the treatment of Parkinson disease: is tyramine a challenge? J Clin Pharmacol 52:620–628
Chen QM, Tu VC, Wu Y, Bahl JJ (2000) Hydrogen peroxide dose dependent induction of cell death or hypertrophy in cardiomyocytes. Arch Biochem Biophys 373:242–248
Coatrieux C, Sanson M, Negre-Salvayre A, Parini A, Hannun Y, Itohara S, Salvayre R, Augé N (2007) MAO-A- induced mitogenic signalling is mediated by reactve oxygen species, MMP-2, and the sphingolipid pathway. Free Rad Biol Med 43:80–89
Coton M, Romano A, Spano G, Ziegler K, Vetrana C, Desmarais C, Lonvaud-Funel A, Lucas P, Coton E (2010) Occurrence of biogenic amine-forming lactic acid bacteria in wine and cider. Food Microbiol 27:1078–1085
de Marcaida JA, Schwid SR, White WB, Blindauer K, Fahn S, Kieburtz K, Stern M, Shoulson I, Coordinators PSGTPTSIa (2006) Effects of tyramine administration in Parkinson’s disease patients treated with selective MAO-B inhibitor rasagiline. Movement Disorders 21:1716–1721
Fankhauser C, Charieras T, Caille D, Rovei V (1994) Interaction of MAO inhibitors and dietary tyramine: a new experimental model in the conscious rat. J Pharmacol Toxicol Methods 32:219–224
Finberg JP (2014) Update on the pharmacology of selective inhibitors of MAO-A and MAO-B: focus on modulation of CNS monoamine neurotransmitter release. Pharmacol Ther 143:133–152
Finberg JP, Gillman K (2011) Selective inhibitors of monoamine oxidase type B and the "cheese effect". Int Rev Neurobiol 100:169–190
Flockhart DA (2012) Dietray restructions and drug interactions with monoamine oxidase inhibitors: an update. J Clin Psychiatry 73(suppl 1):17–24
Giaccari A, Soricea G, Muscogiuria G (2009) Glucose toxicity: the leading actor in the pathogenesis and clinical history of type 2 diabetes – mechanisms and potentials for treatment. Nutr Metab Cardiovasc Dis 19:365–377
Gilliam LK, Palmer JP, Taborsky GJ Jr (2007) Tyramine-mediated activation of sympathetic nerves inhibits insulin secretion in humans. J Clin Endocrinol Metab 92:4035–4038
Gomez-Zorita S, Tréguer K, Mercader J, Carpéné C (2013) Resveratrol directly affects in vitro lipolysis and glucose transport in human fat cells. J Physiol Biochem 69:585–593
Grès S, Bour S, Valet P, Carpéné C (2012) Benzylamine antihyperglycemic effect is abolished by AOC3 gene invalidation in mice but not rescued by semicarbazide-sensitive amine oxidase expression under the control of aP2 promoter. J Physiol Biochem 68:651–662
Harant-Farrugia I, Garcia J, Iglesias-Osma MC, Garcia-Barrado MJ, Carpéné C (2014) Is there an optimal dose for dietary linoleic acid? Lessons from essential fatty acid deficiency supplementation and adipocyte functions in rats. J Physiol Biochem 70:615–627
Humphrey SJ, Curry JT, Turman CN, Stryd RP (2001) Cardiovascular sympathomimetic amine interactions in rats treated with monoamine oxidase inhibitors and the novel oxazolidinone antibiotic linezolid. J Cardiovasc Pharmacol 37:548–563
Jansen SC, van Dusseldorp M, Bottema KC, Dubois AE (2003) Intolerance to dietary biogenic amines: a review. Ann Allergy Asthma Immunol 91:233–240
Khwanchuea R, Mulvany MJ, Jansakul C (2008) Cardiovascular effects of tyramine: adrenergic and cholinergic interactions. Eur J Pharamacol 579:308–317
Lange AB (2009) Tyramine: from octopamine precursor to neuroactive chemical in insects. General Compar Endocrinol 162:18–26
Lino CS, Sales TP, Gomes PB, do Amaral JF, Alexandre FSO, Silveira ER, Ferreira JM, de Sousa DF, de Queiroz MGR, de Sousa FCF, de Castro Brito GA, da Rocha Cipriano Brito SM, de Barros Viana GS (2007) Anti-diabetic activity of a fraction from Cissus verticillata and tyramine, its main bioactive constituent, in alloxan-induced diabetic rats. Am J Pharmacol Toxicol 2:178–188
Maguire JJ, Parker WAE, Foord SM, Bonner TI, Neubig RR, Davenport AP (2009) International Union of Pharmacology. LXXII. Recommendations for Trace Amine Receptor nomenclature. Pharmacol Rev 61:1–8
Marti L, Morin N, Enrique-Tarancon G, Prévot D, Lafontan M, Testar X, Zorzano A, Carpéné C (1998) Tyramine and vanadate synergistically stimulate glucose transport in rat adipocytes by amine oxidase-dependent generation of hydrogen peroxide. J Pharmacol Exp Ther 285:342–349
McCabe-Sellers BJ, Staggs CG, Bogle ML (2006) Tyramine in foods and monoamine oxidase inhibitor drugs: a crossroad where medicine, nutrition, pharmacy, and food industry converge. J Food Composition Anal 19:S58–S65
McDonald GR, Hudson AL, Dunn SMJ, You H, Baker GB, Whittal RM, Martin JW, Jha A, Edmondson DE, Holt A (2008) Bioactive contaminants leach from disposable laboratory plasticware. Science 322:917
Mialet-Perez J, Bianchi P, Kunduzova O, Parini A (2007) New insights on receptor-dependent and monoamine oxidase-dependent effects of serotonin in the heart. J Neural Transm 114:823–827
Miguel F, Augusto AC, Gurgueira SA (2009) Effect of acute vs chronic H2O2-induced oxidative stress on antioxidant enzyme activities. Free Radic Res 43:340–347
Morin N, Visentin V, Calise D, Marti L, Zorzano A, Testar X, Valet P, Fischer Y, Carpéné C (2002) Tyramine stimulates glucose uptake in insulin-sensitive tissues in vitro and in vivo via its oxidation by amine oxidases. J Pharmacol Exp Ther 303:1238–1247
Mourmoura E, Rigaudière JP, Couturier K, Hininger I, Laillet B, Malpuech-Brugère C, Azarnoush K, Demaison L (2015) Long-term abdominal adiposity activates several parameters of cardiac energy function. J Physiol Biochem. doi:10.1007/s13105-015-0427-7
Mundinger TO, Cummings DE, Taborsky GJ Jr (2006) Direct stimulation of ghrelin secretion by sympathetic nerves. Endocrinology 147:2893–2901
Pfudstein B, Tricker AR, Theobald E, Spiegehalder B, Preussmann R (1991) Mean daily intake of primary and secondary amines from foods and beverages in West Germany in 1989-1990. Food Chem Toxicol 29:733–739
Pogula BK, Maharajan MK, Oddepalli DR, Boini L, Arella M, Sabarimuthu DQ (2012) Morin protects heart from beta-adrenergic-stimulated myocardial infarction: an electrographic, biochemical, and histological study in rats. J Physiol Biochem 68:433–446
Ruiz-Capillas C, Jiménez-Colmenero F (2004) Biogenic amines in meat and meat products. Crit Rev Food Sci Nutr 44:489–499
Sandler M, Glover V, Ashford A, Esmail A (1980) The inhibition of tyramine oxidation and the tyramine hypertensive response ("cheese effect") may be independent phenomena. J Neural Transm 48:241–247
Schäfers RF, Poller U, Pönicke K, Geissler M, Daul AE, Michel MC, Brodde O-E (1997) Influence of adrenoceptor and muscarinic receptor blockade on the cardiovascular effects of exogenous noradrenaline and of endogenous noradrenaline released by infused tyramine. Naunyn-Schmiedeberg’s Arch Pharmacol 355:239–249
Stohs SJ, Hartman MJ (2015) A review of the receptor binding and pharmacological effects of N -methyltyramine. Phytotherapy Res 29:14–16
Straub BW, Kicherer M, Schilcher SM, Hammes WP (1995) The formation of biogenic amines by fermentation organisms. Z Lebensm-Unters Forsch 201:79–82
Subra C, Fontana E, Visentin V, Testar X, Carpéné C (2003) Tyramine and benzylamine partially but selectively mimic insulin action on adipose differentiation in 3T3-L1 cells. J Physiol Biochem 59:209–216
Thireau J, Zhang BL, Poisson D, Babuty D (2008) Heart rate variability in mice: a theoretical and practical guide. Exp Physiol 93:83–94
Til HP, Falke HE, Prinsen MK, Willems MI (1997) Acute and subacute toxicity of tyramine, spermidine, spermine, putrescine and cadaverine in rats. Food Chem Toxicol 35:337–348
Tiller JWG, Maguire KP, Davies BM, Dowling JT, Tung LH, Rand MJ (1990) Tyramine-induced cardiac arrhythmias. Human Psychopharmacol 5:313–321
VanDenBerg CM, Blob LF, Kemper EM, Azzaro AJ (2003) Tyramine pharmacokinetics and reduced bioavailability with food. J Clin Pharmacol 43:604–609
Villeneuve C, Guilbeau-Frugier C, Sicard P, Lairez O, Ordener C, Duparc T, De Paulis D, Couderc B, Spreux-Varoquaux O, Tortosa F, Garnier A, Knauf C, Valet P, Borchi E, Nediani C, Gharib A, Ovize M, Delisle MB, Parini A, Mialet-Perez J (2013) p53-PGC-1α pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: role in chronic left ventricular dysfunction in mice. Antioxid Redox Signal 18:5–18
Visentin V, Bour S, Boucher J, Prevot D, Valet P, Ordener C, Parini A, Carpene C (2005) Glucose handling in streptozotocin-induced diabetic rats is improved by tyramine but not by the amine oxidase inhibitor semicarbazide. Eur J Pharmacol 522:139–146
Visentin V, Marq P, Bour S, Subra C, Prévot D, Morin N, Valet P, Monje MC, Nepveu F, Carpéné C (2003) Effect of prolonged treatment with tyramine on glucose tolerance in streptozotocin-induced diabetic rats. J Physiol Biochem 59:225–232
Visentin V, Prévot D, Marti L, Carpéné C (2003) Inhibition of rat fat cell lipolysis by monoamine oxidase and semicarbazide-sensitive amine oxidase substrates. Eur J Pharmacol 466:235–243
Walker SE, Shulman KI, Tailor SAN, Gardner D (1996) Tyramine content of previously restricted foods in monoamine oxidase inhibitor diets. J Clin Psychopharmacol 16:383–388
Acknowledgments
Part of this work was partly supported by “DIOmed project”. S. Schaak was the recipient of a “Bonus Qualité Recherche” funding from Université Paul Sabatier, Toulouse, and J. Mercader was funded owing to the grant INTERREG IVB SUDOE 1/P1/E178. The authors express gratitude to Angelo Parini, Jean-Michel Senard, Philippe Valet and the staff of PA3S (Toulouse) for facilitating scientific exchanges. The authors also thank Delphine Turpin, Danielle Prévot, Estelle Wanecq, Zsuzsa Iffiú-Soltész and the “plateforme Anexplo” (US006INSERM, Toulouse) for invaluable technical help.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Carpéné, C., Schaak, S., Guilbeau-Frugier, C. et al. High intake of dietary tyramine does not deteriorate glucose handling and does not cause adverse cardiovascular effects in mice. J Physiol Biochem 72, 539–553 (2016). https://doi.org/10.1007/s13105-015-0456-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-015-0456-2