
Abstract
The partial sequences of 16S rRNA and cytochrome c oxidase subunit I (COI) mitochondrial genes were analyzed for species identification and phylogenetic relationships among the commercially important Indian sciaenids (Otolithes cuvieri, Otolithes ruber, Johnius dussumieri, Johnius elongatus, Johnieops vogleri, Otolithoides biauritus and Protonibea diacanthus). Sequence analysis of both genes revealed that the seven species fell into three distinct groups, which were genetically distant from each other and exhibited identical phylogenetic resolution. Partial sequences of both the genes provided sufficient phylogenetic information to distinguish the seven sciaenids indicating the usefulness of mtDNA-based approach in species identification.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Family Sciaenidae in the Order Perciformes is widely distributed throughout the world with approximately 70 genera and 300 species including about 30 species from Indian waters [1, 2]. The Sciaenidae is a species rich family of primarily marine fishes with particularly high diversity in estuaries of the Atlantic, Pacific and Indian Oceans [3]. The sciaenids, popularly known as croakers or drums, contribute significantly to the world marine fisheries particularly of the warm shallow seas and estuaries of North and South America, West Africa, South and South-East Asia [4]. The Indian sciaenids, including the present commercially important species (Otolithes cuvieri, Otolithes ruber, Johnius dussumieri, Johnius elongatus, Johnieops vogleri, Otolithoides biauritus and Protonibea diacanthus), contribute approximately 4.6% to the total Indian marine fish production.
Conventionally, sciaenid fishes are identified using morphological, meristic and anatomical characters [5, 6]. However, considerable ambiguity exists due to morphological similarity and overlapping meristic counts [6, 7]. Synonym citations in FishBase indicate the possibility of ambiguous identification with respect to some sciaenid species of genus Johnieops and Johnius [8]. Accurate identification of morphologically similar species is essential for population dynamic assessment and fisheries management. Application of molecular tools can provide valuable information for species identification to complement the taxonomic data and validation of systematic positions, phylogeny and other applications like fish product identification in trade monitoring [9]. Among the earlier studies on Indian sciaenids, allozyme markers have been used to document genetic variation in Johnieops dussumieri, Kathala axillaries, Pennahia macropthalmus and Otoloithes ruber [7]. Recently, RAPD markers have been used for molecular identification of five Indian sciaenids [10].
Mitochondrial DNA has been extensively studied in fish phylogenetics since mitochondrial 16S rRNA gene and the protein coding cytochrome c oxidase subunit I (COI) gene are highly conserved. These mitochondrial genes have been sequenced in various invertebrate and vertebrate taxa [11–17]. The well characterized COI gene has proved to be a robust evolutionary marker for the analysis of intraspecific and interspecific relationships in many marine fish and shellfish [16–19]. The 16S rRNA mitochondrial gene has also been shown to be a good marker to differentiate fish species and has been used in comparative intergeneric and interspecific studies in several families of Perciformes [20–24].
The success of conservation programs and effective management policies depend on the levels of genetic divergence within and between species and developing strategies to maintain the natural genetic diversity. In the present study, 16S rRNA and COI genes were selected to study phylogenetic relationship and genetic relatedness among seven commercially important Indian sciaenids.
Materials and methods
Sample collections
Seven sciaenids (O. cuvieri, O. ruber, J. dussumieri, J. elongatus, J. vogleri, O. biauritus and P. diacanthus) were collected from Versova (Latitude 19 °08′ N and Longitude 72 °50′ E) and New Ferry Wharf, Mumbai (Latitude 18 °96′ N and Longitude 72 °85′ E) on the North Western coast of India. Species identification and nomenclature followed the synopsis of Indian sciaenid fishes [6]. Samples were collected during July 2006 and approximately 100 mg of white muscle tissue from five individuals of each species was preserved in 95% ethanol until used.
DNA isolation
The DNA was isolated following [25] with minor modifications. The concentration of isolated DNA was estimated using a UV spectrophotometer. The DNA was diluted to get a final concentration of 100 ng/μl.
Amplification and sequencing
The mitochondrial 16S rRNA gene was amplified in a 50 μl reaction volume with 5 μl of 10× Taq polymerase buffer, 0.2 mM of each dNTP, 0.4 μM of each primer, 2.5 U of Taq polymerase and 5 μl genomic DNA using the thermal cycler PTC 200 (MJ Research). The primers used for the amplification of the partial 16S rRNA gene were 16SAR (5′-CGCCTGTTTATCAAAAACAT-3′) and 16SBR (5′-CCGGTCTGAACTCAGATCACGT-3′) [26]. The thermal profile used was 36 repetitions of a three step cycle consisting of denaturation at 94°C for 1 min, annealing at 55°C for 1 min and extension at 72°C for 1.5 min including 4 min for initial denaturation at 94°C and 7 min for the final extension at 72°C.
COI gene was also amplified in a final concentration of 50 μl volume with a final concentration of 5 μl of 10× Taq polymerase buffer, 2 μl of MgCl2 (50 mM), 0.25 μl of each dNTP (0.05 mM), 0.5 μl of each primer (0.01 mM), 0.6 U of Taq polymerase and 5 μl of genomic DNA. The primers used for the amplification of the COI gene were FishF1–5′-TCAACCAACCACAAAGACATTGGCAC-3′ and FishR1-5′-TAGACTTCTGGGTGGCCAAAGAATCA-3′ [16]. The thermal regime consisted of an initial step of 2 min at 95°C followed by 35 cycles of 40 s at 94°C, 40 s at 54°C and 1 min 10 s at 72°C followed in turn by final extension of 10 min at 72°C.
PCR products were visualized on 1.2% agarose gels and the most intense products were selected for sequencing. Products were labeled using the BigDye Terminator V.3.1 Cycle sequencing Kit (Applied Biosystems, Inc) and sequenced bidirectionally using an ABI 3730 capillary sequencer following manufacturer’s instructions.
Sequence analysis
Sequences were aligned using ClustalW [27] and submitted to GenBank under the accession numbers (EF051049, EF028701, EF528197–EF528233, EF534109-EF534127, EF536890-EF536896). The extent of sequence differences between species was calculated by averaging pair-wise comparisons of sequence difference across all individuals. The 16S rRNA sequences of the five individuals of each species were aligned to yield a final alignment varying from 577 bp (P. diacanthus) to 590 bp (O. biauritus). The COI sequences of the five individuals of each species were aligned to yield a final alignment of 655 bp. Pair-wise evolutionary distance among haplotypes was determined by the Kimura 2-Parameter method [28] using the software program MEGA 3.1 (Molecular Evolutionary Genetics Analysis) [29]. The number of polymorphic sites and nucleotide diversity (Pi), nucleotide composition and number of transition and transversion between species were determined by DnaSp ver 3 [30]. Gaps were considered as missing data on the phylogenetic reconstructions. Neighbor Joining (NJ) and Maximum Parsimony (MP) trees were constructed using MEGA 3 using Thunnus orientalis from NCBI (GenBank Accession No. AB185022) as an out-group. To verify the robustness of the internal nodes of NJ and MP trees, bootstrap analysis was carried out using 1000 pseudoreplications [31].
Results
A total of 35 individuals from 5 genera (Otolithes, Johnius, Johnieops, Protonibea and Otolithoides) were used for partial sequence analysis of 16S rRNA and COI genes, which yielded 70 sequences. Simplicity and un-ambiguity were observed among the sequences of the both mitochondrial regions.
Sequencing of the 16SrRNA gene produced an average of 583 (range 577–590) nucleotide base pairs per taxon. Multiple alignments resulted in a consensus length of 597 sites including base pairs and gaps. Two haplotypes were observed per species. Of the 597 sites, 427, 170, 165 and 5 were conserved, variable, parsimony informative and singleton respectively. The polymorphic sites are given in Fig. 1. The analysis revealed nucleotide frequencies as A = 28.10%, T = 25.50%, G = 22.50% and C = 24.00%. As expected, average transitional pairs (si = 48) were more frequent than transversional pairs (sv = 23) with an average ratio of 2.10.

Alignment of partial DNA sequences of the mitochondrial gene, 16S rRNA of seven Indian sciaenids (only variable sites are reported) (H1: Haplotype 1, H2: Haplotype 2, Oc: Otolithes cuvieri, Or: Otolithes ruber, Je: Johnius elongatus, Jd: Johnius dussumieri, Ob: Otolithoides biauritus, Pd: Protonibea diacanthus, Jv: Johnieops vogleri)
Intergeneric and interspecies sequence divergences are given in Table 1. Average sequence diversity among the sciaenids was 0.139 whereas average interspecies sequence diversity was 0.137. The highest intergeneric sequence diversity (0.195) was between P. diacanthus and J. dussumieri where as the lowest value (0.0710) was between species J. vogleri and J. elongatus. The highest interspecies sequence diversity with in genera (0.0873) was between O. ruber and O. cuvieri and the lowest value (0.0588) was between J. dussumieri and J. elongatus. Interspecies nucleotide differences ranged from 34 to 51 and intergeneric nucleotide difference ranged from 41 to 111 (Table 1).
Pair-wise genetic distance values (Kimura 2 parameter) based on 16S rRNA using MEGA 3.1 are given in Table 2. The average genetic distance of individuals among sciaenid species was estimated as 0.139 and within species as 0.002. Congeneric interspecies distance ranged from 0.057 to 0.087 and the intergeneric distance ranged from 0.089 to 0.230. The highest congeneric interspecies genetic distance (0.087) was between O. cuvieri and O. ruber and the highest inter generic distance (0.230) was between P. diacanthus and J. dussumieri.
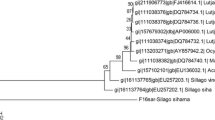
All five sequences for each species were included in the phylogenetic analysis. The NJ (Fig. 2) and MP trees revealed identical phylogenetic relationship among the species. Three major clusters were obtained with the first cluster formed by the congeneric species under the genus Johnius (J. dussumieri and J. elongatus) with Johnieops vogleri a subcluster. The second cluster was formed by two congeneric species under the genus Otolithes (O. cuvieri and O. ruber) and the third cluster formed by P. diacanthus and O. biauritus. In both the trees, these clusters were supported by high bootstrap values (NJ 88–100% and MP 84–99%).

Neighbour Joining (NJ) phylogenetic tree of Indian sciaenids inferred from DNA sequences of mitochondrial gene 16S rRNA
Sequencing the COI gene produced an average of 655 nucleotide base pairs per taxon. No insertions, deletions or stop codons were observed in any sequence and multiple alignments resulted in a consensus length of 655 sites including base pairs and gaps. One to two haplotypes were observed in all the seven species. Of the 655 sites, 436, 219, 217 and 2 were conserved, variable, parsimony informative and singleton respectively. The polymorphic sites are given in Fig. 3. As expected, all variable changes within species were third codon position transitional substitutions. The analysis revealed nucleotide frequencies as A = 23.6%, T = 28.9%, G = 18.8% and C = 28.7%. As expected, average transitional pairs (si = 61) were more frequent than transversional pairs (sv = 30) with an average ratio of 2.00.

Alignment of partial DNA sequences of the mitochondrial gene, COI of seven Indian sciaenids (only variable sites are reported) (H1: Haplotype 1, H2: Haplotype 2, Oc: Otolithes cuvieri, Or: Otolithes ruber, Je: Johnius elongatus, Jd: Johnius dussumieri Ob: Otolithoides biauritus, Pd: Protonibea diacanthus, Jv: Johnieops vogleri)
Average intergeneric and interspecies sequence divergence values are given in Table 3. Average sequence diversity among the sciaenids was 0.163 whereas average congeneric interspecies sequence diversity was 0.147. The highest intergeneric sequence diversity (0.2137) was observed between O. biauritus and J. vogleri and the lowest value (0.1023) was between J. vogleri and J. elongatus. The highest interspecies sequence diversity (0.0580) was between O. ruber and O. cuvieri and the lowest value (0.0259) was between J. dussumieri and J. elongatus. The average inter-species nucleotide differences ranged from 16 to 36 and intergeneric nucleotide difference ranged from 67 to 140 (Table 3).
Pair-wise genetic distance values (K2P) based on COI sequences using MEGA 3.1 were given in Table 4. The average genetic distance between species was estimated as 0.160. The average distance within species was 0.002. The interspecies distance with COI ranged from 0.025 to 0.059 and the intergeneric distance ranged from 0.111 to 0.258. The highest intergeneric distance (0.258) was between Otolithoides biauritus and J. vogleri whereas the highest interspecies distance (0.059) was found between O. cuvieri and O. ruber. All five COI sequences for each species were included in the phylogenetic analysis. The NJ (Fig. 4) and MP tree revealed identical phylogenetic relationship among the species. As with 16S rRNA, three major clusters were obtained with the first cluster formed by the congeneric species under the genus Johnius (J. dussumieri and J. elongatus) with Johnieops vogleri as asubcluster of this first clade. The second clade was formed by two congeneric Otolithes species (O. cuvieri and O. ruber). The third cluster was formed by P. diacanthus and O. biauritus. As with 16S rRNA, the clusters were supported by high bootstrap values (NJ 98–100% and MP 95–99%) except at the node formed by P. diacanthus and other species in the MP tree.

Neighbour Joining (NJ) phylogenetic tree of Indian sciaenids inferred from DNA sequences of mitochondrial gene COI
Discussion
Phylogenetic relationships based on morphological characters and molecules are mostly concordant [16, 22]. The seven species of sciaenids from the Indian coast were found genetically distinct from each other and partitioned into three groups without any haplotypes sharing or overlapping, based on the partial sequence information of both 16S rRNA and COI genes. The sequences of both regions of mtDNA demonstrated simplicity and unambiguity. Stop codons were absent from all amplified sequences of COI and all the amplified sequences were 655 bp in length. This suggests that NUMTs (nuclear DNA sequences originating from mtDNA sequences) were not sequenced (vertebrate NUMTs are typically smaller than 600 bp; [16]). NUMTs in Actinopterygii has not been reported except in the puffer fish, Fugu rubripes [32].
In the present study, the sciaenid species that are morphologically and meristically almost identical,–J. dussumieri and J. elongatus on one hand and O. cuvieri and O. ruber on the other are sister clades in the NJ and MP trees with both 16 Sr RNA and COI gene sequences. Similarly, the large-bodied sciaenids, O. biauritus and P. diacanthus, formed sister clades. Another species J. vogleri was separated into a sister lineage of Johnius with a high bootstrap value. The high genetic divergence values between Johnius and Johnieops (with 16S rRNA, average 7.1% between J. elongatus and J. vogleri and 8.9% between J. dussumieri and J. vogleri; with COI, average 11.1% between J. elongatus and J. vogleri and 10.3% between J. dussumieri and J. vogleri) suggest J. vogleri as a distinct species. By comparing the allozyme and RAPD profiles of nine sciaenids belonging to 6 genera (Johnieops, Johnius, Otolithes, Protonibea, Kathala and Perrahia), Menezes et al. and Lakra et al. have also supported the distinct generic status of Johnieops [7, 10].
In general, the genetic divisions of all the sciaenid species, identified by our data correspond with the taxonomic sub-divisions previously proposed by Mohan [6] and FAO species Identification Sheets [33]. Estimates of genetic divergence with both 16S rRNA and COI genes were sufficient enough to discriminate individuals of different sciaenid species. These values correspond well with the reports in other sciaenids for 16S rRNA (4.8% to 15.3%; [15]) and in other teleosts for COI (1.04 to 20.63%; [16]) partial sequences. In all seven species, levels of intra-specific variation were low (average 0.2% for 16 SrRNA and 0.2% for COI) and this was reflected in the low number of haplotypes (maximum two). This may be due to the high proportion of the identified haplotype in the samples or the limited number of individuals (5 each) collected for the present study. Ward et al. also reported very low within-species genetic diversity in several marine teleosts [16].
The observed transition versus transversion ratios in sciaenids are also comparable to those of many teleosts [16, 34]. Transitions outnumbered transversions in the present study in accordance with the previous reports on mtDNA in fish [13, 15]. Generally for teleost mtDNA, a much larger excess of transitions related to transversion is typically observed [16]. The GC content of the 583 bp 16Sr RNA (49.5%) and 655 bp COI (46.7%) region was on relatively high in all the seven sciaenid species. Ward et al. reported an overall higher GC content in fishes based on complete mtDNA genome ranging from 38.4–43.2% and with COI alone, 42.2–47.1%, which was mostly attributable to 3rd base variation [16]. In our study also, the sciaenids exhibited more nucleotide changes at 3rd position, consistent with most mutations being synonymous.
An interesting observation was the high degree of K2P nucleotide divergence with 16S rRNA gene (inter species 5.9–8.7%; intergeneric 8.9 to 23.0%), indicating its ability to adequately describe interrelationships of sciaenid species. Generally, 16S rRNA is considered more conservative than COI and is used to investigate the relationship among different species and genera. However, sequence analyses of the 16S rRNA gene lacked the ability to resolve relationships in some marine taxa such as sparids and percoid [35]. However, Vinson et al. reported high nucleotide divergence (8.3%) among the sciaenid species in Northern Brazil using 16S r RNA gene sequences and Chakraborty et al. also showed similar results in ribbon fishes and silver biddies, indicating the usefulness of this gene sequence for accurate identification of species [15, 34, 36]. DNA barcoding based on partial sequence information of COI gene has been widely used for species identification of fishes [16, 19]. It provides a framework for the development of simple, low expense, PCR-based assays that can clearly differentiate virtually all fish species. Our results revealed that COI barcoding is an effective marker for identification of sciaenids from Indian seas. The study has also supported the claim of robustness of universal primers for 16S rRNA and COI gene. The primer pairs used in the study following Palumbi et al. and Ward et al. could successfully amplify an approximately 583 bp 16S rRNA gene and 655 bp segment of COI respectively in all sciaenids [16, 26].
In conclusion, our results are congruent with the taxonomic divisions of sciaenids, based on morphological characters as reported by [6]. The species-diagnostic profiles inferred from both 16S rRNA and COI gene sequences were consistent with Johnieops being a distinct genus and Johnieops vogleri being a separate species from Johnius dussumieri and J. elongatus. This is a good starting point for examining the status of sciaenids especially those belonging to the genera Johnieops and Johnius documented in FishBase. The study also supports the earlier results on genetic relationships of sciaenids obtained using allozyme and RAPD markers [7, 10]. It is concluded that partial sequence information of both the mitochondrial genes-16S rRNA and COI can be used as a diagnostic molecular marker in identification and resolution of taxonomic ambiguity of sciaenids.
References
Talwar PK (1995) Fauna of India and adjacent countries: Pisces, Perciformes: Sciaenidae. Zoological Survey of India, Kolkatta, India, 144 pp
Mohanraj G, Batcha H, Gomthy (2003) Sciaenids. In: Mohan Joseph, M, Jayaprakash AA (eds) Status of exploited marine fishery resources of India, Cent. Mar. Fish. Res. Inst., Kochi-682018, India, pp 133–140
Chao LN (1978) A basis for classifying Western Atlantic Sciaenidae (Teleostei, Perciformes). NOAA Technical Report Circular 415:1–64, NOAA, NMFS, USA
Blaber SJM (2003) Biology of the spotted sea trout. In: Lutz PL (ed) CRC marine biology series. Fish and Fisheries 4(4):374–375
Taniguchi N (1970) Comparative osteology of the sciaenid fishes from Japan and its adjacent waters. III. Premaxillary and dentary. Jap J Ichthyol 17:135–140
Mohan RSL (1981) An illustarted synopsis of the fishes of the family Sciaenidae in India. Indian J Fish 28(1,2):1–24
Menezes MR, Naik S, Martins M (1993) Genetic characterization in four sciaenid species from the Arabian Sea. J Fish Biol 43:61–67
Froese R, Pauly D (2006) FishBase. World Wide Web electronic publication. http://www.fishbase.org
Shivji M, Clarke S, Pank M, Natanson L, Kohler N, Stanhope M (2002) Genetic identification of pelagic shark body parts for conservation and trade monitoring. Conserv Biol 16(4):1036–1047
Lakra WS, Goswami M, Mohindra V, Lal KK, Punia P (2007) Molecular identification of five Indian sciaenids (Pisces: Perciformes, Sciaenidae) using RAPD markers. Hydrobiologia 583:359–363
Brown WM (1985) The mitochondrial genome of animals: molecular evolutionary genetics. In: MacIntyre RJ (ed) Molecular evolutionary genetics. Plenum Press, New York. pp 95–130
Bermingham E, Lessios HA (1993) Rate variation of protein and mtDNA evolution as revealed by sea urchins separated by the Isthmus of Panama. Proc Natl Acd Sci USA 90:2734–2738
Santos S, Schneider H, Sampaio I (2003) Genetic differentiation of Macrodon ancylodon (Sciaenidae, Perciformes) populations in Atlantic coastal waters of South America as revealed by mtDNA analysis. Genet Mol Biol 26(2):151–161
Munasinghe H, Burridge C, Austin C (2004) The systematics of freshwater crayfish of the genus Cherax Erichson (Decapoda: Parastacidae) in eastern Australia re-examined using nucleotide sequences from 12S rRNA and 16S rRNA genes. Invertebrate Systematics: Aus J Sci Res 18:215–225
Vinson C, Grazielle G, Schneider H, Sampaio I (2004) Sciaenidae fish of the Caete river estuary, Northern Brazil: mitochondrial DNA suggests explosive radiation for the Western Atlantic assemblage. Genet Mol Biol 27(2):174–180
Ward RD, Zemlak TS, Innes BH, Last PR, Hebert PDN (2005) DNA barcoding Australia’s fish species. Phil Trans Royal Soc B 360:1847–1857
An HS, Jee YJ, Min KS, Kim BL, Han SJ (2005) Phylogenetic analysis of six pacific abalone (Haiotidae) based on DNA sequences of 16S rRNA and cytochrome c oxidase subunit I mitochondrial genes. Mar Biotechnol 7:373–380
Baldwin BS, Black M, Sanjur O, Gustafson R, Lutz RA, Vrijenhoek RC (1996) A diagnostic molecular marker for zebra mussels (Dreessena polymorpha) and potentially co-occurring bivalves: mitochondrial COI. Mol Mar Biol Biotechnol 5:9–14
Spies IB, Gaichas S, Stevenson DE, O JW, Canino MF (2006) DNA based identification of Alaska skates (Amblyraja, Bathyraja and Raja: Rajidae) using cytochrome c oxidase submit (COI) variation. J Fish Bol 69(B):283–292
Ritchie AP, Lavoue S, Lecointre G (1997) Molecular phylogenetics and the evolution of Antarctic notothenoid fishes. Comp Biochem Physiol 118A(4):1009–1025
Tringali MD, Bert TM, Seyoum S, Bermingham E, Bartolacci D (1999) Molecular phylogenetics and ecological diversification of the transisthmian fish genus Centropomus (Perciformes: Centropomidae). Mol Phylogenet Evol 13(1):193–207
Bernardi G, Robertson DR, Clifton KE, Azurro E (2000) Molecular systematics, zoogeography and evolutionary ecology of the Atlantic parrotfish genus Sparisoma. Mol Phylogenet Evol 15(2):292–300
Craig MT, Pondella DJ, Franck JPC, Hafner JC (2001) On the status of the serranid fish genus Epinephelus: evidence for paraphyly based upon 16S DNA sequence. Mol Phylogenet Evol 19(1):121–130
Streelman JT, Alfaro M, Westneat MW, Bellwood DR, Karl SA (2002) Evolutionary history of the parrot fishes: biogeography, ecomorphology and comparative diversity. Evolution 56(5):961–971
Ruzzante DE, Taggart CT, Cook C, Goddard S (1996) Genetic differentiation between inshore and offshore Atlantic cod (Gadus morhua) off Newfoundland: microsatellite DNA variation and antifreeze level. Can J Fish Aquat Sci 53:634–645
Palumbi S, Martin A, Romano S, McMillan WO, Stice L, Grabowski G (1991) The simple fool’s guide to PCR. Version2.0. Honolulu, HL 96822: Department of Zoology and Kewalo Marine Laboratory, University of Hawaii
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgens DG (1997) The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res 24:4876–4882
Kimura MA (1980) Simple method for estimating rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kumar S, Tamura K, Jakobsen IB, Nei M (2004) MEGA3.1: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R (2006) DNA sequence polymorphism version 4.10.9
Felsenstein J (1993) PHYLIP (Phylogeny Inference Package) Version 3.50 Distributed by the author. Department of Genetics, University of Washington, Seattle, USA
Bensasson D, Zhang DX, Hartl DL, Hewitt GM (2001) Mitochondrial pseudogenes: evolution’s misplaced witnesses. Trends Ecol Evol 16:314–321
Fisher W, Bianchi G (1984) FAO species identification sheets for fishery purposes. Western Indian Ocean (Fishing Area 51), vol IV. Food and Agriculture Organization (FAO of the UN), Rome
Chakraborty A, Aranishi F, Iwatsuki W (2006a) Genetic differences among three species of the genus Trichiurus (Perciformes: Trichiuridae) based on mitochondrial DNA analysis. Ichthyol Res 53:93–96
Orrell TM, Carpenter KE (2004) A phylogeny of the fish family Sparidae (porgies) inferred form mitochondrial sequence data. Mol Phylogenet Evol 32:425–434
Chakraborty A, Venugopal MN, Hidaka K, Iwatsuki Y (2006b) Genetic differentiation between two colour morphs of Gerres erythrourus (Perciformes: Gerreidae) from the Indo-Pacific region. Ichthyol Res 53:185–188
Acknowledgements
The authors are grateful to Dr. S. Ayyappan, Deputy Director General (Fisheries), ICAR for the support and encouragement. We thank Dr. R. D. Ward, Senior Principal Scientist, CSIRO Marine and Atmospheric Research, Hobart, Australia for suggestions and critical evaluation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lakra, W.S., Goswami, M. & Gopalakrishnan, A. Molecular identification and phylogenetic relationships of seven Indian Sciaenids (Pisces: Perciformes, Sciaenidae) based on 16S rRNA and cytochrome c oxidase subunit I mitochondrial genes. Mol Biol Rep 36, 831–839 (2009). https://doi.org/10.1007/s11033-008-9252-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-008-9252-1