
Abstract
Pyroptosis has been proven to be responsible for secondary brain injury after intracerebral hemorrhage (ICH). A recent study reported that Raf kinase inhibitor protein (RKIP) inhibited assembly and activation of inflammasome in macrophages. Our present study aimed to investigate the effects of RKIP on inflammasome-mediated neuronal pyroptosis and underlying neuroprotective mechanisms in experimental ICH. Here, we showed that RKIP expression was decreased both in cerebrospinal fluid (CSF) samples from patients with ICH and in the peri-hematoma tissues after experimental ICH. In mouse ICH model, activation of RKIP remarkably improved neurological deficits, reduced brain water content and BBB disruption, and promoted hematoma absorption at 24 h after ICH, as well as alleviated neuronal degeneration, reduced membrane pore formation, and downregulated pyroptotic molecules NLRP3, caspase-1 P20, GSDMD-N, and mature IL-1β. Besides, RKIP activation decreased the number of caspase-1 P20-positive neurons after ICH. However, RKIP inhibitor reserved the neuroprotective effects of RKIP at 24 h following ICH. Moreover, RKIP could bind with ASC, then interrupt the assembly of NLRP3 inflammasome. Mechanistically, inhibiting the caspase-1 by VX-765 attenuated brain injury and suppressed neuronal pyroptosis after RKIP inhibitor-pretreated ICH. In conclusion, our findings indicated that activation of RKIP could attenuate neuronal pyroptosis and brain injury after ICH, to some extent, through ASC/Caspase-1/GSDMD pathway. Thus, RKIP may be a potential target to attenuate brain injury via its anti-pyroptosis effect after ICH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Intracerebral hemorrhage (ICH) is a devastating type of stroke with high mortality and morbidity, for which heretofore no effective treatment is available [1]. Pyroptosis was proven to be implicated in the pathogenesis of secondary brain injury after ICH [2]. To be specific, pyroptosis, a novel type of programmed cell death, is characterized by cell swelling, membrane rupture, formation of nonselective pore caused by cleavage of gasdermin D (GSDMD), extravasation of cellular content, and secretion of interleukin-1β (IL-1β) and IL-18 exacerbating inflammation response [3, 4]. Therefore, inhibition of pyroptosis might be a therapeutic target for attenuating brain injury after ICH [2, 5].
Raf kinase inhibitor protein (RKIP), a prototypical member of the phosphatidylethanolamine-binding protein (PEBP) family, is involved in neural development, cardiac function, and spermatogenesis [6,7,8]. As a signaling switch, RKIP negatively regulates several protein kinases signaling cascades, such as inhibiting MAP kinase (Raf-MEK-ERK), G protein-coupled receptor (GPCR) kinase, and NF-κB signaling cascades [9,10,11]. Previous studies reported that the expression of RKIP was commonly downregulated in multiple cancers, while overexpressing RKIP induced cancer cells to programmed cell death [12, 13]. Besides, decreased expression of RKIP was associated with a wide variety of disorders such as circadian rhythms, synaptic long-term depression (LTD), neural development and differentiation, stress, depression, Alzheimer’s disease, and brain cancer [11, 14,15,16]. Mounting evidence revealed that overexpression of RKIP attenuated brain injury after ischemia stroke, whereas knockdown of RKIP exacerbated the deficits after ischemia stroke [17, 18]. A recent study showed that RKIP was directly bound to apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), thus interrupted the assembly and activation of the nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome in macrophages [19]. However, the effects of RKIP on neuronal pyroptosis after hemorrhagic stroke have not been elucidated.
Classical pyroptosis is initiated by NLRP3 inflammasome, which contains NLRP3, ASC, and Pro-Caspase-1 [20]. NLRP3 inflammasome can amplify the inflammatory response by activating its core component, caspase-1 [21,22,23]. Activated caspase-1 cleaves GSDMD, the N-terminal and C-terminal cleavage fragments (GSDMD-N and GSDMD-C, respectively), and cleaves pro-IL-1β and pro-IL-18 [24]. N-terminal fragment of GSDMD forms pores in the plasma membrane of cells, resulting in its permeability to drainage inflammatory intracellular contents [25]. In contrast, inhibition of caspase-1 exerts neuroprotective impacts on various CNS disorders [26]. In experimental Alzheimer’s disease, VX-765, a caspase-1 selective inhibitor, protected mouse brains from memory deficits through improvement in Aβ clearance [27]. Inhibition of caspase-1 mediated neuroprotective effects through polarizing M2 microglia/macrophage and suppressing NF-κB activation in experimental stroke [28]. Moreover, NLRP3 inflammasome-mediated pyroptosis was involved in the pathogenesis of brain injury after ICH [29]. In recent years, there has been a consensus that NLRP3 inflammasome-mediated pyroptosis played a critical role in the development of brain injury after TBI and stroke [30, 31]. Activation of NLRP3 inflammasome aggravated brain injury after TBI and stroke, while inhibiting information of NLRP3 inflammasome improved brain injury [32, 33]. Therefore, inhibition of inflammasome-mediated pyroptosis might be a feasible approach to attenuate brain injury after ICH.
In the present study, we hypothesized that activation of RKIP binding ASC could alleviate neuronal pyroptosis and improve neurological function after ICH in mice, and the anti-pyroptosis mechanism of RKIP is mediated through Caspase-1/GSDMD signaling pathway.
Materials and Methods
CSF Sample Collection
Patients with basal ganglion ICH admitted to our hospital were screened. Inclusion criteria were spontaneous ICH and age ≥ 18 years, diagnosed with basal ganglia ICH on noncontrast CT within 24 h after symptoms onset. The amount of hematoma was 10–20 mL in volume on CT scan. Exclusion criteria were as follows: (1) Presences of other previous systemic diseases including uremia, liver cirrhosis, malignancy, and chronic heart or lung disease. (2) ICH caused by traumatic brain injury, cerebral aneurysm, arteriovenous malformation, brain tumor stroke, or hemorrhagic transformation of ischemic stroke. (3) History of arteriovenous malformation of the brain and history of ruptured cerebral aneurysm. In the ICH group and the control group, the cerebrospinal fluid (CSF) samples were collected through lumbar puncture. Fresh CSF samples were spun down, and supernatant was collected and stored at − 80 °C until analysis. The informed consents were obtained from patients, controls, or family members in all cases before CSF samples were collected. The study was performed with approval from the Ethics Committee of the Second Affiliated Hospital of Chongqing Medical University.
Animals
The experiments were conducted ini C57BL/6 male mice (weight about 25 g), purchased from and bred at the Animal Center of Chongqing Medical University. All mice were kept at room temperature (22 ± 1 °C) with a 12-h day/night cycle (humidity: 60 ± 5%) with free access to food and water. All mouse experimental protocols were approved by the Animal Ethics Committee of Chongqing Medical University. The study was conducted in accordance with the National Institutes of Health guide for the care and use of Laboratory Animals and the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines and was approved by the Institutional Animal Care and Use Committee of Chongqing Medical University.
Cell Culture
Immortalized mouse hippocampal neuron HT-22 (CL-0595) cells were provided by Cell Bank, Shanghai. The cells were cultured in Dulbecco’s modified eagle medium (DMEM) (4.5 g/L glucose) containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C in a 5% CO2 atmosphere. When the cells reach to approximately 80% confluence, they were digested with trypsin and passaged for additional experiments. The cells were pretreated with lipopolysaccharide (LPS) (1 μg/mL) for 4 h. After stimulation, the cells were disrupted using lysis buffer, finally centrifuged, and collected as previously described.
Experimental Design
Six separate experiments were designed as follows (Fig. 1). A total of 330 mice were used (Supplementary Table S1).

Experimental design and animal groups. Co-IP, co-immunoprecipitation; DMSO, dimethyl sulfoxide; ELISA, enzyme-linked immunosorbent assay; FJC, Fluoro-Jade C staining; ICH, intracerebral hemorrhage; IF staining, immunofluorescence staining; MRI, magnetic resonance imaging; TEM, transmission electron microscope; WB, Western blot
Experiment 1
To detect the expression level of RKIP in patients with ICH and healthy controls, cerebrospinal fluid (CSF) samples were tested by the enzyme-linked immunosorbent assay (ELISA).
Experiment 2
Time course of the expression of endogenous RKIP in the peri-hematoma tissue after ICH was measured by Western blot. The cellular localization of RKIP was detected by double immunofluorescence staining at 24 h after ICH.
Experiment 3
To assess effects of RKIP on brain injury after ICH, three doses of didymin (0.5, 1.5, 4.5 mg/kg, Solarbio, China), a RKIP up-regulator, were administered intraperitoneally at 1 h after ICH insult to upregulate RKIP expression. Locostatin (0.5 mg/kg, Darmstadt, Germany), a RKIP inhibitor, was administered intraperitoneally at 1 h before ICH insult to inhibit endogenous RKIP [34]. Mice were randomly divided into 6 groups: sham, ICH + vehicle (DMSO), ICH + didymin (0.5 mg/kg), ICH + didymin (1.5 mg/kg), ICH + didymin (4.5 mg/kg), ICH + locostatin (0.5 mg/kg). Neurological test, brain water content, Evans blue (EB) extravasation, and magnetic resonance imaging (MRI) were evaluated 24 h after ICH.
To validate the delivery efficiency of intraperitoneal administration of didymin (1.5 mg/kg) and lococstatin (0.5 mg/kg), the expression of RKIP was examined by Western blot and immunofluorescence assay. Mice were randomly assigned to 6 groups: naive + vehicle (DMSO), naive + didymin (1.5 mg/kg), naive + locostatin (0.5 mg/kg), ICH + vehicle (DMSO), ICH + didymin (1.5 mg/kg), ICH + locostatin (0.5 mg/kg).
Experiment 4
To evaluate effects of RKIP on neuronal degeneration and neuronal pyroptosis after ICH. Fluoro-Jade C (FJC) staining, transmission electron microscopy (TEM), Western blot, and immunofluorescence staining were performed after ICH. Based on the results of experiment 3, the optimal dose of didymin was 1.5 mg/kg, which was set as a standard dose for the rest of the experiment. Mice were randomly divided into 4 groups: sham, ICH + vehicle (DMSO), ICH + didymin (1.5 mg/kg), ICH + locostatin (0.5 mg/kg).
Experiment 5
To explore the interaction of RKIP and ASC, co-immunoprecipitation was performed in mice at 24 h after ICH. In addition, to exclude false positive of the interaction of RKIP and ASC, immunofluorescence staining was performed in HT-22 cells after LPS stimulation to verify both RKIP and ASC co-expressed in the cytoplasm.
Experiment 6
To investigate underlying mechanisms of RKIP-mediated anti-pyroptotic effect, locostatin (0.5 mg/kg) was intraperitoneally injected in each group at 1 h before ICH insult, and then a Caspase-1 inhibitor, VX-765 (50 mg/kg, San Diego, CA, USA) [27, 28], was administered intraperitoneally at 1 h after ICH insult. Mice were randomly divided into 3 groups: sham + locostatin (0.5 mg/kg), ICH + locostatin (0.5 mg/kg) + vehicle, ICH + locostatin (0.5 mg/kg) + VX-765 (50 mg/kg). Neurological test, brain water content, EB extravasation, Western blot, and double immunofluorescence staining were performed.
ICH Mouse Model Induction
ICH model was induced by autologous blood injection as previously described [35]. Briefly, the mice were anesthetized with 1% pentobarbital (50 mg/kg, i.p.) and all experimental mice received a total of 30 μl autologous arterial blood drawn from the central artery of the tail, which was injected successively into the basal ganglion without anticoagulation (stereotaxic coordinates: 0.2 mm anterior, 2.3 mm left lateral, and 3.5-mm deep). When the injection was completed, the needle was left in place for more than 10 min to prevent possible backflow. After the withdrawal of the needle, the burr hole was filled with bone wax and the skin was sutured and sterilized. Body temperature was maintained at 37 °C on an electric blanket throughout all of these procedures. After surgeries, the ICH model was evaluated by Bederson score to assess whether the modeling is successful [36]. Unsuccessful ICH models, including asymptomatic and dead mice before sacrifice, were excluded from this study.
Intraperitoneal Drug Administration
Following the manufacturer’s instructions, didymin (0.5, 1.5, 4.5 mg/kg, Solarbio, China), locostatin (0.5 mg/kg, Darmstadt, Germany), and VX-765 (50 mg/kg, San Diego, CA, USA) were dissolved in dimethylsulfoxide (DMSO), respectively, and then administered intraperitoneally.
Neurobehavioral Function Test
Neurobehavioral functions were evaluated using modified Garcia test and corner turn test at 24 h following ICH by a blinded investigator as previously described [37]. In the modified Garcia test, mice were evaluated for seven items including spontaneous activity, axial sensation, vibrissae touch, limb symmetry, lateral turning, forelimb walking, and climbing. Each test was scored as either 0–3 or 1–3, and established a maximum deficit score of 21. A higher indicated a better neurological function. For the corner turn test, the mouse was allowed to proceed freely into a 30° corner and choose either left or right to exit the corner. The choice of direction during 10 repeats was recorded, with at least 30-s break between the trials, and the percentage of ipsilateral turns was calculated. The percentage of right turn to 10 trials was then calculated.
BBB Permeability
BBB permeability was assessed using method of Evans blue extravasation [38]. After injected with Evans blue for 3 h, mice were transcardially perfused with cold phosphate-buffered saline (0.1 M, PBS, pH 7.4) under deep anesthesia. The operator took out mouse brain, and then separated into left or right hemisphere and cerebellum, stored at − 80 °C immediately. The right part of the 1 g brain was homogenized in 100 μl PBS, sonicated, and centrifuged (12,000 g, 4 °C, 30 min). The supernatant was collected and added an equal amount of trichloroacetic acid (TCA) was incubated overnight by 4 °C. After centrifugation (12,000 g, 4 °C, 30 min), Evans blue stain was measured by spectrophotometer (Thermo Fisher Scientific, USA) at 610 nm.
Brain Water Content
Wet/dry method was used to measure ICH-induced brain edema as previously described [38, 39]. Briefly, mice were sacrificed under deep pentobarbital anesthesia at 24 h after ICH. The brain was immediately removed and cut into a 4-mm coronal slice and separated into five parts: ipsilateral and contralateral basal ganglia, ipsilateral and contralateral cortex, and cerebellum. The cerebellum was retained as an internal control. Each part was immediately weighed on an electronic analytical balance (FA2204B, Techcomp, USA) to obtain the wet weight (WW) and then dehydrated at 100 °C for 72 h. The samples were then reweighted to obtain a dry weight (DW). Water content of brain tissue was calculated as follows: brain water content (%) = [(wet weight − dry weight)/wet weight] × 100%.
Fluoro-Jade C Staining
Degenerating neurons were evaluated by Fluoro-Jade C (FJC) staining as previously described [40]. Briefly, tissue sections were immersed in a basic alcohol consisting 1% sodium hydroxide in 80% ethanol for 5 min, followed by 70% ethanol for 2 min, and 0.06% potassium permanganate solution for 10 min. Then, samples were incubated with 0.0001% working solution of FJC for 10 min. Two observers blinded to the experimental group counted the FJC-positive cells in six sections per brain (at 20 × magnification) through the injury’s epicenter. Data were expressed as the ratio of FJC-positive cells (relative to Sham group).
Transmission Electron Microscopy
Brian tissues (1 × 1 × 1 mm) from the ipsilateral cortex were fixed in 2.5% glutaraldehyde and then were postfixed in 1% osmium tetroxide. After being dehydrated in a gradient of ethanol (30%, 50%, 70%, 90%, 100%) and being embedded in araldite, tissues were cut into 50–60-nm sections. Finally, the thin sections were stained with 3% uranyl acetate and lead citrate and were then scanned using an H7500 transmission electron microscope (TEM) (Hitachi, Japan).
Immunofluorescence Staining
Double fluorescence staining was conducted as previously described [41]. The mice were deeply anesthetized and were transcardially perfused with 20 mL ice-cold PBS followed by 20 mL of 4% paraformaldehyde at 24 h post-ICH. The brain was removed and then was fixed overnight in 4% paraformaldehyde, and then the brain was dehydrated in 20% sucrose for 1 day and then replaced in 30% sucrose for another day. The brain was embedded in OCT and frozen in a − 80 ℃ refrigerator and then was cut into 8-μm-thick coronal sections using a cryostat (CM1860; Leica Microsystems, Germany). For double immunohistochemistry staining, the brain sections were incubated at 4 °C overnight with primary antibody: anti-Neun (1:100, Abcam, USA), anti-ionized calcium-binding adaptor molecule 1 (Iba-1, 1:200, Wako, Japan), anti-glial fibrillary acidic protein (GFAP, 1:200, CST, USA), and anti-RKIP (1:50, Abcam, USA), anti-GSDMD (1:50, Sigma, USA). After being washed three times, the sections were incubated with appropriate secondary antibody (1:200, Bioss, China) for 1 h at 37 °C. For each mouse, we acquired images from at least three randomly selected fields per section and chose at least two randomly selected sections from the major hemorrhagic territory using a fluorescence microscope (U-HGLGPS, OLYMPUS, Japan). Microphotographs were analyzed with cellSens Standard software.
The LPS-pretreated HT-22 cells were seeded on glass coverslips in 24-well plates. After various treatments, the cells were fixed in 4% paraformaldehyde at room temperature for 20 min and then washed thrice in PBST. After permeabilization with 0.1% Triton X-100/PBS for 15 min, the cells were washed with PBS, blocked in PBS with 5% BSA at room temperature for 1 h, and then incubated with RKIP (1:100, Abcam, USA) and ASC (1:50, Santa Cruz, USA) primary antibodies at 4 °C overnight. After being washed with PBS, the cells were incubated with FITC-conjugated secondary antibodies (1:1000) for 1 h at room temperature. Finally, the cells were washed with PBS, mounted in Fluoroshield containing DAPI, and analyzed by confocal microscopy.
Western Blot
Western blotting was performed as previously described [42]. After mice were perfused with ice-cold PBS (0.1 M, pH 7.4) at 24 h post-operation, the peri-hematoma tissues were collected and stored in a − 80 °C freezer until use. After sample preparation, 20 μg of total protein of every sample was separated by SDS gel electrophoresis and electro-transfer to a PVDF membrane, and the membrane was blocked for 1 h at 37 °C followed by incubated with the primary antibody overnight at 4 °C. The primary antibodies were anti-RKIP (1:1000, Abcam, USA), anti-ASC (1:1000, Abcam, USA), anti-NLRP3 (1:1000, Abcam, USA), anti-GSDMD (1:1000, Abcam, USA), anti-caspase-1 (1:1000, Abcam, USA), and anti-β-actin (1:5000, proteintech, China). The secondary antibodies (ZSGB-BIO) were incubated for 1 h at 37 °C. The protein bands were visualized using enhanced chemiluminescence (ECL), and the relative protein quantity was determined using ImageJ software (National Institutes of Health, USA).
Co-immunoprecipitation
Tissue protein lysates were obtained with NP40 buffer (150 mM NaCl, 50 mM Tris–HCl [pH 7.5], 1% NP40) containing a protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Take out 60 μl tissue protein as input group. Wash protein G-agarose beads (Thermo) 3 times with washing buffer (50 mm Tris (pH 7.5), 0.1 mm ethylenediaminetetraacetic acid, 1% Triton X-100, 10% glycerol, 1 mm phenylmethylsulfonyl fluoride, and 1 mm DTT). Protein G-agarose (Thermo) beads were added with capturing antibody for 2 h at 4 °C and then washed with the washing buffer. For immunoprecipitation, 1 mg of total protein was used with 40 μl of Protein G Dynabeads pre-coated with 2 μl of capturing antibody. Tissue proteins were incubated with the mixed sample. After overnight incubation, beads were extensively washed 5 times with NP40 buffer. Samples were eluted by incubation with 1 × concentrated electrophoresis sample buffer and the input group was added 2 × concentrated electrophoresis sample buffer, then both were boiled for 10 min. The supernatant was collected to proceed to Western blot.
Enzyme-Linked Immunosorbent Assay
ELISA was performed as previously described [43]. Tissue protein from experimental animals and cerebrospinal fluid samples from patients with ICH and healthy control individuals were obtained and detected by enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, USA), according to the manufacturer’s instruction. At 24 h after ICH, mice were anesthetized and then perfused with 0.1 M PBS. Tissue protein from experimental animals with ICH was obtained and homogenized in lysis buffer containing protease inhibitors (to each 10 mg of tissue, 100 μl of lysis buffer was used); the supernatant without dilution was used to measure IL-1β. Mouse DuoSet ELISA kit to IL-1β was purchased from R&D Systems and performed as instructed by the manufacturer. The protein concentration of the lysate was determined using a Pierce TM bicinchoninic acid assay protein assay kit (Thermo Scientific, Rockford, IL, USA).
Magnetic Resonance Imaging
Mice were under anesthesia by inhalation of 3.5% isoflurane and maintained by inhalation of 1.0–2.0% isoflurane in 70% N2O and 30% O2 by a nose cone. During MRI scan, the animal’s respiration was continually monitored by a small animal monitoring and gating system (SA Instruments, Stoney Brook, NY) via a pillow sensor positioned under the abdomen. Mice were placed on a heated circulating water blanket to maintain normal body temperature (36–37℃) and under a special coil for small animals.. The T2WI parameters were set as follows: the repetition time (TR) was 8000 ms, the echo time (TE) was 85 ms, and the slice thickness was 1.0 mm using a 3.0-T MRI scanner (MAGNETOM Prisma, SIEMENS, Germany). Post-scanning, mice were transferred to a heated cage for 5–10 min in order to recover from anesthesia, and then returned to their home cages. All image analyses were performed by 2 observers blinded to the mouse treatment factors.
Statistics Analysis
All results were expressed as the mean ± SD and analyzed with GraphPad Prism (version 9.0, GraphPad Software, Inc.) and Image J (version 1.8.0) using one-way analysis of variance followed by a Student–Newman–Keuls test. P value of < 0.05 was defined as statistically significant.
Results
Mortality and Exclusion
All sham-operated were survived. The overall mortality of ICH mice was 7.87% (26/330) in this study. There is no significant difference in the operation groups among experiments 2–6. Four mice were ruled out from this study due to no hematoma (Supplementary Table S1).
Changes of RKIP Expression in CSF Samples from ICH Patients and in the Peri-hematoma Tissues After Experimental ICH
ELISA result showed that RKIP expression level was decreased in CSF samples from patients with ICH compared with healthy control individuals (Fig. 2a). Western blot analysis showed that the level of RKIP in the peri-hematoma tissues significantly decreased at 24 h in the ICH group compared with the sham group (Fig. 2b,c). Double immunofluorescence staining showed that RKIP was mainly expressed in microglia, astrocytes, and neurons in the peri-hematoma tissues at 24 h after ICH (Fig. 2d). The above-mentioned results together indicated that decreased expression of RKIP was closely associated with the development of brain injury after ICH.

Changes of RKIP expression in CSF samples from ICH patients and in the peri-hematoma tissues after experimental ICH. a ELISA assay results showed that the expression of RKIP was decreased in CSF from patients with ICH compared with CSF from healthy control individuals. n = 6 for each group. *P < 0.05 vs healthy controls. b Representative Western blot band and quantitative analyses of RKIP time-dependent expression in right hemisphere after ICH. n = 6 per group. *P < 0.05 vs sham. c Representative brain sample with schematic illustration presented four regions in the peri-hematoma area (indicated by black boxes). d Representative images of double immunofluorescence staining showed that RKIP was colocalized with microglia (Iba-1), astrocytes (GFAP), and neurons (NeuN) separately at 24 h after ICH. n = 3 per group. Scale bar = 50 μm
The Neuroprotective Effects of RKIP on Brain Injury After ICH
The neurological deficits and brain edema were worse in ICH + vehicle group, ICH + didymin (0.5 mg/kg) group, and ICH + didymin (4.5 mg/kg) group in contrast with the sham group at 24 h after ICH (Fig. 3a–c). Administration of didymin (1.5 mg/kg) markedly improved the neurological deficits (Fig. 3a), reduced brain edema (Fig. 3b), and decreased BBB permeability assessed by EB extravasation in ipsilateral hemisphere (Fig. 3c) compared with those in the ICH + vehicle group. Based on these results, the optimal dose of didymin was 1.5 mg/kg, which was set as a standard dose for the rest of the experiment. However, inhibition of endogenous RKIP by locostatin significantly aggravated neurological deficits (Fig. 3a), increased brain water content (Fig. 3b), and BBB permeability (Fig. 3c) at 24 h after ICH compared with those in the ICH + vehicle group. To evaluate the role of RKIP in hematoma size and hematoma absorption, brain gross specimens were taken and MRI scan was performed in cerebral hemispheres. Treatment with didymin significantly promoted hematoma absorption, while inhibition of RKIP by locostatin effectively inhibited hematoma absorption (Fig. 3d,e).

The effects of activation of RKIP and inhibition of endogenous RKIP on neurological function, brain edema, blood–brain barrier permeability, and hematoma size after ICH. a–e Activation of RKIP with didymin (1.5 mg/kg) significantly improved neurological deficits (a) and reduced brain water content (b) and EB extravasation (c), as well as promoted hematoma absorption (d, e) at 24 h after ICH. However, administration of locostatin aggravated neurological deficits (a) and increased brain water content (b), EB extravasation (c), and hematoma size (d, e) at 24 h after ICH. n = 6 per group. *P < 0.05 vs sham; #P < 0.05 vs ICH + vehicle. ipsilateral basal ganglia (ipsi-BG), contralateral basal ganglia (contra-BG), ipsilateral cortex (ipsi-CX), contralateral cortex (contra-CX), and cerebellum
In addition, the delivery efficiency of intraperitoneal administration of didymin and lococstatin was validated by Western blot and immunofluorescence assay. Results showed that RKIP expression in ipsilateral hemisphere was significantly increased in the didymin-treated group compared with vehicle group at 23 h after didymin administration, while RKIP expression in ipsilateral hemisphere was significantly decreased in the locostatin-treated group compared with the vehicle group at 25 h after locostatin injection (Supplementary Fig. S1a–c), which indicated that intraperitoneal administration of didymin and lococstatin were successfully delivered into the brain.
Upregulating RKIP Ameliorated Neuronal Injury After ICH
To assess whether upregulating RKIP by didymin treatment can attenuate neuronal injury following ICH, FJC staining was conducted to detect neuronal degeneration in the peri-hematoma tissues. FJC-positive neurons were remarkably increased at 24 h after ICH in the ICH + vehicle group compared with the sham group (Fig. 4a, 4b). Upregulation of RKIP diminished the density of FJC-positive neurons in the peri-hematoma tissues at 24 h after ICH in the ICH + didymin group, compared with the ICH + vehicle group. Conversely, RKIP-inhibitor aggravated the neuronal degeneration in the ICH + locostatin group compared with the ICH + vehicle group (Fig. 4a, 4b). To detect membrane integrity and pores formed by GSDMD-N on the neuronal membrane post-ICH, TEM was employed. As shown in Fig. 4d, the GSDMD membrane pore formation in neurons and neuronal membrane rupture at the ipsilateral basal ganglia region were obvious in the ICH + vehicle group, whereas GSDMD membrane pores were less formed and membrane integrity was to some extent preserved in the ICH + didymin group compared with the ICH + vehicle group. On the contrary, membrane pores were more formed in neurons and its membrane rupture was further aggravated and we could not see the continuous cell membrane structure at the ipsilateral basal ganglia region in the ICH + locostatin group, compared with the ICH + vehicle group (Fig. 4d).

The effects of activation of RKIP and inhibition of endogenous RKIP on neuronal injury at 24 h after ICH. a, b Fluoro-Jade C staining and quantitative analysis of FJC-positive cells in the ipsilateral basal ganglia. Results showed that the number of FJC-positive cells was decreased in the ICH + didymin group and was increased in the ICH + locostatin group compared with the ICH + vehicle group respectively at 24 h after ICH. Scale bar = 50 μm. n = 3 for each group. *P < 0.05 vs sham; #P < 0.05 vs ICH + vehicle. c Representative brain sample with schematic illustration presented four regions in the peri-hematoma area (indicated by black boxes). d Representative transmission electron microscopy images of neurons in ipsilateral basal ganglia. Results showed that plasma membrane pore formation was reduced in the ICH + didymin group compared with the ICH + vehicle group. The damage of membrane integrity was markedly aggravated in the ICH + lococstatin group compared with the ICH + vehicle group. The red box features discontinuous cell membrane structure and red bar features membrane pores. Scale bar = 1 μm. n = 3 for each group
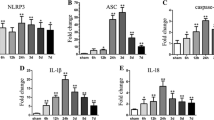
Activation of RKIP Decreased the Expression of Pyroptotic Molecules After ICH
Western blot analysis demonstrated the expression of pyroptotic molecules such as NLRP3, cleaved caspase-1, and GSDMD-N was significantly increased at 24 h after ICH, which was evidently diminished by RKIP activation after administration of didymin (Fig. 5a, 5b). Consistently, ELISA result showed that mature IL-1β was significantly increased at 24 h after ICH, whereas RKIP upregulation effectively decreased the protein level of mature IL-1β (Fig. 5c). In contrast, opposite results on the expression of pyroptotic molecules were observed with RKIP-inhibitor treatment in the ICH + locostatin group compared with the ICH + vehicle group (Fig. 5a–c). Immunofluorescence staining results showed that limited Caspase-1 P20 immunoreactivity was observed in sham mice; however, intense Caspase-1 P20 immunostaining accumulated in the peri-hematoma tissues in ICH mice. The amount of Caspase-1 P20-positive neurons was decreased in the peri-hematoma tissues at 24 h in the ICH + didymin group compared to the ICH + vehicle group, which was increased by RKIP-inhibitor treatment (Fig. 5d).

The effect of RKIP activation on expression of pyroptotic molecules following ICH. a Representative Western blot bands of downstream pyroptotic molecules. b Densitometric quantification suggested the expression of NLRP3, Caspase-1 P20, and GSDMD-N was prominently downregulated in the ICH + didymin group compared with the ICH + vehicle group at 24 h after ICH. The expression of pyroptotic molecules was increased in the ICH + locostatin group compared with the ICH + vehicle group at 24 h after ICH. c ELISA assay results showed that IL-1β production was decreased in the ICH + didymin group and was increased in the ICH + locostatin group compared with the ICH + vehicle group separately. n = 6 for each group. *P < 0.05 vs sham; #P < 0.05 vs ICH + vehicle. d Double immunofluorescence staining and quantitative analysis showed the amount of Caspase-1 P20-positive neurons was decreased in the ICH + didymin group and was increased in the ICH + locostatin group compared with the ICH + vehicle group, respectively. Scale bar = 50 μm. n = 3 for each group. *P < 0.05 vs sham; #P < 0.05 vs ICH + vehicle
RKIP Directly Bound ASC
To further explore the interaction of RKIP and ASC, co-immunoprecipitated (co-IP) was conducted. The result showed that RKIP was associated with ASC (Fig. 6a). Besides, immunofluorescence staining in HT-22 cells after LPS stimulation showed both RKIP and ASC expressed in the cytoplasm and excluded false positive of the interaction (Fig. 6b).

The interaction of RKIP and ASC. a Co-immunoprecipitation showed that RKIP can interact with ASC. n = 3 for each group. b Immunofluorescence staining in HT-22 cells after LPS stimulation showed both RKIP and ASC co-expressed in the cytoplasm. Scale bar = 20 μm. n = 3 for each group
Inhibition of Caspase-1 Alleviated Neuronal Pyroptosis and Brain Injury After RKIP Inhibitor-Pretreated ICH
To investigate whether RKIP-mediated neuronal pyroptosis is related to Caspase-1/GSDMD pathway, a Caspase-1 selective inhibitor, VX-765, was administered intraperitoneally after RKIP inhibitor-pretreated ICH. As manifested in Fig. 7a–c, neurological deficits and brain edema were significantly alleviated and destroy of BBB permeability was mitigated in the ICH + locostatin + VX-765 group compared with the ICH + locostatin + vehicle group. Western blot and ELISA results showed expression of caspase-1 P20, GSDMD-N, and mature IL-1β was downregulated in the ICH + locostatin + VX-765 group compared with the ICH + locostatin + vehicle group (Fig. 7d–f). Consitent effect of VX-765 on RKIP-inhibitor treatment was observed by the immunofluorescence staining assay. In the peri-hematoma tissue at 24 h after ICH, the number of GSDMD-N-positive neurons was increased in the ICH + locostatin + vehicle group compared with sham + locostatin group, while the number of GSDMD-N-positive neurons was decreased in the ICH + locostatin + VX-765 group compared with the ICH + locostatin + vehicle group (Fig. 7g).

Inhibitor of Caspase-1, VX-765, ameliorated neuronal pyroptosis, and brain injury after locostatin-pretreated ICH. a–c VX-765 significantly improved neurological deficits (a), decreased brain water content (b), and EB extravasation (c) at 24 h after ICH compared with those in the ICH + locostatin + vehicle group. d Representative Western blot bands of the downstream pyroptotic molecules. e Densitometric quantification suggested that the expression of GSDMD-N and Caspase-1 P20 at 24 h after ICH was prominently downregulated in the ICH + locostatin + VX-765 group compared with the ICH + locostatin + vehicle group. f ELISA assay showed that IL-1β production was decreased in the ICH + locostatin + VX-765 compared with the ICH + locostatin + vehicle group. n = 6 for each group. *P < 0.05 vs sham; #P < 0.05 vs ICH + locostatin + vehicle. g GSDMD/Neun double immunofluorescence staining and quantitative analysis of GSDMD-positive neurons showed that the number of GSDMD-positive neurons was decreased in the ICH + locostatin + VX-765 group compared with the ICH + locostatin group. Scale bar = 50 μm. n = 3 for each group. *P < 0.05 vs sham; #P < 0.05 vs ICH + locostatin + vehicle
Discussion
The novel findings in the present study were as follows: (1) RKIP was significantly decreased both in CSF samples from patients with ICH and in the peri-hematoma tissues at 24h after experimental ICH; (2) Upregulating RKIP attenuated brain edema and BBB disruption, improved neurological deficits, and promoted hematoma absorption following ICH; (3) Activation of RKIP alleviated neuronal degeneration and neuronal membrane rupture, and inhibited the expression of pyroptotic molecules such as NLRP3, cleaved caspase-1, and GSDMD-N after ICH; (4) Inhibition of endogenous RKIP by locostatin significantly increased the expression of pyroptotic molecules, thus aggravated neurological impairments at 24h post-ICH; (5) Caspase-1 inhibitor ameliorated neuronal pyroptosis and brain injury after RKIP inhibitor-pretreated ICH; (6) ASC/Caspase-1/GSDMD pathway was a potential mechanism of RKIP-mediated neuroprotection. Taken together, our findings indicated that RKIP could alleviate neuronal pyroptosis and improve neurological impairments, which were at least in part mediated by ASC/Caspase-1/GSDMD signaling pathway (Fig. 8).

Schematic diagram of potential molecular mechanisms of the anti-pyroptosis effect of RKIP activation through ASC/Caspase-1/GSDMD signaling pathway following ICH
Pyroptosis, which is characterized with plasma membrane pore formation and causes massive leakage of cytosolic contents, was considered as one of the crucial pathological processes of brain injury following ICH [4, 44, 45]. The latest evidence has shown that NLRP3 inflammasome mediated cell pyroptosis following ICH by evoking the activation of caspase-1 to trigger the cleavage of GSDMD and maturation of potent proinflammatory mediators (IL-1β and IL-18) [46,47,48]. The cleaved GSDMD (GSDMD-N) forms nonselective pores in membrane to accelerate osmosis of inflammatory intracellular contents, leading to amplified inflammatory response [25]. Recent studies showed that neuronal pyroptosis served as a crucial mechanism of brain injury after ICH [5]. In the present study, neuronal membrane pores formed by GSDMD-N and severe membrane rupture were observed at the acute stage of ICH and expressions of pyroptotic molecules were increased after ICH, indicating that neuronal pyroptosis was involved in the brain injury after ICH in morphology and molecule biology. Therefore, suppressing neuronal pyroptosis might be beneficial to prevent subsequent inflammation response and brain injury after ICH.
RKIP plays various roles in different pathological conditions [19, 49]. As a multifunctional protein, RKIP could control cellular growth, motility, differentiation, and tumor metastasis [12, 13]. Previous studies showed that the expression of RKIP was decreased in various cancers and neurodegenerative diseases, such as AD and PD [13, 15, 50, 51]. Consistent with previous studies in the cerebral ischemia model [17, 18], we also observed a significant decrease level of RKIP in CSF samples from patients with ICH, as well in the peri-hematoma tissues at 24 h after experimental ICH. These results indicated that RKIP downregulation is associated with the pathogenesis of brain injury after ICH. Previous studies revealed that overexpression of RKIP played a protective role in cancers and neurodegenerative diseases [13, 15, 19]. In the present study, upregulating RKIP by didymin attenuated neurological deficits, brain water content, and BBB permeability; promoted hematoma absorption; and decreased the number of degenerative neurons after ICH. However, inhibiting RKIP with locostatin exacerbated neurological dysfunction, brain edema, and BBB breakdown following ICH. These results suggested that RKIP exerted neuroprotective property in secondary brain injury after ICH.
A recent study reported that RKIP binding with ASC negatively regulated the formation and activation of inflammasome in macrophages [19]. Consistent with the previous study, Co-IP assays showed that RKIP directly bound ASC. Furthermore, upregulating RKIP by didymin significantly decreased the expression of pyroptotic molecules including NLRP3, Caspase-1, GSDMD-N, and inflammatory molecules IL-1β, as well as the number of Caspase-1 P20-positive-neurons after ICH. Conversely, inhibiting the expression of RKIP with locostatin increased the expression of pyroptotic molecules. These results indicated that RKIP blocked NLRP3 inflammasome assembly through competing with NLRP3 to interact with ASC, therefore suppressed ICH-induced neuronal pyroptosis.
Activated Caspase-1 executes pyroptosis in macrophages by recruitment to an inflammasome in response to intracellular bacteria [26, 52]. It also cleaved GSDMD, the key protein of pyroptosis [53]. Previous studies demonstrated that inhibition of caspase-1 inhibited inflammasome activation and pyroptosis after middle cerebral artery occlusion (MCAO) [28]. In the current study, Caspase-1 level was increased after ICH, which was consistent with the previous findings in ischemic stroke [54]. After upregulating RKIP by didymin, we observed that decreased expression of cleaved caspase-1 preserved neuronal membrane integrity and reduced neuronal pyroptosis. On the contrary, inhibition of RKIP by locostatin greatly increased caspase-1 expression and aggravated neuronal pyroptosis and neurological deficits after ICH. Moreover, inhibition of caspase-1 with VX-765 could ameliorate neuronal pyroptosis and brain injury after locostatin-pretreated ICH. Thus, these findings strengthened our hypothesis that the anti-pyroptosis effect of RKIP might be mediated at least in part through inhibiting caspase-1 activation.
There are some limitations in the present study. RKIP is a multifunctional protein. In this study, we just concentrated on its anti-pyroptosis effect after ICH. Therefore, further studies are needed to elucidate other neuroprotective roles of RKIP and its underlying signaling mechanisms in brain injury after ICH. Additionally, since RKIP was also expressed in microglia and astrocytes, its anti-pyroptosis effect on microglia and astrocytes following ICH requires further investigation. Finally, the long-term outcome and underlying mechanisms of RKIP need to be investigated in brain injury after ICH in the future.
Conclusions
Activation of RKIP binding ASC could alleviate neuronal pyroptosis and improve neurological deficits through Caspase-1/GSDMD signaling pathway after ICH in mice. Therefore, RKIP might be a promising target to attenuate brain injury for ICH patients.
Data Availability
The data used in the present study are available from the corresponding author on reasonable request.
References
Cordonnier C, Demchuk A, Ziai W, Anderson CS. Intracerebral haemorrhage: current approaches to acute management. Lancet. 2018;392:1257–68. https://doi.org/10.1016/S0140-6736(18)31878-6.
Fang Y, Gao S, Wang X, Cao Y, Lu J, Chen S, Lenahan C, Zhang JH, Shao A, Zhang J. Programmed cell deaths and potential crosstalk with blood-brain barrier dysfunction after hemorrhagic stroke. Front Cell Neurosci. 2020;14:68. https://doi.org/10.3389/fncel.2020.00068.
Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. https://doi.org/10.1038/nrmicro2070.
Bobinger T, Burkardt P. H BH, Manaenko A: Programmed cell death after intracerebral hemorrhage. Curr Neuropharmacol. 2018;16:1267–81. https://doi.org/10.2174/1570159X15666170602112851.
Chen S, Zuo Y, Huang L, Sherchan P, Zhang J, Yu Z, Peng J, Zhang J, Zhao L, Doycheva D, et al. The MC4 receptor agonist RO27-3225 inhibits NLRP1-dependent neuronal pyroptosis via the ASK1/JNK/p38 MAPK pathway in a mouse model of intracerebral haemorrhage. Br J Pharmacol. 2019;176:1341–56. https://doi.org/10.1111/bph.14639.
Al-Mulla F, Bitar MS, Taqi Z, Yeung KC. RKIP: much more than Raf kinase inhibitory protein. J Cell Physiol. 2013;228:1688–702. https://doi.org/10.1002/jcp.24335.
Keller ET, Fu Z, Brennan M. The role of Raf kinase inhibitor protein (RKIP) in health and disease. Biochem Pharmacol. 2004;68:1049–53. https://doi.org/10.1016/j.bcp.2004.04.024.
Bernier I, Jolles P. Purification and characterization of a basic 23 kDa cytosolic protein from bovine brain. Biochim Biophys Acta. 1984;790:174–81. https://doi.org/10.1016/0167-4838(84)90221-8.
Rajkumar K, Nichita A, Anoor PK, Raju S, Singh SS, Burgula S. Understanding perspectives of signalling mechanisms regulating PEBP1 function. Cell Biochem Funct. 2016;34:394–403. https://doi.org/10.1002/cbf.3198.
Granovsky AE, Rosner MR. Raf kinase inhibitory protein: a signal transduction modulator and metastasis suppressor. Cell Res. 2008;18:452–7. https://doi.org/10.1038/cr.2008.43.
Ling HH, Mendoza-Viveros L, Mehta N, Cheng HY. Raf kinase inhibitory protein (RKIP): functional pleiotropy in the mammalian brain. Crit Rev Oncog. 2014;19:505–16. https://doi.org/10.1615/critrevoncog.2014011899.
Zaravinos A, Bonavida B, Chatzaki E, Baritaki S: RKIP: a key regulator in tumor metastasis initiation and resistance to apoptosis: therapeutic targeting and impact. Cancers (Basel) 2018, 10. https://doi.org/10.3390/cancers10090287
Gabriela-Freitas M, Pinheiro J, Raquel-Cunha A, Cardoso-Carneiro D, Martinho O: RKIP as an inflammatory and immune system modulator: implications in cancer. Biomolecules 2019, 9. https://doi.org/10.3390/biom9120769
Penas C, Apraiz A, Munoa I, Arroyo-Berdugo Y, Rasero J, Ezkurra PA, Velasco V, Subiran N, Bosserhoff AK, Alonso S, et al: RKIP regulates differentiation-related features in melanocytic cells. Cancers (Basel) 2020, 12. https://doi.org/10.3390/cancers12061451
Zuo H, Liu X, Wang D, Li Y, Xu X, Peng R, Song T. RKIP-mediated NF-kappaB signaling is involved in ELF-MF-mediated improvement in AD rat. Int J Med Sci. 2018;15:1658–66. https://doi.org/10.7150/ijms.28411.
Maki M, Matsukawa N, Yuasa H, Otsuka Y, Yamamoto T, Akatsu H, Okamoto T, Ueda R, Ojika K. Decreased expression of hippocampal cholinergic neurostimulating peptide precursor protein mRNA in the hippocampus in Alzheimer disease. J Neuropathol Exp Neurol. 2002;61:176–85. https://doi.org/10.1093/jnen/61.2.176.
Su L, Zhao H, Zhang X, Lou Z, Dong X. UHPLC-Q-TOF-MS based serum metabonomics revealed the metabolic perturbations of ischemic stroke and the protective effect of RKIP in rat models. Mol Biosyst. 2016;12:1831–41. https://doi.org/10.1039/c6mb00137h.
Su L, Zhang R, Chen Y, Zhu Z, Ma C. Raf kinase inhibitor protein attenuates ischemic-induced microglia cell apoptosis and activation through NF-kappaB pathway. Cell Physiol Biochem. 2017;41:1125–34. https://doi.org/10.1159/000464119.
Qin Q, Liu H, Shou J, Jiang Y, Yu H, Wang X. The inhibitor effect of RKIP on inflammasome activation and inflammasome-dependent diseases. Cell Mol Immunol. 2021;18:992–1004. https://doi.org/10.1038/s41423-020-00525-3.
Liu YG, Chen JK, Zhang ZT, Ma XJ, Chen YC, Du XM, Liu H, Zong Y, Lu GC. NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis. 2017;8: e2579. https://doi.org/10.1038/cddis.2016.460.
Ma Q, Chen S, Hu Q, Feng H, Zhang JH, Tang J. NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann Neurol. 2014;75:209–19. https://doi.org/10.1002/ana.24070.
Yuan B, Shen H, Lin L, Su T, Zhong S, Yang Z. Recombinant adenovirus encoding NLRP3 RNAi attenuate inflammation and brain injury after intracerebral hemorrhage. J Neuroimmunol. 2015;287:71–5. https://doi.org/10.1016/j.jneuroim.2015.08.002.
Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–7. https://doi.org/10.1038/ni.1703.
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8. https://doi.org/10.1038/nature18629.
Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, Li Z, Wang Y, Zhao Q, Shao F, Ding J. Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell. 2020;180(941–955): e920. https://doi.org/10.1016/j.cell.2020.02.002.
Israelov H, Ravid O, Atrakchi D, Rand D, Elhaik S, Bresler Y, Twitto-Greenberg R, Omesi L, Liraz-Zaltsman S, Gosselet F, et al. Caspase-1 has a critical role in blood-brain barrier injury and its inhibition contributes to multifaceted repair. J Neuroinflammation. 2020;17:267. https://doi.org/10.1186/s12974-020-01927-w.
Flores J, Noel A, Foveau B, Lynham J, Lecrux C, LeBlanc AC. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat Commun. 2018;9:3916. https://doi.org/10.1038/s41467-018-06449-x.
Li Q, Dai Z, Cao Y, Wang L. Caspase-1 inhibition mediates neuroprotection in experimental stroke by polarizing M2 microglia/macrophage and suppressing NF-kappaB activation. Biochem Biophys Res Commun. 2019;513:479–85. https://doi.org/10.1016/j.bbrc.2019.03.202.
Li X, Wang T, Zhang D, Li H, Shen H, Ding X, Chen G. Andrographolide ameliorates intracerebral hemorrhage induced secondary brain injury by inhibiting neuroinflammation induction. Neuropharmacology. 2018;141:305–15. https://doi.org/10.1016/j.neuropharm.2018.09.015.
Hu X, Chen H, Xu H, Wu Y, Wu C, Jia C, Li Y, Sheng S, Xu C, Xu H, et al. Role of pyroptosis in traumatic brain and spinal cord injuries. Int J Biol Sci. 2020;16:2042–50. https://doi.org/10.7150/ijbs.45467.
Barrington J, Lemarchand E, Allan SM. A brain in flame; do inflammasomes and pyroptosis influence stroke pathology? Brain Pathol. 2017;27:205–12. https://doi.org/10.1111/bpa.12476.
Bai R, Lang Y, Shao J, Deng Y, Refuhati R, Cui L. The role of NLRP3 inflammasome in cerebrovascular diseases pathology and possible therapeutic targets. ASN Neuro. 2021;13:17590914211018100. https://doi.org/10.1177/17590914211018100.
Xu P, Hong Y, Xie Y, Yuan K, Li J, Sun R, Zhang X, Shi X, Li R, Wu J, et al. TREM-1 exacerbates neuroinflammatory injury via NLRP3 inflammasome-mediated pyroptosis in experimental subarachnoid hemorrhage. Transl Stroke Res. 2021;12:643–59. https://doi.org/10.1007/s12975-020-00840-x.
Lin X, Wei J, Nie J, Bai F, Zhu X, Zhuo L, Lu Z, Huang Q. Inhibition of RKIP aggravates thioacetamide-induced acute liver failure in mice. Exp Ther Med. 2018;16:2992–8. https://doi.org/10.3892/etm.2018.6542.
Rodriguez JA, Sobrino T, Lopez-Arias E, Ugarte A, Sanchez-Arias JA, Vieites-Prado A, de Miguel I, Oyarzabal J, Paramo JA, Campos F, et al: CM352 reduces brain damage and improves functional recovery in a rat model of intracerebral hemorrhage. J Am Heart Assoc 2017, 6. https://doi.org/10.1161/JAHA.117.006042
Fu S, Luo X, Wu X, Zhang T, Gu L, Wang Y, Gao M, Cheng Y, Xie Z. Activation of the melanocortin-1 receptor by NDP-MSH attenuates oxidative stress and neuronal apoptosis through PI3K/Akt/Nrf2 pathway after intracerebral hemorrhage in mice. Oxid Med Cell Longev. 2020;2020:8864100. https://doi.org/10.1155/2020/8864100.
Xie Z, Huang L, Enkhjargal B, Reis C, Wan W, Tang J, Cheng Y, Zhang JH. Recombinant Netrin-1 binding UNC5B receptor attenuates neuroinflammation and brain injury via PPARgamma/NFkappaB signaling pathway after subarachnoid hemorrhage in rats. Brain Behav Immun. 2018;69:190–202. https://doi.org/10.1016/j.bbi.2017.11.012.
Wu X, Fu S, Liu Y, Luo H, Li F, Wang Y, Gao M, Cheng Y, Xie Z. NDP-MSH binding melanocortin-1 receptor ameliorates neuroinflammation and BBB disruption through CREB/Nr4a1/NF-kappaB pathway after intracerebral hemorrhage in mice. J Neuroinflammation. 2019;16:192. https://doi.org/10.1186/s12974-019-1591-4.
Zhu Q, Enkhjargal B, Huang L, Zhang T, Sun C, Xie Z, Wu P, Mo J, Tang J, Xie Z, Zhang JH. Aggf1 attenuates neuroinflammation and BBB disruption via PI3K/Akt/NF-kappaB pathway after subarachnoid hemorrhage in rats. J Neuroinflammation. 2018;15:178. https://doi.org/10.1186/s12974-018-1211-8.
Gao YY, Tao T, Wu D, Zhuang Z, Lu Y, Wu LY, Liu GJ, Zhou Y, Zhang DD, Wang H, et al. MFG-E8 attenuates inflammation in subarachnoid hemorrhage by driving microglial M2 polarization. Exp Neurol. 2021;336: 113532. https://doi.org/10.1016/j.expneurol.2020.113532.
Xie Z, Enkhjargal B, Reis C, Huang L, Wan W, Tang J, Cheng Y, Zhang JH: Netrin-1 preserves blood-brain barrier integrity through deleted in colorectal cancer/focal adhesion kinase/RhoA signaling pathway following subarachnoid hemorrhage in rats. J Am Heart Assoc 2017, 6. https://doi.org/10.1161/JAHA.116.005198
Zheng X, Zhang L, Kuang Y, Venkataramani V, Jin F, Hein K, Zafeiriou MP, Lenz C, Moebius W, Kilic E, et al. Extracellular vesicles derived from neural progenitor cells–a preclinical evaluation for stroke treatment in mice. Transl Stroke Res. 2021;12:185–203. https://doi.org/10.1007/s12975-020-00814-z.
Mertens JC, Blanc-Guillemaud V, Claesen K, Cardona P, Hendriks D, Tyl B, Molina CA. Carboxypeptidase U (TAFIa) is rapidly activated and deactivated following thrombolysis and thrombectomy in stroke patients. Transl Stroke Res. 2021. https://doi.org/10.1007/s12975-021-00962-w.
Chen S, Mei S, Luo Y, Wu H, Zhang J, Zhu J. Gasdermin family: a promising therapeutic target for stroke. Transl Stroke Res. 2018;9:555–63. https://doi.org/10.1007/s12975-018-0666-3.
Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265:130–42. https://doi.org/10.1111/imr.12287.
Heinisch O, Zeyen T, Goldmann T, Prinz M, Huber M, Jung J, Arik E, Habib S, Slowik A, Reich A, et al. Erythropoietin abrogates post-ischemic activation of the NLRP3, NLRC4, and AIM2 inflammasomes in microglia/macrophages in a TAK1-dependent manner. Transl Stroke Res. 2021. https://doi.org/10.1007/s12975-021-00948-8.
Xiao L, Zheng H, Li J, Wang Q, Sun H. Neuroinflammation mediated by NLRP3 inflammasome after intracerebral hemorrhage and potential therapeutic targets. Mol Neurobiol. 2020;57:5130–49. https://doi.org/10.1007/s12035-020-02082-2.
Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, Ghetti B, Koller BH, LeBlanc AC. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015;22:1676–86. https://doi.org/10.1038/cdd.2015.16.
Lin W, Ma C, Su F, Jiang Y, Lai R, Zhang T, Sun K, Fan L, Cai Z, Li Z, et al. Raf kinase inhibitor protein mediates intestinal epithelial cell apoptosis and promotes IBDs in humans and mice. Gut. 2017;66:597–610. https://doi.org/10.1136/gutjnl-2015-310096.
Odabaei G, Chatterjee D, Jazirehi AR, Goodglick L, Yeung K, Bonavida B. Raf-1 kinase inhibitor protein: structure, function, regulation of cell signaling, and pivotal role in apoptosis. Adv Cancer Res. 2004;91(91):169–200. https://doi.org/10.1016/S0065-230x(04)91005-6.
Wen Z, Shu Y, Gao C, Wang X, Qi G, Zhang P, Li M, Shi J, Tian B. CDK5-mediated phosphorylation and autophagy of RKIP regulate neuronal death in Parkinson’s disease. Neurobiol Aging. 2014;35:2870–80. https://doi.org/10.1016/j.neurobiolaging.2014.05.034.
Denes A, Lopez-Castejon G, Brough D. Caspase-1: is IL-1 just the tip of the ICEberg? Cell Death Dis. 2012;3: e338. https://doi.org/10.1038/cddis.2012.86.
Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, Ruan J, Luo X, Lou X, Bai Y, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21:736–45. https://doi.org/10.1038/s41590-020-0669-6.
Li J, Hao JH, Yao D, Li R, Li XF, Yu ZY, Luo X, Liu XH, Wang MH, Wang W. Caspase-1 inhibition prevents neuronal death by targeting the canonical inflammasome pathway of pyroptosis in a murine model of cerebral ischemia. CNS Neurosci Ther. 2020;26:925–39. https://doi.org/10.1111/cns.13384.
Acknowledgements
We thank Prof. Hong Wang, from Key Laboratory of Diagnostic Medicine Designated by the Ministry of Education, Chongqing Medical University, for providing experimental help.
Funding
This work was supported by the Kuanren Talents Program of the Second Affiliated Hospital of Chongqing Medical University (No. 201959), Venture & Innovation Support Program for Chongqing Overseas Returnees (No. CX2019156), Chongqing Science and Health Joint Medical Research Project (No. 2020GDRC006), and Chongqing Postgraduate Scientific Research Innovation Project (No.CYS20198).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Conceptualization was performed by LG and ZX; material preparation, data collection, and analysis were performed by LG, MS, and YY; methodology and software were performed by LG, YT, and RL. The first draft of the manuscript was written by LG and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. All experimental procedures were implemented according to the National Institutes of Health guide for the care and use of Laboratory Animals and the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines and were approved by the Institutional Animal Care and Use Committee of Chongqing Medical University.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
12975_2022_1009_MOESM1_ESM.pdf
Supplementary file1 (PDF 1581 kb) Supplementary Fig.S1 The delivery efficiency of intraperitoneal administration of Didymin and Lococstatin
12975_2022_1009_MOESM2_ESM.pdf
Supplementary file2 (PDF 79 kb) Supplementary Table S1 Summary of experimental groups, sample sizes, and mortality rate in the study
Rights and permissions
About this article

Cite this article
Gu, L., Sun, M., Li, R. et al. Activation of RKIP Binding ASC Attenuates Neuronal Pyroptosis and Brain Injury via Caspase-1/GSDMD Signaling Pathway After Intracerebral Hemorrhage in Mice. Transl. Stroke Res. 13, 1037–1054 (2022). https://doi.org/10.1007/s12975-022-01009-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-022-01009-4