
Abstract
Stroke was the 2nd leading cause of death and a major cause of morbidity. Unfortunately, there are limited means to promote neurological recovery post-stroke, but research has unearthed potential targets for therapies to encourage post-stroke neurogenesis and neuroplasticity. The occurrence of neurogenesis in adult mammalian brains, including humans, was not widely accepted until the 1990s. Now, adult neurogenesis has been extensively studied in human and mouse neurogenic brain niches, of which the subventricular zone of the lateral ventricles and subgranular zone of the dentate gyrus are best studied. Numerous other niches are under investigation for neurogenic potential. This review offers a basic overview to stroke in the clinical setting, a focused summary of recent and foundational research literature on cortical neurogenesis and post-stroke brain plasticity, and insights regarding how the meninges and choroid plexus have emerged as key players in neurogenesis and neuroplasticity in the context of focal cerebral ischemia disrupting the anterior circulation. The choroid plexus and meninges are vital as they are integral sites for neuroimmune interactions, glymphatic perfusion, and niche signaling pertinent to neural stem cells and neurogenesis. Modulating neuroimmune interactions with a focus on astrocyte activity, potentially through manipulation of the choroid plexus and meningeal niches, may reduce the exacerbation of stroke by inflammatory mediators and create an environment conducive to neurorecovery. Furthermore, addressing impaired glymphatic perfusion after ischemic stroke likely supports a neurogenic environment by clearing out inflammatory mediators, neurotoxic metabolites, and other accumulated waste. The meninges and choroid plexus also contribute more directly to promoting neurogenesis: the meninges are thought to harbor neural stem cells and are a niche amenable to neural stem/progenitor cell migration. Additionally, the choroid plexus has secretory functions that directly influences stem cells through signaling mechanisms and growth factor actions. More research to better understand the functions of the meninges and choroid plexus may lead to novel approaches for stimulating neuronal recovery after ischemic stroke.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction to Stroke
Stroke is the third leading cause of years of life lost [1] and remains a leading cause of serious long-term disability in the USA [2]. Each year about 795,000 Americans experience a new or recurrent stroke [3]. Unfortunately, therapies for stroke recovery are limited and survivors often exhibit a high degree of morbidity and long-term disability. Stroke is the disruption of blood supply to the brain, and there are two main classifications: (1) ischemic stroke, where there is cessation of blood flow, and which is typically caused by thrombus at the site of the brain vessel or embolus from the heart; and (2) hemorrhagic stroke, which is caused by the rupture of a blood vessel, aneurysm, or other vascular lesion, and which not only disrupts flow to the normal brain parenchyma, but the hemorrhage also compresses brain tissue, compounding the injury. Acute ischemic stroke (AIS), the focus of this review, is commonly differentiated into five subtypes based on the Trial of Org 10172 in Acute Stroke Treatment (TOAST) classification system [4] (Fig. 1). Ultimately, both acute ischemic and hemorrhagic stroke result in neurological impairment due to tissue damage in the brain from the loss in blood flow and reduced oxygenation of the tissue.

TOAST classification of stroke etiology. Vascular causes of stroke include cardioembolic disease (CE), small vessel disease (SVD) in the brain vessels, and large artery atherosclerosis (LAA) as depicted in the internal carotid artery (ICA) or the first segment of the middle cerebral artery (MCA). Yellow represents core infarct and blue represents the penumbra, which is threatened brain that can be salvaged by revascularization
Stroke affects 16.9 million people worldwide [5]. Approximately 5.5 million people die of stroke per year, making it the second leading cause of death [6], but there are more than 80 million stroke survivors across the globe. The prevalence of stroke is such that 1 in 4 adults will suffer a stroke in their lifetime. It is estimated that stroke-related medical costs will increase from 61.5 billion US dollars (USD) in 2015 to 183 billion USD in 2035, due to both acute care and the fact that approximately 50% of stroke survivors have residual deficits at 6 months and beyond [7].
The main causal and modifiable risk factors associated with stroke include hypertension, hypercholesterolemia, carotid stenosis, and atrial fibrillation [5]. In addition, tobacco abuse, diabetes mellitus, alcohol abuse, obesity, poor nutrition or high-risk diets, sedentary lifestyle, sleep apnea, chronic kidney disease, chronic inflammation, stress, and depression are likely also causal factors. Non-modifiable causal risk factors include age, sex, and genetic predisposition. The risk of stroke increases with age, doubles for every subsequent decade in life, and approximately ¾ of all strokes occur in people age 65 years or older [8]. Stroke has historically been thought of as a disease predominantly affecting men, however, of the seven million survivors of stroke in the USA, 54.2% are women [7]. The genetic predisposition to ischemic stroke is 37.9%, calculated using genome-wide complex trait analysis [6].
Given the striking clinical risks of stroke, there is a demand for more research on mechanisms in neurorecovery following stroke-induced brain damage. The discovery and acceptance of adult neurogenesis through the turn of the twenty-first century has generated interest in promoting neural repair, plasticity, and neurogenesis to improve post-stroke neurological recovery and to reduce morbidity. Of the almost 800,000 patients who suffer a stroke each year, 87% are ischemic [3], and almost 75% of these occur in the anterior circulation [9]. The first section of this review covers the clinical aspects of stroke and the second section covers research literature on cortical neurogenesis, post-stroke brain plasticity, and known or novel factors that could be useful therapeutic targets for treatment after focal cerebral ischemia disrupting anterior circulation.
Clinical Observations in the Care of Acute Ischemic Stroke
Current Treatments for AIS
Patients who present with stroke have a sudden onset of focal neurological deficit including headache, dizziness, blurred vision, facial weakness or drooping, arm or leg weakness, and changes in balance and speech. Upon arrival to the hospital, suspected stroke patients are evaluated for these symptoms by the National Institute of Health Stroke Scale (NIHSS). After an initial NIHSS exam, stroke patients are taken immediately for a non-contrast computerized tomography (CT) scan of the brain, to determine if the stroke is ischemic or hemorrhagic. Following this, a CT of brain and neck is performed with injection of dye, called a CT angiogram (CTA), which identifies if there is a narrowing or occlusion in the carotid arteries leading to the brain, or in the brain vessels themselves. Perfusion scans have been validated to identify patients who present with large vessel occlusion (LVO) strokes to undergo endovascular thrombectomy (EVT) [10, 11]. These days, a CT perfusion of the brain may be performed and analyzed by artificial intelligence to delineate the ischemic core infarct from the penumbra (that is, brain tissue irreversibly affected by the stroke versus tissue around the core infarct that is at risk of infarction but can be salvaged if blood flow is re-established).
Without resolution of blood supply, cells in the penumbra lose proper ion homeostasis, structural integrity, and ability to complete transcription and protein synthesis required for apoptotic cell death. Thus, they are more likely to die by necrosis and elicit extensive neuroinflammation. It may be therapeutic to block this progression of events to counteract cell death by necrosis, reduce inflammation, and optimally promote post-stroke neurogenesis. However, no such treatment is currently used in the standard of care for AIS.
Two primary treatments exist for patients who present with AIS. The first is pharmacological thrombolysis with recombinant tissue plasminogen activator (rtPA). Patients who present with AIS within 3–4.5 h, and meet safety criteria for drug administration, can improve after rtPA treatment [12, 13]. More recently, in patients who present with LVO stroke, performing an endovascular thrombectomy using catheters and stents can improve outcomes up to 24 h after onset [10, 11, 14,15,16,17,18,19]. An LVO stroke is defined as a clot in the intracranial portion of the internal carotid artery (ICA), the first segment of the middle cerebral artery (MCA) or the anterior cerebral artery (ACA).
Despite these two available treatment options, almost 80% of patients who present with AIS do not receive acute treatment. The limited time window of rtPA administration, and the patients who either do not have an LVO stroke or present after the time window for EVT, or those in whom the stroke has already occurred, renders treatment futile and potentially dangerous. This has resulted in the large majority of patients being left with debilitating effects related to their stroke.
Clinical Evidence for Neurological Recovery After AIS
Past clinical trials have targeted key steps in the pathophysiology of ischemic stroke for therapeutic adjustment. These pharmacological therapies were intended to optimally promote post-stroke neurological recovery, a process that involves both neurogenesis and neuroplasticity, by counteracting cell death and reducing inflammation (Table 1) [20,21,22,23,24,25,26,27,28,29,30,31].
Without oxygen, glycolytic metabolism in neural tissue generates cellular acidosis that leads to loss of substrates and low pH, which halts ATP production. Subsequently, Na+-K+ ATPases fail to maintain homeostatic membrane potential [32, 33]. Increased intracellular [Na+] leads to depolarization that allows an influx of Ca2+ into cells [34]. High intracellular [Ca2+] activates calpain proteases, phospholipases, and endonucleases which degrade the integrity of cellular structures, membranes, and nuclear contents, thus leading to cell death [35, 36]. Calcium channel blockers have been tested in clinical trials and show variable effects on stroke outcome [25,26,27].
Widespread depolarization causes neurons to fire in the core infarct and release neurotransmitters. Synaptic release of glutamate is particularly significant as it acts as an agonist on N-methyl-D-aspartate (NMDA) receptors and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate receptors to further disrupt calcium homeostasis [37,38,39,40]. Glutamate also acts as an agonist on metabotropic receptors that increase proteolysis and lipolysis, and stimulate protein kinase activity, which alters the function of receptors, membrane channels, and ion translocases by promoting protein phosphorylation [40]. NMDA and AMPA antagonists have been tested in clinical trials and either did not affect or worsened outcomes [31,32,33].
Mitochondria sequester the high levels of Ca2+ in hypoxic cells, damaging the organelles that are already failing to supply the cell with energy due to a lack of oxygen, leading to release of damaging reactive oxygen species (ROS). Due to high [Ca2+], mitochondrial depolarization and swelling ensue and the mitochondrial transition pore opens. Subsequently, organellar contents such as cytochrome C are released and initiate apoptosis [34]. Furthermore, the activity and expression of enzymes like nitric oxide synthase (NOS) are induced, favoring the production of reactive oxygen species (ROS) such as peroxynitrite [41]. Free radical scavengers have been tested in clinical trials and have no effect on stroke outcomes [23, 24].
When pushed out of ion homeostasis, cells in the penumbra lose structural integrity, lose transcription and protein synthesis abilities required for controlled cell death, and are more likely to elicit neuroinflammation. The above pharmacological strategies to promote post-stroke neurological recovery largely aimed to make an environment conducive to neurogenesis and neuroplasticity after injury by interrupting neuroinflammation caused by cell death. Largely, these pharmacological approaches were unsuccessful, potentially because they only modified one pathological feature of stroke in isolation. Future treatments could ideally modify multiple features of the complex pathophysiology of stroke to greater effect. Interestingly, the choroid plexus and meninges emerge as key sites for this therapeutic adjustment and require new research approaches to uncover new therapeutic strategies.
The Current Clinical Relevance of Choroid Plexus and Meninges in AIS
Due to their important roles in neuroinflammation, discussed in later sections, the choroid plexus and meninges are clinically relevant to the pathophysiology of AIS but have not yet been targeted in standard clinical therapies. Anti-inflammatory drugs to treat stroke have been tested but are largely administered orally or by the intravenous route, and enter the brain through the blood-brain barrier (BBB), bypassing the meninges and choroid plexus [42]. Drug delivery through the brain-cerebrospinal fluid (CSF) barrier has been implicated in transmeningeal placement of drug-delivering devices in people with focal epilepsy [43], and this could be a model for drug delivery modified for stroke treatment. Efforts to harness the choroid plexus for potential drug delivery, biotherapeutics, and gene therapy to increase beneficial biological agents in the CSF are early in development, but have merit [44]. The ability to modulate what choroid plexus epithelial cells secrete into the CSF would be particularly useful if it could reduce the detrimental immune cell entry to the central nervous system (CNS) through the meninges and choroid plexus, which is so damaging in ischemic injury [45]. Most of the work to understand the roles of the meninges and choroid plexus in brain disease have been conducted in pre-clinical mouse models. More translational studies are necessary to understand how the meninges and choroid plexus can be targeted for therapy after stroke in humans.
New Mechanistic Insights for the Care of AIS
Adult Neurogenesis Under Physiological Conditions
Only in the past few decades has it become widely accepted that the CNS retains plasticity throughout adulthood. Previously, convention dictated the vertebrate brain to be fixed, nonregenerative, and unchanging in adults, as described by renowned histologist and Nobel Prize laureate Ramón y Cajal [46]. The first evidence of neurogenesis in adults was provided by Josef Altman who detected the creation of adult-born neurons and glia in the rat brain by labeling proliferating cells with tritiated thymidine. Subsequent in vitro evidence for stem cells in the subventricular zone (SVZ) and subgranular zone (SGZ) later followed [47,48,49]. Since then, a variety of factors have been demonstrated to upregulate neurogenesis in mammals, including exercise, environmental enrichment, antidepressants, pregnancy, stroke, and injury [50,51,52,53,54,55,56,57], while other factors have the opposite effect and impede adult neurogenesis, such as stress, mental illness, aging, excess alcohol and opioid use, and neurodegenerative diseases [58,59,60,61,62,63].
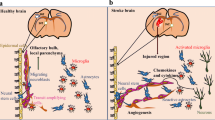
In healthy basal conditions, adult neurogenesis has been demonstrated in several brain regions, some of which are better described than others. Currently, there are two well-described neurogenic niches in the adult mouse and human brains where adult neural stem cells (NSCs) reside: the dentate gyrus (DG) of the hippocampus and the walls of the lateral ventricles, including the ventricular-subventricular zone (V-SVZ; Fig. 2). Adult NSCs or precursor cells in the DG occur in the two- to three-cell layer thick SGZ and give rise to excitatory glutamatergic neurons that end up in the DG granule cell layer and play an important role in spatial and temporal memory processing [64]. In the walls of the lateral ventricles, adult NSCs occur in the SVZ and give rise to neuroblasts that travel through the rostral migratory stream (RMS) to the olfactory bulb (OB), particularly in mice, where they can become interneurons that play an important role in olfaction-related behaviors [65].

Ventricular neurogenesis in the adult mouse and human brain. Subventricular zone (SVZ) neurogenesis in the mouse (left) and human (right) brain. Sagittal (top) or coronal (bottom) sections are shown to illustrate how neural stem cells (NSCs) residing in the SVZ give rise to neuroblasts that migrate along the rostral migratory stream (RMS) to the olfactory bulb (OB) and then differentiate into mature neurons. The final destination of SVZ-derived neurons is different between mice and humans. In mice, the SVZ more prominently gives rise to neurons in the OB (thick arrow, top left) but can contribute new neurons elsewhere as well (thin arrows, top left). In humans, the SVZ does not contribute many new neurons to the OB (thin arrow, top right) but does contribute to new neurons in the nearby striatum (thick arrows, top right).
In humans, the patterns of adult neurogenesis are different, but NSCs are believed to reside in the adult human SGZ and SVZ. While SGZ neurogenesis does contribute new neurons to the human hippocampus as in mice, SVZ neurogenesis contributes more new neurons to the human striatum than the olfactory bulb, unlike the mouse [66, 67]. For more information, the reader is referred to two review articles that summarize the literature and controversy on adult neurogenesis in humans [68, 69], including the ongoing debate about hippocampal neurogenesis in humans. Importantly, neurogenesis is relatively rare under basal conditions for adult humans, but glial and other cell types are regularly replaced in the brain. Some glia are likely generated from multipotent stem cells capable of giving rise to neurons, glia, and other cells in the brain [70, 71]. Regardless of the true nature of neurogenesis in adult humans, neuroplastic processes aside from neurogenesis, such as gliogenesis, axonogenesis, and synaptogenesis, are implicated in stroke recovery [72]. Outside of the murine SGZ and SVZ, adult neurogenesis has been proposed to occur and, in some cases, extensively demonstrated in regions, such as the hypothalamus. There is disagreement in the field about how extensively other niches contribute to adult neurogenesis due to a lack of consistent markers for NSC detection, but neural stem and progenitor cells have been located in the mouse cortex, striatum, cerebellum, and elsewhere [70].
Cortical Neurogenesis in Response to Stroke and Other Injury
In normal conditions, the SGZ and SVZ are the most significant sites of neurogenesis in the adult mouse and human brain, but injury induces neurogenesis in other regions, like the cortex, to varying degrees and according to severity of injury [73,74,75]. While in basal conditions mouse neuroblasts normally differentiate from NSCs in the SVZ and migrate along the RMS to the OB, in response to cerebral ischemia the neuroblasts display increased presence in the RMS and stray from the traditional route to the surrounding parenchyma and striatal ischemic penumbra [76,77,78,79]. The same is true in human brains after cerebral ischemia [57, 73, 80,81,82,83], indicating that this niche may be significant for post-stroke neurorecovery in humans.
Proliferating cells in the mouse SGZ can be found after ischemia, and they migrate to the granule cell layer of the DG as in normal conditions, but do not migrate outside of the hippocampus [76]. Since SGZ neuroblast migration is regionally restricted in this manner, the SVZ is a more prominent source of the neuronal precursors that migrate to injured brain areas after focal cerebral ischemia. While hippocampal neurogenesis does not contribute new neurons or glia to sites of ischemia outside of the hippocampus, it has been associated with post-stroke cognitive impairment and recovery as new neurons contribute to remodeling in the hippocampus that may preserve short-term, but impair long-term memory [80].
Very few new cortical neurons are created in adulthood under normal conditions. Previous reports have demonstrated that newborn cortical neurons make up 0.005–0.03% of the existing neurons in the cerebral cortex [81,82,83,84,85]. However, brain injury increases that figure by 0.06–1% of total cortical neurons [79, 86,87,88,89]. Data indicate that the newborn neurons that are generated in the cortex do not derive solely from precursor cells of the SVZ [79]. NSCs capable of giving rise to cortical neurons and glia have also been detected within the cortex itself upon induction of neurogenesis, and we have described a novel adult quiescent stem cell in the brain, marked by telomerase reverse transcriptase (TERT), which we also observe in small numbers in the adult mouse cortex [70]. Below we will discuss cortical NSCs which have been detected after induction by various techniques aside from just ischemia. These data are worth considering in the stroke context, because stroke may induce neurogenesis from cortical NSCs in a similar manner to their induction by other lesions.
In the cortical white matter of humans suffering from epilepsy, arterial aneurysm, or traumatic injury, NSCs have been isolated and demonstrated to make neurospheres that can give rise to fully differentiated neurons and glia, even when transplanted to fetal rat brains [90]. NSCs and neural progenitor cells (NPCs) of adult rat cortical white matter were also induced by laser lesions to the brain [91].
In cortical gray matter, there are cells expressing NG2, an integral membrane proteoglycan expressed by glial precursor cells, that are reported to generate new cortical GABAergic interneurons [82]. While NG2+ cells have been shown to give rise to neurons and glia in other neurogenic niches [92,93,94,95,96,97,98,99], it will be necessary to figure out whether NG2+ cells of the cortex are progenitors capable of generating neurons and glia, or glia alone. NSCs and NPCs distributed in the first and most superficial cortical layer of rats were discovered upon focal laser-lesion of the visual cortex. These NSCs were defined as nestin+ or vimentin+ cells since many NSCs express these markers, but direct evidence of neurogenesis from these cells was not shown [91]. However, separate studies were able to directly label putative proliferating NPCs with a GFP-expressing retroviral vector in the first cortical layer of adult rodents and show they were capable of producing subclasses of GABAergic interneurons [79].
In the perivascular regions of the cerebral cortex, NSCs were located after stroke and were positive for nestin and some pericyte markers [100], indicating that stem/progenitor cells may be using the vasculature as guides for migration. Nestin+ NSCs were also discovered in the leptomeninges surrounding the cerebral cortex where cortical inhibitory and pyramidal neurons were generated after electrical stimulation of the amygdala [101]. Taken together, these studies favor the presence of distributed NSC populations in the cortical parenchyma, but do not adequately address the lineage relationship these precursor cells might share with each other and to NSCs of the SVZ. It is also not clear if these NSCs are true multipotent stem cells or committed NPCs. Lineage tracing experiments may help delineate whether some of these observed NSC/NPCs are tissue resident stem cells or migrating cells that share a common origin.
NSCs may give rise to cortical neurons in the cortex and appear to be differentially activated by injury according to type and severity. Minor injury (via 10 min of carotid artery occlusion) elicited the first cortical layer and leptomeningeal NSC/NPCs to generate new neurons. However, major injuries (focal cerebral ischemia from 90-min or 7–90-day permanent clamp, aspiration, laser-lesion, or chromophore targeted neuronal degeneration) caused neurogenesis specifically from the SVZ, perivascular regions, gray and white matter, and the first cortical layer [76, 79, 87, 89, 91, 102]. The widespread distribution of NSCs that can be differentially activated according to severity and type of injury may be beneficial for achieving a tailored and robust neurogenic response to ischemic injury, and may be able to be exploited or “pushed” to promote regeneration in a clinical setting.
Novel Roles for the Meninges and Choroid Plexus as Adult Stem Cell Niches
The meninges are a novel niche for neural precursor cells, and this population is particularly attractive as a therapeutic target given the unique qualities of being widespread across the brain surfaces, closely associated with cortical tissue and vasculature, and amenable to cell trafficking and migration [103]. The meninges are composed of the dura mater, arachnoid mater, and pia mater (superficial to deep layers) with a CSF-filled subarachnoid space. The leptomeninges are the deeper two meningeal layers previously described to harbor nestin+ NSCs, some of which are proliferative in rat brains from development through adulthood [104]. Other studies have described stem cell-like populations in adult meninges that activate upon CNS injury by increasing proliferation and migration to the lesion [101, 105,106,107,108]. Nakagomi and colleagues were able to show that nestin+ cells taken from meninges could differentiate to neurons and glia in vitro and in vivo after transplantation [106]. Meningeal NSCs reside in a widespread niche that encompasses the brain and spinal cord, possibly allowing this tissue to serve as a pathway for delivery of neural precursors generated in classical neurogenic niches to distant sites. Supporting this idea, the meningeal extracellular matrix (ECM) is known to have fractones, which are specialized structures that bind extracellular regulators of proliferation and migration and concentrate them to increase their potency [109, 110].
In both rodent and human brains, N-sulfated heparin sulfate structures play an important role in cell migration and proliferation. They are highly associated with the SVZ and hippocampal SGZ and form continuous connections between these two neurogenic niches and the OB, RMS, sub-callosum, subcapsule zones, and importantly, also the meninges [111]. Altogether, these characteristics of the meninges and their association with neurogenic niches indicate that the meninges may be able to guide stem cell migration across various brain regions, an important process for injury response and regeneration. Recent work also indicates that the meninges are involved in stroke-induced local brain inflammation [112]. Further studies could trace the fate of meningeal NPCs and other regenerative cell types in order to clarify the functional significance of this niche, and to determine the potential role of meninges in brain neurogenesis and recovery after stroke. Data have shown that progenitors originating from the meninges can migrate into the cortex and give rise to neurons in neonatal mice, but this has not yet been demonstrated in adults [113]. More work will be required to confirm if there are resident stem/progenitor cells in the adult meninges that can migrate into parenchymal tissues and contribute to neurogenesis, or if these detectable stem/progenitor cells are just migrating through the meninges from other brain regions.
The choroid plexus (ChP), like the meninges, is also proposed to be a stem cell niche in the adult brain. The ChP is a vascularized organ that is present within the ventricular cavity and serves to secrete CSF into all the interconnected ventricles of the brain. In mice and humans, CSF circulates unidirectionally from the lateral ventricles, to the third, and then fourth ventricle until it is reabsorbed into circulation. Reports show the ChP contains putative neural stem cells capable of giving rise to neural cells in vitro and after transplantation into injured spinal cords [114,115,116]. We also observe TERT+ adult stem cells in the meninges and ChP, both regions of significant cell proliferation and differentiation following lineage tracing of these progenitor cells [70]. Some in vivo evidence shows that ChP cells are potential neural precursor cells in the adult mammalian brain, due to their potential to proliferate and generate cells expressing neuronal nuclear antigen (NeuN), a mature neuronal cell marker, and glial fibrillary acidic protein (GFAP), a glial/astrocytic cell marker [117]. Beyond their potential roles as stem cell niches, the ChP and meninges are vital for processes that greatly impact post-stroke neurogenesis, including neuroinflammation, immune trafficking, glial activation, and glymphatic circulation. The ChP also secretes factors that can promote neural recovery and regeneration, such as nerve growth factors [118].
Neuroinflammation and Astrocyte Involvement After Stroke Impacts Neuroplasticity
Astrocytes are critical players in carrying out adaptive responses to neuroinflammation after acute stage stroke by restricting extensive tissue damage and helping return cells to homeostasis [119,120,121]. Reactive astrocytes surround ischemic stroke lesions and upregulate intermediate filament proteins GFAP or nestin, which serve as platforms for signaling in cell stress responses [122, 123]. Astrocytes in the penumbra proliferate and some migrate toward the infarct border to form a glial scar. This is important in acute phase stroke because reactive astrocytes secrete ECM molecules to form a scar, which closes off the area of damage and prevents infiltrating leukocytes from leaving the infarct to invade healthy tissue, where neuroinflammation would be damaging. Unfortunately, reactive astrocytes also limit the ability of new neurons to migrate or regenerate in damaged tissue. For instance, ephrin-A5 produced by reactive astrocytes is known to limit axonal sprouting and functional recovery after injury [124]. Since reactive gliosis may become detrimental at later stages of injury, inhibition of growth-inhibiting chondroitin sulfate proteoglycans produced by astrocytes and oligodendrocyte precursors can improve sprouting of axons after trauma [125,126,127,128,129,130,131]. Reactive astrocytes can also disrupt neurogenesis purely by mechanical means rather than through signaling mechanisms. In the SVZ after ischemic stroke, reactive astrocytes disrupt neuroblast migration scaffolding with their processes that cause aberrant reorganization of the niche [132]. In response to neuroinflammation, reactive astrogliosis is beneficial for immediately restricting the site of damage but may be detrimental if it persists beyond the acute phase by blocking endogenous repair via neuroplasticity and neurogenesis.
The complement system may be a good target for modulating astrocyte response to ischemic injury. Mice deficient in C3 and subjected to focal cerebral ischemia showed smaller infarcts and reduced neurological impairment 1 to 7 days after transient ischemia, likely resulting from decreased neutrophil chemotaxis and oxidative stress brought on by C3a, one of the cleavage products of C3 [133, 134]. Furthermore, mice treated with antagonism of C3aR, the receptor through which the anaphylatoxin C3a activates endothelial cells and recruits leukocytes to the brain, displayed enhanced endogenous neurogenesis [135]. While this suggests that C3aR signaling is deleterious in the acute phase after stroke for its inflammatory effect, there are some positive effects of C3a and C3aR signaling specifically in astrocytes.
After permanent focal cerebral ischemia in adult mice, the number of newborn neurons around the infarct was decreased due to C3aR deficiency, but increased from C3a overexpression [136]. In response to C3a, astrocytes with increased C3aR expression after ischemia alter their intracellular signaling [137], express cytokines IL-6, IL-8, and nerve growth factor [138,139,140], and promote their own survival by inhibiting apoptosis [141]. Astrocytes are demonstrably required for C3a to promote neurorecovery in the context of excitotoxicity, which is similar to the stroke environment [142], and C3aR signaling can activate NSCs to undergo neuronal differentiation and contribute to neurogenesis in adult mice [143, 144]. Together, these data suggest that C3aR signaling in astrocytes can improve neurological outcome after stroke. Thus, targeting this complement pathway to promote post-stroke neurorecovery will have to balance the neuroprotective effects of C3a/C3aR mediated by astrocytes, NSCs, and possibly other cells with the inflammatory effect of C3a/C3aR signaling in the classical complement pathway.
Targeting the Meninges and Choroid Plexus for Modulating Neuroinflammation
Traditionally, the meninges and ChP are studied less for their potential roles as NSC niches than for their regulatory power over post-stroke neurogenesis. After stroke, protective inflammatory processes are initiated in response to damage-associated molecular patterns, or markers of cellular damage generated from necrotic or apoptotic cells and cell debris. Activation of resident microglia and astrocytes gives way to expression of cytokine and adhesion molecules in the cerebrum to recruit peripheral leukocytes, such as neutrophils and other granulocytes, monocyte-derived macrophages, and eventually lymphocytes. Leukocyte infiltration peaks 3 days after permanent and transient ischemia but continues for about 1 week [145, 146]. Hypoxia causes damage to the capillary endothelium, allowing neutrophils to be among the first to infiltrate through the blood-brain barrier (BBB) and generate free radicals that are neurotoxic if over-generated [147, 148]. Acute neuroinflammation can be beneficial by facilitating phagocytosis and clearance of cellular damage, but if it persists too long or is inadequately controlled, it inhibits neurogenesis and recovery [149]. Accordingly, blocked lymphocyte trafficking causes infarct volume in cerebral ischemia models to decrease while improving outcome [150].
Highly significant routes of immune cell entry to the cerebrum include the meningeal compartment and the ChP. Proinflammatory T-cells accumulate in the meninges early after stroke, and lymphocytes from the ChP are recruited to peri-infarct brain tissue in response to cerebral ischemia [151]. Cortical stroke is also accompanied by elevated macrophages in the ChP and CSF [152] and by elevated expression of adhesion molecules, chemokines, and other mediators of monocyte-derived macrophage chemotaxis in the ChP [152]. Macrophages migrate to the ischemic hemisphere when administered into the CSF, and when primed toward an anti-inflammatory phenotype before administration they improved recovery of motor and cognitive function after stroke with no effect on infarct volume [152]. A better understanding of peripheral immune access to the CNS via meninges, ChP, and brain vasculature and glymphatics will advise strategies for modulating inflammation to optimize neurogenesis in the recovery of stroke victims.
In addition to regulating immune trafficking, the ChP regulates processes of neuroplasticity. The ChP actively secretes factors into the CSF that when sensed by cells, serve as migratory cues for newly generated neurons [153]. CSF factors are also implicated in actively maintaining stem cell quiescence [154]. Other ChP-secreted factors support proliferation of SVZ cells in vitro to form neurospheres consisting of multipotent cells [155]. The ChP is highly important in its secretory role because it makes up the blood-CSF barrier, which determines what soluble factors from the blood may enter the CSF. The stringency of the blood-CSF gateway is influenced by whole body signals where the ChP acts as a sensor integrating and responding to physiological signals from the circulation, nervous system, and immune system [156,157,158]. The ChP serves as an important secretory tissue that regulates aspects of both neuroinflammation and neuroplasticity. Thus, potentially altering ChP secretory behavior and gene expression to control neuroinflammation and promote neurogenesis could be a useful therapy for the stroke patient.
Targeting the Meninges and Choroid Plexus to Restore Glymphatic Perfusion and Improve Stroke Recovery
Astrocytes are particularly crucial for their role in the glymphatic system (GS) and in maintaining the BBB. Here, the most salient points pertaining to the glymphatic system as a target for stroke treatment will be discussed. For more depth, the reader is referred to Lv et al. which provides novel insights into the roles of GS impairment in stroke [159], but we will specifically cover its relevance to post-stroke neurogenesis and neuroplasticity. In brief, the glymphatic system begins at the ChP which produces CSF by setting up an osmotic gradient that facilitates the movement of water from blood, across the choroid plexus epithelium, and into the ventricles. CSF flows through the four interconnected ventricles of the brain and permeates the subarachnoid space of the meninges [160,161,162,163]. The middle and anterior cerebral arteries run through the subarachnoid space and their arterioles penetrate the parenchyma. These arterioles are surrounded by CSF-filled perivascular spaces that narrow and disappear as arterioles give way to capillaries thinly covered by astrocytic endfeet, which help maintain the BBB. The perivascular space CSF is separated from the brain parenchyma by the leptomeninges, a basal lamina, and astrocytic endfeet through which CSF may flow out with little resistance. Postcapillary venules have perivenous spaces into which interstitial fluid (ISF) may flow, carrying metabolites or other wastes to be drained out of the brain toward the cervical lymphatic system [164, 165]. A more comprehensive description of the glymphatic system can be found in Jessen et al. [166].
Impaired glymphatic perfusion in the acute phase of ischemic stroke in mice has been demonstrated by contrast-enhanced MRI [167]. Furthermore, embolic ischemic stroke has been shown to transiently inhibit glymphatic flow [167], which could exacerbate tissue injury due to accumulation of metabolic waste and debris from the injury. In stroke, the innate inflammatory response and edema formation initiates in the perivascular spaces [168], but it is unclear whether impaired glymphatic perfusion results from increased CSF influx, a backlog of ISF, or both [169].
Astrocytes are critical to the exchange of CSF and ISF because their endfeet have aquaporin AQP4 channels and other ion transporters that facilitate the movement of water between the CSF and the parenchymal ISF. The lack of AQP4 polarization in astrocytic cell membranes after stroke disrupts glymphatic functioning, which relies on the proper transport of water [170]. More recent data highlights the controversy over the role of AQP4 in ischemic stroke with some showing knockout, siRNA knockdown, or inhibition of the channel reduces edema and occasionally infarct size [171,172,173], whereas others show the opposite role where AQP4 knockout aggravates injury and increases infarct size [174]. Proper CSF circulation and glymphatic perfusion is vital for post-ischemic stroke neurogenesis and neuroplasticity to be efficacious. For instance, neuronal survival was reduced due to microinfarcts in the parietal cortex of mice, but was ameliorated by overexpression of Slit2, which improves glymphatic clearance [175]. In order for therapies to improve neuroplasticity and neurogenesis after stroke, they must address impaired glymphatic perfusion to ensure survival of new or existing neurons and glia.
Conclusions
Over the past 30 years, the evidence of neuroplasticity and neurogenesis in humans under basal conditions and in response to injury has identified the exciting possibility of improving neurological recovery after ischemic stroke (Fig. 3). Over time, the focus has shifted from a neuron-centric view of stroke treatment to a niche-centric view. Attention is increasingly focused on glial and other cell types that modify the environment of neurons to salvage them and promote plasticity. Furthermore, the goal of pharmacologically modifying a single step of a vastly complex ischemic stroke pathophysiology loses favor because this approach is not sufficient to restore a diseased niche to healthy conditions for neurons and glia to survive. Thus, modulating astrocyte activity in the face of neuroinflammation has become paramount to creating an environment conducive to neurorecovery. Various immunomodulation strategies have also been explored to reduce the exacerbation of stroke by inflammatory mediators. More recently, addressing impaired glymphatic perfusion after ischemic stroke poses a way to clean up the environment by clearing out inflammatory mediators, neurotoxic metabolites, and other accumulated wastes. Importantly, the ChP and the meninges are vital brain niches for each of these functions, as they are integral sites for neuroimmune interactions, glymphatic perfusion, and niche signaling pertinent to NSCs and neurogenesis. Furthermore, they are niches for adult tissue stem cells in mice and possibly humans. Much more is known about the roles of meninges and ChP after stroke in mice than in humans, and thus more translational research is essential to fully appreciate the value of the meninges and ChP in post-stroke therapy for human patients.

Impacts of ischemic stroke on adult brain plasticity. Acute ischemic stroke (AIS) initiates neurogenesis, neuroinflammation, gliosis, and disrupts glymphatic perfusion. Coronal section of a mouse (left) and human (right) brain illustrate how ischemia in the cerebral cortex causes tissue damage that activates astrocytes and other glial cells, and also recruits immune cells to the site of tissue damage. Neurogenesis is induced in mice and humans and NSCs give rise to new neurons in the penumbra. Glymphatic perfusion is dysfunctional, causing accumulation of toxic metabolites in the interstitial fluid, which is associated with larger infarct size (white metabolites, white infarct). Improved glymphatic perfusion in AIS is associated with smaller infarct size (gray metabolites, gray infarct). In mice, targeting glial cells to modulate gliosis decreases infarct size (white to gray infarct), as does enhancing neurogenesis and modulating neuroinflammation
Abbreviations
- TOAST:
-
Trial of Org 10172 in Acute Stroke Treatment
- AIS:
-
Acute ischemic stroke
- NIHSS:
-
National Institute of Health Stroke Scale
- ChP:
-
Choroid plexus
- CT:
-
Computerized tomography
- CTA:
-
CT angiogram
- LVO:
-
Large vessel occlusion
- EVT:
-
Endovascular thrombectomy
- rtPA:
-
Recombinant tissue plasminogen activator
- ICA:
-
Internal carotid artery
- ACA:
-
Anterior cerebral artery
- ROS:
-
Reactive oxygen species
- BBB:
-
Blood-brain barrier
- CSF:
-
Cerebral spinal fluid
- CNS:
-
Central nervous system
- SVZ:
-
Subventricular zone
- SGZ:
-
Subgranular zone
- NSCs:
-
Neural stem cells
- DG:
-
Dentate gyrus
- V-SVZ:
-
Ventricular-subventricular zone
- RMS:
-
Rostral migratory stream
- OB:
-
Olfactory bulb
- TERT:
-
Telomerase reverse transcriptase
- NPCs:
-
Neural progenitor cells
- ECM:
-
Extracellular matrix
- GFAP:
-
Glial fibrillary acidic protein
- GS:
-
Glymphatic system
- ISF:
-
Interstitial fluid
- USD:
-
US dollars
- NMDA:
-
N-methyl-D-aspartate
- AMPA:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
References
Collaborators GBDCoD. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: a systematic analysis for the global burden of disease study 2017. Lancet. 2018;392:1736–88.
Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2020 update: a report from the american heart association. Circulation. 2020;141:e139–596.
Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, et al. Heart disease and stroke statistics-2021 update: a report from the american heart association. Circulation. 2021;143:e254–743.
Adams HP, Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. Toast. Trial of org 10172 in acute stroke treatment. Stroke. 1993;24:35–41.
Hankey GJ. Stroke. Lancet. 2017;389:641–54.
Campbell BCV, De Silva DA, Macleod MR, Coutts SB, Schwamm LH, Davis SM, et al. Ischaemic stroke. Nat Rev Dis Primers. 2019;5:70.
Nimjee SM, Akhter A, Zakery A, Herson P. Sex differences in thrombosis as it affects acute ischemic stroke. Neurobiol Dis. 2022;165:105647.
Bushnell CD, Chaturvedi S, Gage KR, Herson PS, Hurn PD, Jiménez MC, et al. Sex differences in stroke: challenges and opportunities. J Cereb Blood Flow Metab. 2018;38:2179–91.
Merwick A, Werring D. Posterior circulation ischaemic stroke. BMJ. 2014;348:g3175.
Albers GW, Marks MP, Kemp S, Christensen S, Tsai JP, Ortega-Gutierrez S, et al. Thrombectomy for stroke at 6 to 16 hours with selection by perfusion imaging. N Engl J Med. 2018;378:708–18.
Nogueira RG, Jadhav AP, Haussen DC, Bonafe A, Budzik RF, Bhuva P, et al. Thrombectomy 6 to 24 hours after stroke with a mismatch between deficit and infarct. N Engl J Med. 2018;378:11–21.
Hacke W, Kaste M, Bluhmki E, Brozman M, Dávalos A, Guidetti D, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317–29.
National Institute of Neurological D, Stroke rt PASSG. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333:1581-1587.
Berkhemer OA, Fransen PS, Beumer D, van den Berg LA, Lingsma HF, Yoo AJ, et al. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. 2015;372:11–20.
Saver JL, Goyal M, Bonafe A, Diener HC, Levy EI, Pereira VM, et al. Stent-retriever thrombectomy after intravenous t-pa vs. T-pa alone in stroke. N Engl J Med. 2015;372:2285–95.
Goyal M, Demchuk AM, Menon BK, Eesa M, Rempel JL, Thornton J, et al. Randomized assessment of rapid endovascular treatment of ischemic stroke. N Engl J Med. 2015;372:1019–30.
Campbell BC, Mitchell PJ, Kleinig TJ, Dewey HM, Churilov L, Yassi N, et al. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N Engl J Med. 2015;372:1009–18.
Jovin TG, Chamorro A, Cobo E, de Miquel MA, Molina CA, Rovira A, et al. Thrombectomy within 8 hours after symptom onset in ischemic stroke. N Engl J Med. 2015;372:2296–306.
Turk AS III, Siddiqui A, Fifi JT, De Leacy RA, Fiorella DJ, Gu E, et al. Aspiration thrombectomy versus stent retriever thrombectomy as first-line approach for large vessel occlusion (compass): a multicentre, randomised, open label, blinded outcome, non-inferiority trial. The Lancet. 2019;393:998–1008.
Davis SM, Lees KR, Albers GW, Diener HC, Markabi S, Karlsson G, et al. Selfotel in acute ischemic stroke: possible neurotoxic effects of an nmda antagonist. Stroke. 2000;31:347–54.
Albers GW, Goldstein LB, Hall D, Lesko LM, Aptiganel Acute Stroke I. Aptiganel hydrochloride in acute ischemic stroke: a randomized controlled trial. JAMA. 2001;286:2673-2682.
Elting JW, Sulter GA, Kaste M, Lees KR, Diener HC, Hommel M, et al. Ampa antagonist zk200775 in patients with acute ischemic stroke: possible glial cell toxicity detected by monitoring of s-100b serum levels. Stroke. 2002;33:2813–8.
Shuaib A, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, et al. Nxy-059 for the treatment of acute ischemic stroke. N Engl J Med. 2007;357:562–71.
A randomized trial of tirilazad mesylate in patients with acute stroke (RANTTAS). The RANTTAS Investigators. Stroke. 1996;27:1453-1458.
Clinical trial of nimodipine in acute ischemic stroke. The American Nimodipine Study Group. Stroke. 1992;23:3–8.
Horn J, de Haan RJ, Vermeulen M, Limburg M. Very early nimodipine use in stroke (VENUS): a randomized, double-blind, placebo-controlled trial. Stroke. 2001;32:461–5.
Liu X, Wang L, Wen A, Yang J, Yan Y, Song Y, et al. Ginsenoside-rd improves outcome of acute ischaemic stroke - a randomized, double-blind, placebo-controlled, multicenter trial. Eur J Neurol. 2012;19:855–63.
Enlimomab Acute Stroke Trial I. Use of anti-icam-1 therapy in ischemic stroke: results of the enlimomab acute stroke trial. Neurology. 2001;57:1428-1434.
Hess DC, Wechsler LR, Clark WM, Savitz SI, Ford GA, Chiu D, et al. Safety and efficacy of multipotent adult progenitor cells in acute ischaemic stroke (MASTERS): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2017;16:360–8.
Kikuchi K, Tanaka E, Murai Y, Tancharoen S. Clinical trials in acute ischemic stroke. CNS Drugs. 2014;28:929–38.
Veltkamp R, Gill D. Clinical trials of immunomodulation in ischemic stroke. Neurotherapeutics. 2016;13:791–800.
Siesjo BK. Pathophysiology and treatment of focal cerebral ischemia. Part II: mechanisms of damage and treatment. J Neurosurg. 1992;77:337-354.
Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–34.
Smith WS. Pathophysiology of focal cerebral ischemia: a therapeutic perspective. J Vasc Interv Radiol. 2004;15:S3-12.
Mazala DA, Grange RW, Chin ER. The role of proteases in excitation-contraction coupling failure in muscular dystrophy. Am J Physiol Cell Physiol. 2015;308:C33-40.
Weber JT. Altered calcium signaling following traumatic brain injury. Front Pharmacol. 2012;3:60.
Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–568.
Simon RP, Swan JH, Griffiths T, Meldrum BS. Blockade of <i>n</i>-methyl-d-aspartate receptors may protect against ischemic damage in the brain. Science. 1984;226:850–2.
Liu X, Hunter C, Weiss HR, Chi OZ. Effects of blockade of ionotropic glutamate receptors on blood-brain barrier disruption in focal cerebral ischemia. Neurol Sci. 2010;31:699–703.
Willard SS, Koochekpour S. Glutamate, glutamate receptors, and downstream signaling pathways. Int J Biol Sci. 2013;9:948–59.
Van Hemelrijck A, Hachimi-Idrissi S, Sarre S, Ebinger G, Michotte Y. Post-ischaemic mild hypothermia inhibits apoptosis in the penumbral region by reducing neuronal nitric oxide synthase activity and thereby preventing endothelin-1-induced hydroxyl radical formation. Eur J Neurosci. 2005;22:1327–37.
Smith CJ, Denes A, Tyrrell PJ, Di Napoli M. Phase II anti-inflammatory and immune-modulating drugs for acute ischaemic stroke. Expert Opin Investig Drugs. 2015;24:623–43.
Madhavan D, Mirowski P, Ludvig N, Carlson C, Doyle W, Devinsky O, et al. Effects of subdural application of lidocaine in patients with focal epilepsy. Epilepsy Res. 2008;78:235–9.
Gonzalez AM, Leadbeater WE, Burg M, Sims K, Terasaki T, Johanson CE, et al. Targeting choroid plexus epithelia and ventricular ependyma for drug delivery to the central nervous system. BMC Neurosci. 2011;12:4.
Solar P, Zamani A, Kubickova L, Dubovy P, Joukal M. Choroid plexus and the blood-cerebrospinal fluid barrier in disease. Fluids Barriers CNS. 2020;17:35.
Cajal RS. Degeneration and regeneration of the nervous system. London: Oxford University Press; 1928.
Altman J. Are new neurons formed in the brains of adult mammals? Science. 1962;135:1127–8.
Altman J, Das GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. 1965;124:319–35.
Altman J. The discovery of adult mammalian neurogenesis. In: Seki T, Sawamoto K, Parent JM, Alvarez-Buylla A, editors. Neurogenesis in the adult brain. Tokyo: Springer Inc; 2011. p. 3–46.
Ma CL, Ma XT, Wang JJ, Liu H, Chen YF, Yang Y. Physical exercise induces hippocampal neurogenesis and prevents cognitive decline. Behav Brain Res. 2017;317:332–9.
Clemenson GD, Deng W, Gage FH. Environmental enrichment and neurogenesis: from mice to humans. Curr Opin Behav Sci. 2015;4:56–62.
Ahlfeld J, Filser S, Schmidt F, Wefers AK, Merk DJ, Glass R, et al. Neurogenesis from sox2 expressing cells in the adult cerebellar cortex. Sci Rep. 2017;7:6137.
Fabel K, Wolf SA, Ehninger D, Babu H, Leal-Galicia P, Kempermann G. Additive effects of physical exercise and environmental enrichment on adult hippocampal neurogenesis in mice. Front Neurosci. 2009;3:50.
Anacker C, Zunszain PA, Cattaneo A, Carvalho LA, Garabedian MJ, Thuret S, et al. Antidepressants increase human hippocampal neurogenesis by activating the glucocorticoid receptor. Mol Psychiatry. 2011;16:738–50.
Shingo T, Gregg C, Enwere E, Fujikawa H, Hassam R, Geary C, et al. Pregnancy-stimulated neurogenesis in the adult female forebrain mediated by prolactin. Science. 2003;299:117–20.
Lindvall O, Kokaia Z. Neurogenesis following stroke affecting the adult brain. Cold Spring Harb Perspect Biol. 2015;7(11):a019034.
Zheng W, ZhuGe Q, Zhong M, Chen G, Shao B, Wang H, et al. Neurogenesis in adult human brain after traumatic brain injury. J Neurotrauma. 2013;30:1872–80.
Schoenfeld TJ, McCausland HC, Morris HD, Padmanaban V, Cameron HA. Stress and loss of adult neurogenesis differentially reduce hippocampal volume. Biol Psychiatry. 2017;82:914–23.
Schoenfeld TJ, Cameron HA. Adult neurogenesis and mental illness. Neuropsychopharmacology. 2015;40:113–28.
Babcock KR, Page JS, Fallon JR, Webb AE. Adult hippocampal neurogenesis in aging and Alzheimer’s disease. Stem Cell Rep. 2021;16:681–93.
Golub HM, Zhou QG, Zucker H, McMullen MR, Kokiko-Cochran ON, Ro EJ, et al. Chronic alcohol exposure is associated with decreased neurogenesis, aberrant integration of newborn neurons, and cognitive dysfunction in female mice. Alcohol Clin Exp Res. 2015;39:1967–77.
Zhang Y, Loh HH, Law PY. Effect of opioid on adult hippocampal neurogenesis. ScientificWorldJournal. 2016;2016:2601264.
Zhang Y, Xu C, Zheng H, Loh HH, Law PY. Morphine modulates adult neurogenesis and contextual memory by impeding the maturation of neural progenitors. PLoS One. 2016;11:e0153628.
Aimone JB, Deng W, Gage FH. Resolving new memories: a critical look at the dentate gyrus, adult neurogenesis, and pattern separation. Neuron. 2011;70:589–96.
Sakamoto M, Kageyama R, Imayoshi I. The functional significance of newly born neurons integrated into olfactory bulb circuits. Front Neurosci. 2014;8:121.
Moreno-Jimenez EP, Terreros-Roncal J, Flor-Garcia M, Rabano A, Llorens-Martin M. Evidences for adult hippocampal neurogenesis in humans. J Neurosci. 2021;41:2541–53.
Ernst A, Alkass K, Bernard S, Salehpour M, Perl S, Tisdale J, et al. Neurogenesis in the striatum of the adult human brain. Cell. 2014;156:1072–83.
Beckervordersandforth R, Rolando C. Untangling human neurogenesis to understand and counteract brain disorders. Curr Opin Pharmacol. 2020;50:67–73.
Kumar A, Pareek V, Faiq MA, Ghosh SK, Kumari C. Adult neurogenesis in humans: a review of basic concepts, history, current research, and clinical implications. Innov Clin Neurosci. 2019;16:30–7.
Jensen^ G, Beaulieu A^, Curtis C, Blaszkiewicz M, Passarelli J, Caron L, Thomas S, Morin T, Aniapam C, Brennan C, Ford C, Stevenson O, Lynes M, Carlone D, Breault D, Townsend* K. Telomerase reverse transcriptase (TERT) as a novel and unique marker for quiescent stem cells in the adult mouse brain. (2022) Under Revised Resubmission
Feliciano DM, Bordey A, Bonfanti L. Noncanonical sites of adult neurogenesis in the mammalian brain. Cold Spring Harb Perspect Biol. 2015;7:a018846.
Dietrichs E. Brain plasticity after stroke—implications for post-stroke rehabilitation. Tidsskr Nor Laegeforen. 2007;127:1228–31.
Nakayama D, Matsuyama T, Ishibashi-Ueda H, Nakagomi T, Kasahara Y, Hirose H, et al. Injury-induced neural stem/progenitor cells in post-stroke human cerebral cortex. Eur J Neurosci. 2010;31:90–8.
Kreuzberg M, Kanov E, Timofeev O, Schwaninger M, Monyer H, Khodosevich K. Increased subventricular zone-derived cortical neurogenesis after ischemic lesion. Exp Neurol. 2010;226:90–9.
Ohira K, Hagihara H, Miwa M, Nakamura K, Miyakawa T. Fluoxetine-induced dematuration of hippocampal neurons and adult cortical neurogenesis in the common marmoset. Mol Brain. 2019;12:69.
Jin K, Sun Y, Xie L, Peel A, Mao XO, Batteur S, et al. Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol Cell Neurosci. 2003;24:171–89.
Hou SW, Wang YQ, Xu M, Shen DH, Wang JJ, Huang F, et al. Functional integration of newly generated neurons into striatum after cerebral ischemia in the adult rat brain. Stroke. 2008;39:2837–44.
Bendel O, Bueters T, von Euler M, Ove Ogren S, Sandin J, von Euler G. Reappearance of hippocampal ca1 neurons after ischemia is associated with recovery of learning and memory. J Cereb Blood Flow Metab. 2005;25:1586–95.
Ohira K, Furuta T, Hioki H, Nakamura KC, Kuramoto E, Tanaka Y, et al. Ischemia-induced neurogenesis of neocortical layer 1 progenitor cells. Nat Neurosci. 2010;13:173–9.
Cuartero MI, de la Parra J, Perez-Ruiz A, Bravo-Ferrer I, Duran-Laforet V, Garcia-Culebras A, et al. Abolition of aberrant neurogenesis ameliorates cognitive impairment after stroke in mice. J Clin Invest. 2019;129:1536–50.
Bernier PJ, Bedard A, Vinet J, Levesque M, Parent A. Newly generated neurons in the amygdala and adjoining cortex of adult primates. Proc Natl Acad Sci U S A. 2002;99:11464–9.
Dayer AG, Cleaver KM, Abouantoun T, Cameron HA. New gabaergic interneurons in the adult neocortex and striatum are generated from different precursors. J Cell Biol. 2005;168:415–27.
Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ. Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci. 1999;2:260–5.
Gould E, Vail N, Wagers M, Gross CG. Adult-generated hippocampal and neocortical neurons in macaques have a transient existence. Proc Natl Acad Sci U S A. 2001;98:10910–7.
Koketsu D, Mikami A, Miyamoto Y, Hisatsune T. Nonrenewal of neurons in the cerebral neocortex of adult macaque monkeys. J Neurosci. 2003;23:937–42.
Ohira K, Takeuchi R, Shoji H, Miyakawa T. Fluoxetine-induced cortical adult neurogenesis. Neuropsychopharmacology. 2013;38:909–20.
Chen J, Magavi SS, Macklis JD. Neurogenesis of corticospinal motor neurons extending spinal projections in adult mice. Proc Natl Acad Sci U S A. 2004;101:16357–62.
Jiang W, Gu W, Brannstrom T, Rosqvist R, Wester P. Cortical neurogenesis in adult rats after transient middle cerebral artery occlusion. Stroke. 2001;32:1201–7.
Magavi SS, Leavitt BR, Macklis JD. Induction of neurogenesis in the neocortex of adult mice. Nature. 2000;405:951–5.
Nunes MC, Roy NS, Keyoung HM, Goodman RR, McKhann G 2nd, Jiang L, et al. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med. 2003;9:439–47.
Sirko S, Neitz A, Mittmann T, Horvat-Brocker A, von Holst A, Eysel UT, et al. Focal laser-lesions activate an endogenous population of neural stem/progenitor cells in the adult visual cortex. Brain. 2009;132:2252–64.
Aguirre A, Gallo V. Postnatal neurogenesis and gliogenesis in the olfactory bulb from ng2-expressing progenitors of the subventricular zone. J Neurosci. 2004;24:10530–41.
Robins SC, Trudel E, Rotondi O, Liu X, Djogo T, Kryzskaya D, et al. Evidence for ng2-glia derived, adult-born functional neurons in the hypothalamus. PLoS One. 2013;8:e78236.
Aguirre AA, Chittajallu R, Belachew S, Gallo V. Ng2-expressing cells in the subventricular zone are type c-like cells and contribute to interneuron generation in the postnatal hippocampus. J Cell Biol. 2004;165:575–89.
Belachew S, Chittajallu R, Aguirre AA, Yuan X, Kirby M, Anderson S, et al. Postnatal ng2 proteoglycan-expressing progenitor cells are intrinsically multipotent and generate functional neurons. J Cell Biol. 2003;161:169–86.
Dimou L, Simon C, Kirchhoff F, Takebayashi H, Gotz M. Progeny of olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J Neurosci. 2008;28:10434–42.
Komitova M, Zhu X, Serwanski DR, Nishiyama A. Ng2 cells are distinct from neurogenic cells in the postnatal mouse subventricular zone. J Comp Neurol. 2009;512:702–16.
Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (ng2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009;10:9–22.
Platel JC, Gordon V, Heintz T, Bordey A. Gfap-gfp neural progenitors are antigenically homogeneous and anchored in their enclosed mosaic niche. Glia. 2009;57:66–78.
Tatebayashi K, Tanaka Y, Nakano-Doi A, Sakuma R, Kamachi S, Shirakawa M, et al. Identification of multipotent stem cells in human brain tissue following stroke. Stem Cells Dev. 2017;26:787-797.
Ninomiya S, Esumi S, Ohta K, Fukuda T, Ito T, Imayoshi I, et al. Amygdala kindling induces nestin expression in the leptomeninges of the neocortex. Neurosci Res. 2013;75:121–9.
Sundholm-Peters NL, Yang HK, Goings GE, Walker AS, Szele FG. Subventricular zone neuroblasts emigrate toward cortical lesions. J Neuropathol Exp Neurol. 2005;64:1089–100.
Decimo I, Dolci S, Panuccio G, Riva M, Fumagalli G, Bifari F. Meninges: a widespread niche of neural progenitors for the brain. Neuroscientist. 2021;27:506–28.
Bifari F, Berton V, Pino A, Kusalo M, Malpeli G, Di Chio M, et al. Meninges harbor cells expressing neural precursor markers during development and adulthood. Front Cell Neurosci. 2015;9:383.
Decimo I, Bifari F, Rodriguez FJ, Malpeli G, Dolci S, Lavarini V, et al. Nestin- and doublecortin-positive cells reside in adult spinal cord meninges and participate in injury-induced parenchymal reaction. Stem Cells. 2011;29:2062–76.
Nakagomi T, Molnar Z, Nakano-Doi A, Taguchi A, Saino O, Kubo S, et al. Ischemia-induced neural stem/progenitor cells in the pia mater following cortical infarction. Stem Cells Dev. 2011;20:2037-2051.
Nakagomi T, Molnar Z, Taguchi A, Nakano-Doi A, Lu S, Kasahara Y, et al. Leptomeningeal-derived doublecortin-expressing cells in poststroke brain. Stem Cells Dev. 2012;21:2350-2354.
Kumar M, Csaba Z, Peineau S, Srivastava R, Rasika S, Mani S, et al. Endogenous cerebellar neurogenesis in adult mice with progressive ataxia. Ann Clin Transl Neurol. 2014;1:968–81.
Mercier F, Kitasako JT, Hatton GI. Anatomy of the brain neurogenic zones revisited: fractones and the fibroblast/macrophage network. J Comp Neurol. 2002;451:170–88.
Kerever A, Schnack J, Vellinga D, Ichikawa N, Moon C, Arikawa-Hirasawa E, et al. Novel extracellular matrix structures in the neural stem cell niche capture the neurogenic factor fibroblast growth factor 2 from the extracellular milieu. Stem Cells. 2007;25:2146–57.
Mercier F, Arikawa-Hirasawa E. Heparan sulfate niche for cell proliferation in the adult brain. Neurosci Lett. 2012;510:67–72.
Herisson F, Frodermann V, Courties G, Rohde D, Sun Y, Vandoorne K, et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat Neurosci. 2018;21:1209–17.
Bifari F, Decimo I, Pino A, Llorens-Bobadilla E, Zhao S, Lange C, et al. Neurogenic radial glia-like cells in meninges migrate and differentiate into functionally integrated neurons in the neonatal cortex. Cell Stem Cell. 2017;20:360-373.e367.
Kitada M, Chakrabortty S, Matsumoto N, Taketomi M, Ide C. Differentiation of choroid plexus ependymal cells into astrocytes after grafting into the pre-lesioned spinal cord in mice. Glia. 2001;36:364–74.
Itokazu Y, Kitada M, Dezawa M, Mizoguchi A, Matsumoto N, Shimizu A, et al. Choroid plexus ependymal cells host neural progenitor cells in the rat. Glia. 2006;53:32–42.
Huang SL, Shi W, Jiao Q, He XJ. Change of neural stem cells in the choroid plexuses of developing rat. Int J Neurosci. 2011;121:310–5.
Li Y, Chen J, Chopp M. Cell proliferation and differentiation from ependymal, subependymal and choroid plexus cells in response to stroke in rats. J Neurol Sci. 2002;193:137–46.
Stopa EG, Berzin TM, Kim S, Song P, Kuo-LeBlanc V, Rodriguez-Wolf M, et al. Human choroid plexus growth factors: what are the implications for CSF dynamics in Alzheimer’s disease? Exp Neurol. 2001;167:40–7.
Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, et al. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab. 2008;28:468–81.
Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–34.
Pekny M, Pekna M, Messing A, Steinhauser C, Lee JM, Parpura V, et al. Astrocytes: a central element in neurological diseases. Acta Neuropathol. 2016;131:323–45.
Pekny M, Lane EB. Intermediate filaments and stress. Exp Cell Res. 2007;313:2244–54.
Pekny M, Pekna M. Reactive gliosis in the pathogenesis of CNS diseases. Biochim Biophys Acta. 2016;1862:483–91.
Overman JJ, Clarkson AN, Wanner IB, Overman WT, Eckstein I, Maguire JL, et al. A role for ephrin-a5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc Natl Acad Sci U S A. 2012;109:E2230-2239.
Bradbury EJ, Carter LM. Manipulating the glial scar: chondroitinase abc as a therapy for spinal cord injury. Brain Res Bull. 2011;84:306–16.
Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, et al. Chondroitinase abc promotes functional recovery after spinal cord injury. Nature. 2002;416:636–40.
Lee H, McKeon RJ, Bellamkonda RV. Sustained delivery of thermostabilized chabc enhances axonal sprouting and functional recovery after spinal cord injury. Proc Natl Acad Sci U S A. 2010;107:3340–5.
Sharma K, Selzer ME, Li S. Scar-mediated inhibition and CSPG receptors in the CNS. Exp Neurol. 2012;237:370–8.
Zhou X, Ren Y, Han L, Mei M, Xu P, Zhang CZ, et al. Role of the akt pathway in microrna expression of human u251 glioblastoma cells. Int J Oncol. 2010;36:665–72.
Wang X, Hasan O, Arzeno A, Benowitz LI, Cafferty WB, Strittmatter SM. Axonal regeneration induced by blockade of glial inhibitors coupled with activation of intrinsic neuronal growth pathways. Exp Neurol. 2012;237:55–69.
Yick LW, Wu W, So KF, Yip HK, Shum DK. Chondroitinase abc promotes axonal regeneration of Clarke’s neurons after spinal cord injury. Neuroreport. 2000;11:1063–7.
Young CC, van der Harg JM, Lewis NJ, Brooks KJ, Buchan AM, Szele FG. Ependymal ciliary dysfunction and reactive astrocytosis in a reorganized subventricular zone after stroke. Cereb Cortex. 2013;23:647–59.
Mocco J, Mack WJ, Ducruet AF, Sosunov SA, Sughrue ME, Hassid BG, et al. Complement component c3 mediates inflammatory injury following focal cerebral ischemia. Circ Res. 2006;99:209–17.
Wu F, Zou Q, Ding X, Shi D, Zhu X, Hu W, et al. Complement component c3a plays a critical role in endothelial activation and leukocyte recruitment into the brain. J Neuroinflammation. 2016;13:23–23.
Ducruet AF, Zacharia BE, Sosunov SA, Gigante PR, Yeh ML, Gorski JW, et al. Complement inhibition promotes endogenous neurogenesis and sustained anti-inflammatory neuroprotection following reperfused stroke. PLoS One. 2012;7:e38664.
Stokowska A, Pekna M. Complement c3a: shaping the plasticity of the poststroke brain. In: Lapchak PA, Zhang JH, editors. Cellular and molecular approaches to regeneration & repair. Cham: Springer; 2018. p. 521–41.
Sayah S, Jauneau AC, Patte C, Tonon MC, Vaudry H, Fontaine M. Two different transduction pathways are activated by c3a and c5a anaphylatoxins on astrocytes. Brain Res Mol Brain Res. 2003;112:53–60.
Sayah S, Ischenko AM, Zhakhov A, Bonnard AS, Fontaine M. Expression of cytokines by human astrocytomas following stimulation by c3a and c5a anaphylatoxins: specific increase in interleukin-6 mrna expression. J Neurochem. 1999;72:2426–36.
Jauneau AC, Ischenko A, Chan P, Fontaine M. Complement component anaphylatoxins upregulate chemokine expression by human astrocytes. FEBS Lett. 2003;537:17–22.
Jauneau AC, Ischenko A, Chatagner A, Benard M, Chan P, Schouft MT, et al. Interleukin-1beta and anaphylatoxins exert a synergistic effect on ngf expression by astrocytes. J Neuroinflammation. 2006;3:8.
Shinjyo N, de Pablo Y, Pekny M, Pekna M. Complement peptide c3a promotes astrocyte survival in response to ischemic stress. Mol Neurobiol. 2016;53:3076–87.
van Beek J, Nicole O, Ali C, Ischenko A, MacKenzie ET, Buisson A, et al. Complement anaphylatoxin c3a is selectively protective against nmda-induced neuronal cell death. NeuroReport. 2001;12:289–93.
Rahpeymai Y, Hietala MA, Wilhelmsson U, Fotheringham A, Davies I, Nilsson AK, et al. Complement: a novel factor in basal and ischemia-induced neurogenesis. EMBO J. 2006;25:1364–74.
Shinjyo N, Ståhlberg A, Dragunow M, Pekny M, Pekna M. Complement-derived anaphylatoxin c3a regulates in vitro differentiation and migration of neural progenitor cells. Stem Cells. 2009;27:2824–32.
Miro-Mur F, Perez-de-Puig I, Ferrer-Ferrer M, Urra X, Justicia C, Chamorro A, et al. Immature monocytes recruited to the ischemic mouse brain differentiate into macrophages with features of alternative activation. Brain Behav Immun. 2016;53:18–33.
Wattananit S, Tornero D, Graubardt N, Memanishvili T, Monni E, Tatarishvili J, et al. Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci. 2016;36:4182–95.
Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–57.
Garcia-Bonilla L, Moore JM, Racchumi G, Zhou P, Butler JM, Iadecola C, et al. Inducible nitric oxide synthase in neutrophils and endothelium contributes to ischemic brain injury in mice. J Immunol. 2014;193:2531–7.
Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation. 2019;16:142.
Liesz A, Zhou W, Mracsko E, Karcher S, Bauer H, Schwarting S, et al. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. 2011;134:704–20.
Llovera G, Benakis C, Enzmann G, Cai R, Arzberger T, Ghasemigharagoz A, et al. The choroid plexus is a key cerebral invasion route for T cells after stroke. Acta Neuropathol. 2017;134:851–68.
Ge R, Tornero D, Hirota M, Monni E, Laterza C, Lindvall O, et al. Choroid plexus-cerebrospinal fluid route for monocyte-derived macrophages after stroke. J Neuroinflammation. 2017;14:153.
Sawamoto K, Wichterle H, Gonzalez-Perez O, Cholfin JA, Yamada M, Spassky N, et al. New neurons follow the flow of cerebrospinal fluid in the adult brain. Science. 2006;311:629–32.
Delgado AC, Ferron SR, Vicente D, Porlan E, Perez-Villalba A, Trujillo CM, et al. Endothelial NT-3 delivered by vasculature and CSF promotes quiescence of subependymal neural stem cells through nitric oxide induction. Neuron. 2014;83:572–85.
Silva-Vargas V, Maldonado-Soto AR, Mizrak D, Codega P, Doetsch F. Age-dependent niche signals from the choroid plexus regulate adult neural stem cells. Cell Stem Cell. 2016;19:643–52.
Baruch K, Schwartz M. CNS-specific T cells shape brain function via the choroid plexus. Brain Behav Immun. 2013;34:11–6.
Marques F, Sousa JC. The choroid plexus is modulated by various peripheral stimuli: implications to diseases of the central nervous system. Front Cell Neurosci. 2015;9:136.
Strazielle N, Ghersi-Egea JF. Potential pathways for CNS drug delivery across the blood-cerebrospinal fluid barrier. Curr Pharm Des. 2016;22:5463–76.
Lv T, Zhao B, Hu Q, Zhang X. The glymphatic system: a novel therapeutic target for stroke treatment. Front Aging Neurosci. 2021;13:689098.
Damkier HH, Brown PD, Praetorius J. Cerebrospinal fluid secretion by the choroid plexus. Physiol Rev. 2013;93:1847–92.
Brown PD, Davies SL, Speake T, Millar ID. Molecular mechanisms of cerebrospinal fluid production. Neuroscience. 2004;129:957–70.
Ames A 3rd, Higashi K, Nesbett FB. Effects of pco2 acetazolamide and ouabain on volume and composition of choroid-plexus fluid. J Physiol. 1965;181:516–24.
Johanson CE. Choroid plexus–cerebrospinal fluid circulatory dynamics: impact on brain growth, metabolism, and repair. In: Conn, P. M. (eds) Neuroscience in medicine. Humana Press, Totowa, NJ, 2008. https://doi.org/10.1007/978-1-60327-455-5_11
Johnston M, Zakharov A, Papaiconomou C, Salmasi G, Armstrong D. Evidence of connections between cerebrospinal fluid and nasal lymphatic vessels in humans, non-human primates and other mammalian species. Cerebrospinal Fluid Res. 2004;1:2.
Murtha LA, Yang Q, Parsons MW, Levi CR, Beard DJ, Spratt NJ, et al. Cerebrospinal fluid is drained primarily via the spinal canal and olfactory route in young and aged spontaneously hypertensive rats. Fluids Barriers CNS. 2014;11:12.
Jessen NA, Munk AS, Lundgaard I, Nedergaard M. The glymphatic system: a beginner’s guide. Neurochem Res. 2015;40:2583–99.
Gaberel T, Gakuba C, Goulay R, Martinez De Lizarrondo S, Hanouz JL, Emery E, et al. Impaired glymphatic perfusion after strokes revealed by contrast-enhanced MRI: a new target for fibrinolysis? Stroke. 2014;45:3092-3096.
del Zoppo GJ. The neurovascular unit in the setting of stroke. J Intern Med. 2010;267:156–71.
Mestre H, Du T, Sweeney AM, Liu G, Samson AJ, Peng W, et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science. 2020;367:6843.
Schroth G, Klose U. Cerebrospinal fluid flow I. Physiology of cardiac-related pulsation. Neuroradiology. 1992;35:1–9.
Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–63.
Yang C, Liu Z, Li H, Zhai F, Liu J, Bian J. Aquaporin-4 knockdown ameliorates hypoxic-ischemic cerebral edema in newborn piglets. IUBMB Life. 2015;67:182–90.
Pirici I, Balsanu TA, Bogdan C, Margaritescu C, Divan T, Vitalie V, et al. Inhibition of aquaporin-4 improves the outcome of ischaemic stroke and modulates brain paravascular drainage pathways. Int J Mol Sci. 2017;19(1):46.
Zeng XN, Xie LL, Liang R, Sun XL, Fan Y, Hu G. Aqp4 knockout aggravates ischemia/reperfusion injury in mice. CNS Neurosci Ther. 2012;18:388–94.
He XF, Li G, Li LL, Li MY, Liang FY, Chen X, et al. Overexpression of slit2 decreases neuronal excitotoxicity, accelerates glymphatic clearance, and improves cognition in a multiple microinfarcts model. Mol Brain. 2020;13:135.
Acknowledgements
The authors wish to thank Sabrina DiPietro for technical assistance on this manuscript.
Funding
KLT was funded by an NSF-CAREER GR124254, a research grant from the Lisa Dean Moseley Foundation, and start-up funding from The Ohio State University. SMN was funded by NINDS 1R01NS123687-01 and NHLBI 1R44HL152869-01.
Author information
Authors and Affiliations
Contributions
JP, SN, and KT all wrote, revised, and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Passarelli, J.P., Nimjee, S.M. & Townsend, K.L. Stroke and Neurogenesis: Bridging Clinical Observations to New Mechanistic Insights from Animal Models. Transl. Stroke Res. 15, 53–68 (2024). https://doi.org/10.1007/s12975-022-01109-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-022-01109-1