
Abstract
A series of Ag/NiO nanocomposite electrocatalysts, with a general molecular formula of Ag x Ni1 − xO, was synthesised employing the citrate sol-gel route and tested for the oxygen evolution reaction (OER) in 0.1 M KOH solution. Crystal structure, morphology and stoichiometry of the catalysts were evaluated by X-ray diffraction (XRD), scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX). The face-centred cubic (fcc) crystalline structure of NiO was revealed as being dominant, having an average crystallite size of 9.6 nm. SEM revealed a non-uniform, cotton-like surface for NiO showing aggregation of particles. Crystallinity of the different synthesised compounds decreased as the Ag content increased. The maximum OER activity was observed for pristine NiO, without any Ag additive, requiring an overpotential of only 263 mV to obtain a current density of 10 mA cm−2 at 25 °C. The addition of Ag inhibited the OER electrocatalytic activity, which might be due to Ag oxidation being observed at 1.431 V.

ᅟ
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Water electrolysis, employing metal oxide electrocatalysts, is to date the most convenient way to generate hydrogen gas (H2) for energy conversion and storage applications [1]. In this process, efficiency of the electrochemical production of hydrogen is linked to the activity of the oxygen evolution reaction (OER) that occurs at the anode. Typically, slow kinetics, due to the four electron catalytic reactions, and large overpotential related to the OER process impede the large-scale viability of the electrolysis process [2]. This has resulted in extensive research efforts to develop efficient OER electrocatalysts that possess both high electrocatalytic activity and stability under harsh conditions [3]. Metal oxides, based on noble metals, for example RuO2 and IrO2, exhibit good electrocatalytic performance for the OER; however, their scarcity, poor durability and high cost limit their commercial application [4]. These issues could be addressed employing metal oxide electrocatalysts that are based on less expensive, environmentally benign as well as naturally abundant transition metals (TM), like Ni, Co, Fe, Mn, Cu, and Sn [5,6,7,8,9,10]. These oxides have high thermal and mechanical stability; however, their poor electronic conductivity notably impedes their catalytic activity, which can be tuned either by doping with other metal oxides or by altering the microstructure. Obtaining optimum metal oxide doping significantly enhances the active surface area, porosity and pore morphology as well as electronic conductivity of the catalyst [11]. These features enable electrocatalysts to generate a high current density at low overpotentials, as well as improving electrode kinetics for the OER [12]. Similarly, the performance of these electrocatalysts are markedly influenced by the synthesis procedure, calcined temperatures and calcination time. Sardar et al. reported that the high temperature solid-state synthesis of mixed metal oxides enhanced the probability for the formation of increased particle sizes, as well as increased crystalline structures that impede the catalytic activity of the material. Hence, soft chemical routes for the synthesis of oxide materials have to be applied to obtain fine grains, accompanied with less crystallinity, in order to obtain improved catalytic activity [13]. The aforementioned statement was also confirmed by Yang et al. who reported that oxides with amorphous surface structures, and distortion in their lattices, have reduced the activation energy of the OER and resulted in improved performance compared to their crystalline counterparts [14].
Within the TM series, Ni-based oxides have received special attention for the OER in alkaline media owing to their catalytic activity and abundance, as well as low cost. The most plausible proposed explanation for the high catalytic activity of NiO is the formation of a β-NiOOH phase, with a decrease in activity (under certain circumstances) being related to Ni(IV) formation [15]. To date, various Ni-based mixed oxide catalysts, with different microstructures and grain sizes, have been synthesised using different chemical synthesis routes. For instance, McCrory et al. reported the OER activity of NiO x , NiCeO x , NiCoO x , NiCuO x , NiFeO x and NiLaO x in acidic and alkaline solutions at similar operating conditions [16]. In these systems, overpotentials ranged from 0.35 to 0.43 V in achieving a current density of 10 mA cm−2. Fominykh et al. also reported on an Fe(III)-doped NiO nanocrystal, with a composition of Fe0.1Ni0.9O, exhibiting a current density of 10 mA cm−2 at an overpotential of 297 mV, which is better than the benchmark IrO2 [17]. In pursuit of high electrocatalytic activity (at low overpotentials), we prepared Ag/NiO nanocomposite electrocatalysts by the citrate sol-gel method and subjected it to physical and electrochemical characterisation for the OER in alkaline medium. The structural and electrochemical properties of these compounds were explored as a function of their composition.
Experimental
Synthesis of Oxides
A series of Ag x Ni1 − xO (x = 0.0, 0.2, 0.4 and 0.6) nanocomposite electrocatalysts was prepared by the citrate sol-gel method [18]. Briefly, metal nitrates (AgNO3 and Ni(NO3)2.H2O from Merck), in the ratio of Ag:Ni 0:1, 0.2:0.8, 0.4:0.6 and 0.6:0.4 mol, were dissolved in 50 ml of deionised water and mixed under constant stirring. Subsequently, a 2.0 M aqueous solution of citric acid (Merck) was added to the solution. The pH of the resulting solution was adjusted to ~ 7 using a 5.0 M aqueous solution of ammonia (Merck). The solution was then heated to 70 °C under constant stirring to obtain a gel. A xerogel was achieved after drying this gel overnight at 120 °C in an oven (Ecotherm, Labotech). Finally, the xerogel was grinded using a pestle and mortar to obtain a homogeneous powder and calcined at 400 °C for 5 h in a furnace (Carbolite ELF 11/14B). The sample was subsequently again grounded to a powder form using a pestle and mortar and stored for further use.
Structural and Electrochemical Characterisation
X-ray diffractograms of the electrocatalysts were recorded in the 2θ range of 20°–80° employing a Bruker D2 phaser desktop diffractometer employing a sealed tube Co X-ray source equipped with Bruker Lynxeye PSD detector. The surface and composition of the electrocatalysts were examined employing SEM (FEI Quanta FEG 250) with an integrated XMax 20 EDX system (Oxford Instruments). Cyclic voltammetry (CV), linear sweep voltammetry (LSV) and chronopotentiometry (CP) techniques were carried out for electrochemical characterisation of the electrocatalysts employing a conventional three-electrode electrochemical cell. A platinum wire (Pine Research Instrumentation) and a Hg/HgO electrode (Radiometer) were used as a counter and reference electrode respectively. Prior to starting each experiment, a glassy carbon (GC) electrode disk insert (Sigradur G, HTW Germany), with a geometrical surface area of 0.196 cm2, was polished with 0.05 μm alumina suspension (Gamma Micropolish II, Buehler). The GC electrode was subsequently washed using ethanol (Merck), Milli-Q water and isopropanol (Merck), and dried employing nitrogen gas. The GC disk inserts were employed as substrates for the prepared electrocatalyst inks. The catalyst ink was prepared by the dispersion of 16 mg of oxide material and 5 mg of Vulcan carbon XC 72 into the solution. In order to do this, 4 ml of a 4:1 volume ratio of water and isopropanol was first ultrasonicated for 10 min, and thereafter, catalyst and carbon were added to this solution. The resulting mixture was sonicated for 20 min to obtain a homogeneous dispersion. Subsequently, 0.2 ml of Nafion (5 wt% in mixture of lower aliphatic alcohols and H2O, from Aldrich) was added to the solution mixture and sonicated for 15 min. Twenty microlitres of the produced ink was dropped onto the top surface of a GC electrode using an electronic micropipette and left overnight at room temperature to dry. The catalyst loading was calculated to be ~ 0.38 mg cm−2. The working electrode was assembled into a rotating disk electrode (RDE) setup from Pine Research Instrumentation, and thereafter, current potential curves of the samples were recorded employing a VSP double-channel potentiostat from Bio-Logic Science Instruments. All the measurements were carried out in N2-saturated 0.1 M KOH (Merck, uniVAR) solutions at 25 °C and 1600 rpm. Calibration of the Hg/HgO electrode vs. RHE electrode was conducted in a H2 saturated 0.1 M KOH solution and was measured as being − 0.946 V vs. RHE. The potentials were IR-corrected employing the equation given below:
where I is the current and R is the ohmic resistance of the electrochemical cell. The ohmic resistance of the cell was measured as being 43 Ω employing an impedance measurement technique (ZIR).
Results and Discussion
The X-ray diffraction technique was used in order to ascertain the effect of Ag addition on the NiO crystal structure. The obtained X-ray diffractograms, in the 2θ range of 10°–80° for the Ag x Ni1 − xO samples, are presented in Fig. 1. The pristine NiO sample exhibits several peaks at 2θ values of 39.00°, 43.50°, 50.05° and 74.00°, which point towards a face-centred cubic (fcc) crystalline structure. These values are quite analogous to those already reported [19, 20].

XRD diffractograms of Ag x Ni1 − xO electrocatalysts
In the case of the Ag dispersed samples, Ag peaks along with NiO peaks were observed at 2θ values of 44.5°, 52.0° and 77.0°, which indicate to the formation of cubic crystalline Ag. Subsequent to Ag dispersion, the intensity of the NiO peaks significantly decreased confirming a homogeneous distribution. According to the XRD diffractogram, each phase was separately present at their own position. Based on the aforementioned, we concluded that there was only an interaction between Ag and NiO, hence confirming the formation of a composite. Moreover, peak broadness and zigzag base lines were also realised as a function of Ag concentration, which is associated with the decrease in crystallinity of the catalysts. The average crystallite size of the Ag x Ni1 − xO catalysts was estimated using the Debye-Scherrer equation and was found to be 9.60, 34.4, 34.3 and 28.4 nm for NiO, Ag0.2Ni0.8O, Ag0.4Ni0.6O and Ag0.6Ni0.4O respectively.
The surface morphology of the prepared samples was examined using SEM analysis and shown in Fig. 2. It is clear from Fig. 2a that pure NiO exhibits a non-uniform, cotton-like surface with aggregation of particles. The accumulation of grains formed many small particles with different sizes, which is of high surface energy, which is a key factor in being responsible for higher catalytic activity [19]. The surface morphology of Ag x Ni1 − xO (e.g. x = 0.2; Fig. 2b) reflects a more porous structure as well as agglomeration of particles, mainly at lower concentrations of Ag content, with the SEM image of Ag0.2Ni0.8O being representative of all prepared composites. To confirm the stoichiometry of the prepared samples, EDX analysis was conducted. The obtained EDX results of the samples are tabulated in Table 1. It is evident from the table that the samples exposed a good distribution of Ni and Ag across the scanned section with a considerable amount of oxygen distributed in the metal-associated regions.

SEM images of electrocatalysts for (a) pure NiO, and (b) Ag0.2Ni0.8O
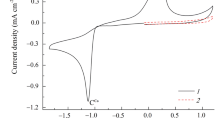
Cyclic voltammograms of the pristine and Ag dispersed NiO samples recorded at a scan rate of 20 mV s−1 are shown in Fig. 3. In the case of the pure NiO sample, two well-defined redox peaks were observed—one at 1.300 V vs. RHE, for the oxidation of Ni(II) to Ni(III), and the other at 1.163 V, for the reduction of Ni(III) to Ni(II). These peaks, in the cyclic voltammogram of pure NiO, can be attributed to the following reaction:

Cyclic voltammograms of Ag x Ni1 − xO electrocatalysts in 0.1 M KOH electrolyte at 20 mV s−1 and 25 °C
In the case of the dispersed samples, all cyclic voltammograms exhibit a clear anodic peak at 1.430 V, which represents the oxidation of Ag(I) to Ag(II) [21]. The reduction peak of Ag(II) to Ag(I) merged into the peak of Ni(II) by manifesting a higher potential shift in the cyclic voltammogram [22]. The addition and increase of Ag content limited the current peak(s) of Ni(II)/Ni(III) and shifted the Ag(I)/Ag(II) current peak to a higher potential region, which brings about the high overpotential required for oxygen evolution. The variation observed in the current peaks for Ni(II)/Ni(III) and Ag(I)/Ag(II) is linked to the Ag x Ni1 − xO catalysts having different electrocatalytic activities.
Moreover, to gain an improved understanding of the differences in electrochemical activity of these electrocatalysts, the oxidation peaks of both Ni and Ag were examined. Generally, capacitance measurements are employed to determine the charge accumulation at the catalyst/electrolyte interface for crystalline materials [23]. Hence, the specific capacitance for each sample was obtained by integrating the peaks of Ni- as well as Ag-oxidation under the CV curves according to the following equation:
where v is the scan rate (V s−1), m is the mass in milligrams of the oxide species deposited on the working electrode, E1 and E2 are the lower and upper potential limits for the integration curve, and j(E) is the current density at electrode potential E. The calculated values of these electrocatalysts are presented in Table 2. The capacitance value for Ni-oxidation decreases with increasing Ag content in the composite sample, while it shows an almost opposite trend for Ag-oxidation. The Ni-oxidation in the pristine NiO sample possessed the maximum capacitance value due to the larger peak area available in the cyclic voltammograms, which points towards the high capacitive nature of the material. As we know that NiO is a far better OER electrocatalyst compared to Ag or Ag2O, due to its low overpotential required, our main concern was to examine the effect of Ag on Ni-oxidation and its OER activity, and not the effect Ni has on Ag-oxidation. In this context, the voltammetric charges (q∗) of the electrocatalysts were also calculated employing the following relation:
where q∗ is the charge (C g−1 cm−2) and S is the geometrical surface area of the working electrode in square centimetre. E1 and E2 are the lower and upper potential limits for the integration curve, and j(E) is the current density at electrode potential E. The calculated voltammetric charge (Table 2) is representative of the active surface area or the number of active sites existing within the catalytic layer [24]. The highest charge value for Ni-oxidation was obtained for pure NiO, compared to the other electrocatalysts reported in this study, which is most likely due to the higher concentration of active sites that exist on the surface of the metal oxide and should therefore point towards NiO being the most active OER electrocatalyst. In the case of the Ag0.6Ni0.8O electrocatalyst, the Ni-oxidation peak is not clear, and it was therefore difficult to calulate the associated capacitance and charge.
Linear sweep voltammetry was used to study the OER activity of the Ag x Ni1 − xO electrocatalysts at 25 °C with a scan rate of 5 mV s−1 (Fig. 4). The activity of the metal oxide electrocatalyst for the OER in alkaline solution is due to the adsorption of OH− ions on the surface of metal ions [25]. This process helps to boost the electron transfer process during which two adsorbed oxygen atoms combine to produce an O2 molecule via the intermediates of metal-OH (M–OH) or metal-hydroperoxo species (M–OOH) [1]. Pure NiO displayed maximum catalytic activity for the OER exhibiting the lowest overpotential for the studied samples.

LSV curves for Ag x Ni1 − xO electrocatalysts in 0.1 M KOH electrolyte at a scan rate of 5 mV s−1 and 25 °C
The overpotentials determined for all samples are presented in Table 3, with the pure NiO electrode requiring an overpotential of only 263 mV to reach a current density of 10 mA cm−2. It is widely accepted that Ni-oxides form Ni-oxy-hydroxides (NiOOH) through oxidation, which is the characteristic feature for high catalytic activity [20, 26] (Eqs. 5–9), with the superscript “*” denoting the surface adsorbed species.
The activity of the NiO electrocatalyst (prepared as part of this investigation) surpasses the activity of various NiO electrocatalysts reported in literature (Table 4). In addition, the obtained OER overpotential of NiO at 10 mA cm−2 is lower when compared to the benchmark RuO2 (380 mV) and IrO2 (380 mV) electrocatalysts as reported by Jung et al. [33] as well as that of IrO2 (320 mV) as reported by McCrory et al. [16]. If we compare the OER activity of NiO prepared by Manivasakan et al. [30] having different morphologies, i.e. nanowires and nanoparticles, the NiO-nanowire electrocatalyst attained a higher OER activity compared to the NiO-nanoparticles due to the ultra-fine nanocrystals and high density of mesopores. Work conducted by Liang et al. [20], on a NiO/Ni foam electrocatalyst, clearly showed that the calcination temperature influences the OER activity of the electrocatalysts. An increase in calcination temperature (up to a specific point) significantly enhanced the catalytic efficiency by producing more NiO. However, the prolonged heating and high calcination temperature generated the largest amount and domain size of the NiO, which constraints charge transport that is owed to the low conductivity and larger domain contact. As such, an optimal calcination temperature is required to accomplish high OER activity. It is evident that the synthesis method has a direct effect on the electrocatalytic activity with overpotentials varying from 324 to 510 mV (Table 4). These different synthesis methods result in different dominant crystal planes as well as different crystallite sizes, with NiO, synthesised as part of this investigation (employing a citrate sol-gel route), exhibiting a (200) dominant crystal plane (Fig. 7) and a crystallite size of 9.6 nm. It is this combination of different crystal planes, crystallite size and inherent electronic effects that give rise to electrocatalytic activity.
The addition of Ag to NiO did not improve the electrocatalytic activity as a drop in current density was observed in the OER potential region. This is due to a silver oxide layer growing on the surface of the NiO electrodes, which is not as efficient as NiO itself. In fact, the composition with the highest Ag content, i.e. Ag0.6Ni0.4O, exhibited the lowest electrocatalytic performance as part of this study. In addition, the increased concentration of Ag inhibits the crystallinity as well as covers the surfaces of NiO particles as we can easily see from the XRD graphs and SEM images, which results in a reduction in electrocatalytic activity.
The electrode kinetics related to the specific electrocatalyst for the OER is explained by the Tafel plot, which is obtained by fitting the LSV curve for the Tafel equation η = b(logj) + a. Figure 5 shows the Tafel plots of pristine and Ag dispersed NiO samples at low potentials. The Tafel slope values for electrocatalysts were determined from the linear part of the η vs. logj curve, and the calculated values of the slopes for the Ag x Ni1 − xO electrocatalysts are displayed in Table 3. Pure NiO exhibits the lowest Tafel slope value (65 mV dec−1) compared to the Ag-dispersed samples, and points towards favourable electrode kinetics and good catalytic activity of NiO compared to that of Ag0.2Ni0.8O, Ag0.4Ni0.6O and Ag0.6Ni0.4O. Nonetheless, this Tafel slope value (for NiO) is comparatively higher than that of the noble metal oxide electrocatalysts: RuO2 (47.7 mV dec−1) and IrO2 (64.6 mV dec−1) as well as transition metal oxide electrocatalysts: NiO nanoparticle (54 mV dec−1) and NiO bulk (50 mV dec−1) [28, 33]. The higher Tafel slope value obtained in this study points towards the electrocatalyst exhibiting slower kinetics compared to the aforementioned electrocatalysts. The differences observed in Tafel slope values might be either due to the intrinsic ability of the adsorbed hydroxyl groups to form oxyhydroxide or variations in the resistance of the catalysts to transfer charge on the surface of the metal oxide [27]. As we know, water oxidation includes several mechanisms with different rate determining steps because of the four electron transfer and complex reaction pathway. Therefore, it has different Tafel slope values 120, 40, 60 and 15 mV dec−1 for different rate determining steps [24]. Our calculated value for pure NiO is close to the theoretical value (60 mV dec−1) suggesting that a second-order reaction, reaction 5, is the rate determining step [34].

Tafel plots for the Ag x Ni1 − xO electrocatalysts generated from the LSV data in Fig. 4
In order to assess the (short-term) durability of all prepared electrocatalysts, CP measurements were conducted at 10 mA cm−2, 25 °C and 1600 rpm for 8 h, with the results presented in Fig. 6. It is clear that the stability plots of all electrocatalysts followed almost a linear horizontal line subsequent to 2 h of testing. Apart from NiO, slight changes/instability are observed in the potential of all Ag-doped nickel oxide electrocatalysts, which might be due to surface passivation of the electrocatalysts under an oxidising regime [35]. The slight initial increase in potential of especially Ag0.4Ni0.6O and Ag0.2Ni0.8O can be attributed to either selective leaching or oxidative degradation of the catalytic layer. This phenomenon, however, has not been explored further. It is clear that pure NiO exhibits not only the best stability, but also the best activity.

Chronopotentiometry plots attesting to the durability of Ag x Ni1 − xO electrocatalysts (recorded at 10 mA cm−2 at 1600 rpm and 25 °C)
At the time of finalising this paper, we learnt that (i) nickel has a very strong affinity for iron [32] and (ii) iron as impurity, both related to the synthesis of the electrocatalyst as well as to the electrolyte used for studying the OER, enhances the electrocatalytic activity of NiO [36]. It is a well-known fact that NiO is chemically unstable and transforms into NiOOH under alkaline conditions. The intermediate NiOOH species absorbs the Fe-impurity from the electrolyte with subsequent enhancement of the OER activity, with Ni0.9Fe0.1OOH being regarded as one of the better OER electrocatalysts [37]. Upon closer inspection, however, we realised that the ammonia solution, used to alter the pH during synthesis of the composites, contained 0.002% of iron as impurity. This constitutes a small molar percentage of 0.6% Fe (relative to the Ni content of the synthesised composites) and we did not venture in quantifying the iron content experimentally. However, the presence of an iron phase was not observed as part of the XRD diffractogram of pure NiO (Fig. 7). Despite this apparent observation of no iron being present, a “significant” electrocatalytic presence of iron cannot be discounted and demands further investigation. In this vain, it has to be stated that the presence of iron in the NiO electrocatalysts listed in Table 4 cannot be discounted either.

XRD diffractogram highlighting the major crystal planes of pure NiO as well as lines for Fe and FeO
Furthermore, to ascertain the presence of iron in our electrolyte solution, we followed a cyclic voltammetry procedure as reported by Stevens et al. [36], who stated that an observed increase in OER current (during cyclic voltammetry) is due to the presence of iron in the electrolyte. From Fig. 8, an initial increase in OER current is evident, which is ascribed to iron (being present in the 0.1 M KOH electrolyte) depositing onto the surface of the Au-electrode. Initially, the deposition of iron is high (during the first 5 cycles), resulting in an increase in OER current, followed by an incremental decrease in OER current, which is reported as being the result of oxidative dissolution that decreases the amount of iron on the surface [38, 39].

Effect of electrolyte impurities on cyclic voltammograms of an Au-electrode in 0.1 M KOH solution recorded at a scan rate of 20 mV s−1 and 25 °C
Conclusions
In this study, we synthesised a series of Ag x Ni1 − xO nanocomposite electrocatalysts employing a citrate sol-gel route and tested it for the OER in alkaline medium. The structure and morphology as well as composition of these catalysts were elucidated employing XRD, SEM and EDX techniques. The addition of silver had no effect in enhancing the electrocatalytic activity of the Ag x Ni1 − xO nanocomposites; in fact, the presence of silver had a detrimental effect. Pure NiO, on the other hand, exhibited exceptional electrocatalytic activity towards the OER, with an overpotential of only 263 mV at a current density of 10 mA cm−2 and a Tafel slope of 65 mV dec−1. The electrocatalytic activity could be attributed to a combination of the dominant (200) crystal plane, the small crystallite size (of 9.6 nm) and inherent electronic effects. The presence of iron, as impurity, positively affecting the electrocatalytic activity towards the OER, cannot be discounted and demands further investigation.
References
V. Maruthapandian, T. Pandiarajan, V. Saraswathy, S. Muralidharan, RSC Adv. 6, 48995–49002 (2016)
Y. Yang, H. Fei, G. Ruan, L. Li, G. Wang, N.D. Kim, J.M. Tour, ACS Appl. Mater. Interfaces 7, 20607–20611 (2015)
Y. Cheng, S.P. Jiang, Progress in Natural Science: Materials International 25, 545–553 (2015)
Y. Meng, W. Song, H. Huang, Z. Ren, S.-Y. Chen, S.L. Suib, J. Am. Chem. Soc. 136, 11452–11464 (2014)
R.B. Moghaddam, C. Wang, J.B. Sorge, M.J. Brett, S.H. Bergens, Electrochem. Commun. 60, 109–112 (2015)
J.A. Bau, E.J. Luber, J.M. Buriak, ACS Appl. Mater. Interfaces 7, 19755–19763 (2015)
R.G. Gonzalez-Huerta, G. Ramos-Sanchez, P.B. Balbuena, J. Power Sources 268, 69–76 (2014)
X. Song, T. Yang, H. Du, W. Dong, Z. Liang, J. Electroanal. Chem. 760, 59–63 (2016)
D.J. Davis, T.N. Lambert, J.A. Vigil, M.A. Rodriguez, M.T. Brumbach, E.N. Coker, S.J. Limmer, J. Phys. Chem. C 118, 17342–17350 (2014)
G. Li, H. Yu, X. Wang, S. Sun, Y. Li, Z. Shao, B. Yi, Phys. Chem. Chem. Phys. 15, 2858–2866 (2013)
G. Liu, J. Xu, Y. Wang, X. Wang, J. Mater. Chem. A 3, 20791–20800 (2015)
L. Trotochaud, S.W. Boettcher, Scr. Mater. 74, 25–32 (2014)
K. Sardar, S.C. Ball, J.D.B. Sharman, D. Thompsett, J.M. Fisher, R.A.P. Smith, P.K. Biswas, M.R. Lees, R.J. Kashtiban, J. Sloan, R.I. Walton, Chem. Mater. 24, 4192–4200 (2012)
Y. Yang, H. Fei, G. Ruan, C. Xiang, J.M. Tour, ACS Nano 8, 9518–9523 (2014)
J. Ramon, G. Mascaros, Chem. Electro. Chem 2, 37–50 (2015)
C.C.L. McCrory, S. Jung, J.C. Peters, T.F. Jaramillo, J. Am. Chem. Soc. 135, 16977–16987 (2013)
K. Fominykh, P. Chernev, I. Zaharieva, J. Sicklinger, G. Stefanic, M. Doblinger, A. Muller, A. Pokharel, S. Bocklein, C. Scheu, T. Bein, D. Fattakhova-Rohlfing, ACS Nano 9, 5180–5188 (2015)
S. Beg, S. Hafeez, N.A.S. Al-Areqi, Solid State Ionics 261, 125–130 (2014)
A.S. Danial, M.M. Saleh, S.A. Salih, M.I. Awad, J. Power Sources 293, 101–108 (2015)
J. Liang, Y.-Z. Wang, C.-C. Wang, S.-Y. Lu, J. Mater. Chem. A 4, 9797–9806 (2016)
L. Shahriary, A.A. Athawale, J. Solid State Electrochem. 19, 2255–2263 (2015)
Y. Ding, Y. Wang, L. Su, H. Zhang, Y. Lei, J. Mater. Chem. 20, 9918–9926 (2010)
T. Audichon, W.T. Napporn, C. Canaff, C. Morais, C. Comminges, K.B. Kokoh, J. Phys. Chem. C 120, 2562–2573 (2016)
T. Audichon, B. Guenot, S. Baranton, M. Cretin, C. Lamy, C. Coutanceau, Appl. Catal. B Environ. 200, 493–502 (2017)
D.M. Jang, I.H. Kwak, E.L. Kwon, C.S. Jung, H.S. Im, K. Park, J. Park, J. Phys. Chem. C 119, 1921–1927 (2015)
H.-Y. Su, Y. Gorlin, I.C. Man, F. Calle-Vallejo, J.K. Nørskov, T.F. Jaramillo, J. Rossmeisl, Phys. Chem. Chem. Phys. 14, 14010–14022 (2012)
G. Liu, X. Gao, K. Wang, D. He, J. Li, Int. J. Hydrog. Energy 41, 17976–17986 (2016)
L.-A. Stern, X. Hu, Faraday Discuss. 176, 363–379 (2014)
N. Cheng, Q. Liu, J. Tian, X. Sun, Y. He, S. Zhai, A.M. Asiri, Int. J. Hydrog. Energy 40, 9866–9871 (2015)
P. Manivasakan, P. Ramasamy, J. Kim, RSC Adv. 5, 33269–33274 (2015)
F. Chekin, H. Tahermansouri, M.R. Besharat, J. Solid State Electrochem. 18, 747–753 (2013)
L. Trotochaud, J.K. Ranney, K.N. Williams, S.W. Boettcher, J. Am. Chem. Soc. 134, 17253–17261 (2012)
S. Jung, C.C.L. McCrory, I.M. Ferrer, J.C. Peters, T.F. Jaramillo, J. Mater. Chem. A 4, 3068–3076 (2016)
J. Ponce, J.-L. Rehspringer, G. Poillerat, J.L. Gautier, Electrochim. Acta 46, 3373–3380 (2001)
W. Song, Z. Ren, S.-Y. Chen, Y. Meng, S. Biswas, P. Nandi, H.A. Elsen, P.-X. Gao, S.L. Suib, ACS Appl. Mater. Interfaces 8, 20802–20813 (2016)
M.B. Stevens, L.J. Enman, A.S. Batchellor, M.R. Cosby, A.E. Vise, C.D.M. Trang, S.W. Boettcher, Chem. Mater. 29, 120–140 (2017)
L. Trotochaud, S.L. Young, J.K. Ranney, S.W. Boettcher, J. Am. Chem. Soc. 136, 6744–6753 (2014)
K.L. Nardi, N. Yang, C.F. Dickens, A.L. Strickler, S.F. Bent, Adv. Energy Mater. 5, 1500412 (2015)
B. Mei, A.A. Permyakova, R. Frydendal, D. Bae, T. Pedersen, P. Malacrida, O. Hansen, I.E.L. Stephens, P.C.K. Vesborg, B. Seger, I. Chorkendorff, J. Phys. Chem. Lett 5, 3456–3461 (2014)
Acknowledgements
Our thanks and appreciation is extended to Mr. Adam Schnier and Prof. Dave Billing of the Department of Chemistry at the Witwatersrand University for conducting the XRD analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Iqbal, M.Z., Kriek, R.J. Silver/Nickel Oxide (Ag/NiO) Nanocomposites Produced Via a Citrate Sol-Gel Route as Electrocatalyst for the Oxygen Evolution Reaction (OER) in Alkaline Medium. Electrocatalysis 9, 279–286 (2018). https://doi.org/10.1007/s12678-018-0455-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-018-0455-5