
Abstract
Background
Existing studies have already revealed the involvement of C–C chemokine receptor type 7 (CCR7) in diverse human cancers, including esophageal cell squamous carcinoma (ESCA). Our current study, aims to explore the relevant mechanisms implicated.
Methods
ESCA cell lines were collected for CCR7 expression quantification using western blot. Following the transfection, the viability, migration and invasion of ESCA cells were evaluated via cell counting kit-8 and Transwell assays. The specific molecular mechanisms underlying the effects of CCR7 in ESCA cells were explored via calculating the expressions of proteins related to metastasis and Janus kinase 2/signal transduction and transcription activation 3 (JAK2/STAT3) signaling pathway via western blot. The correlation between CCR7 and metastasis-related proteins was explored via Pearson’s correlation test.
Results
CCR7 was high-expressed in ESCA cells and CCR7 knockdown repressed the viability, migration and invasion of ESCA cells, concurrent with the increased expression of E-cadherin (E-cad, which was also known as CDH1 and lowly expressed in ESCA cells) and the decreased expressions of vimentin (Vim, which was highly expressed in ESCA cells) and matrix metalloproteinase-9 (MMP-9, which was also highly expressed in ESCA cells). Meanwhile, CCR7 was positively correlated with Vim and MMP-9 yet negatively correlated with E-cad in ESCA cells, which indicated that CCR7 has a role in promoting tumor progression in ESCA cells. Besides, the phosphorylation of STAT3 and JAK2 in ESCA cells was elevated, which was diminished following CCR7 knockdown.
Conclusion
This study proves the modulation of CCR7 on ESCA in vitro, which was achieved via JAK2/STAT3 signaling pathway. Our discovery will provide new therapeutic basis and insights for ESCA.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
As one of the major categories of esophageal cancer, esophageal cancer (ESCA) is a prevalent malignancy in the developing world and carries significant morbidity and mortality, with significant alcohol and tobacco use as the major risk factors [1, 2]. ESCA primarily consists of two separate diseases from an epidemiological and pathological standpoint: esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC). Notably, ESCC makes up about 90% of these cases [3]. ESCC is a rapidly growing, aggressive cancer with a high rate of lymph node metastasis that often impacts the upper two-thirds of the esophagus [4]. Dysphagia and cervical lymph node enlargement typically do not present until the disease has advanced significantly, leading to a poor prognosis and a 5-year survival rate that remains low [5]. Over the past few decades, there has been a significant amount of research conducted on the molecular processes responsible for ESCC pathogenesis [6, 7]. Genetic factors, such as mutations in TP53 and NFE2L2 tumor suppressor genes [8, 9], alongside epigenetic changes like DNA methylation and non-coding RNAs [10, 11], have been identified as crucial contributors to the initiation and progression of ESCC. The complex imbalance in gene expression, including amplification of proto-oncogenes, mutations, deletions, and epigenetic alterations, may act collectively to sustain the malignant biological features of ESCC [9, 12]. Nonetheless, the comprehension of ESCC's oncogenic process continues to be elusive, thereby restricting the development of effective therapy.
Chemokines release their effects through binding to the cell surface chemokine receptors that belong to the highly druggable class A G-protein coupled receptor (GPCR) family [13]. As a member of the chemokine receptor family, C–C chemokine receptor type 7 (CCR7) has been already discussed in various biological processes including cancer [13, 14]. For instance, cervical lymph node metastasis of tongue squamous cell carcinoma (TSCC) could be promoted by CCR7, which therefore has the potential to serve as a biomarker for TSCC [15]. Also, CCR7 overexpression causes cetuximab resistance by cross-talking with Epidermal Growth Factor receptor (EGFR) in colorectal cancer [16]. Besides, CCR7 signaling could promote microglia/macrophage recruitment and chemotherapy resistance in glioblastoma [17]. A review on the biomarkers has addressed the involvement of CCR7 in ESCC, whilst the detailed mechanisms have not been expounded, which will be uncovered in this study [18].
Janus kinase 2/signal transduction and transcription activation 3 (JAK2/STAT3) signaling pathway is a downstream of the JAK/STAT pathway and its dysfunction has been demonstrated to assist the progression and development of cancer [19]. Certain scientists have suggested that this specific pathway is responsible for nearly all immune regulation processes, including those related to the identification of tumor cells and the evasion of the immune system by tumors [20]. The activation of both STAT1 and STAT2 plays a crucial role in generating immune responses against tumors through the production of type I and type II interferons (IFNs) and the subsequent enhancement of IFN-related activities. On the contrary, STAT3 has been widely associated with tumor cell survival, immune system suppression, and persistent inflammation within the tumor environment [21, 22]. The identification of reciprocal regulatory mechanisms, modification of proteins after translation, and alternative methods of signal transmission have increased the complexity of the control exerted by JAK-STAT on the initiation and advancement of tumors [20, 23]. An integrated bulk and single-cell gene expression profile has identified Chemokine C–C motif ligand 18 (CCL18) could promote ESCC cell proliferation via JAK2/STAT3 pathway [24]. The activation of JAK2/STAT3 pathway and malignant transformation of ESCA could be induced by sex‑determining region Y box 12 (SOX12) [25]. Besides, B7-H4 promotes the proliferation of ESCC cells via JAK2/STAT3 pathway [26]. Additionally, CCR7 regulates squamous cell carcinoma of the head and neck cells via JAK2/STAT3 pathway [27]. Nonetheless, such interaction has not been explored in ESCC so far, which brings this study about. Here, we utilized ESCC cell lines to validate the expression of CCR7, and knocked down CCR7 in order to assess its effect on cancer cell function. Finally, the phosphorylation levels of JAK2 and STAT3 in ESCC cells were detected by the western blotting to further explore the mechanism of action of CCR7. In conclusion, our study provides new ideas for the clinical diagnosis and treatment of ESCC.
2 Materials and methods
2.1 Cell culture
Human ESCC cell lines (KYSE410, KYSE510, KYSE520 and KYSE150) and normal esophageal epithelial cell line HEEC were purchased from the American Type Culture Collection (ATCC). We used high-sugar Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin mixture to culture all cells. The medium was changed every 2–3 days in a humidified incubator at 37°C with 5% CO2, and the cells were passaged at appropriate times according to their growth. Prior to the experiments, the cells need to reach the logarithmic growth phase, and serum starvation can be performed as needed to synchronize the cell cycle or reduce the basal metabolic level. These detailed culture methods ensure that the cells grow under optimal conditions and lay the foundation for subsequent experiments.
2.2 Cell transfection
The small interfering RNAs (siRNAs) against CCR7 (si-CCR7 #1 and si-CCR7#2) and the negative control siRNAs were synthesized and ordered from RiboBio. These siRNAs were transfected into ESCC cells following the protocol for Lipofectamine 2000 reagent (Invitrogen) provided by Invitrogen. Cells were inoculated in six-well plates the day before transfection to ensure that the cell density reached 50% confluence the next day. During transfection, siRNA and Lipofectamine 2000 were diluted with serum-free medium Opti-MEM, respectively, and left to stand for 5 min before being mixed and incubated at room temperature for 20 min to form siRNA-Lipofectamine complexes. Subsequently, the complex was added to the cell culture medium and gently shaken. After transfection, the cells were incubated in an incubator at 37 °C with 5% CO2 for 6 h, after which they were replaced with normal medium containing 10% FBS. All cells were harvested 48 h after transfection and used for subsequent analysis in various experiments.
2.3 Cell counting kit-8 (CCK-8) assay
In the CCK-8 assay, logarithmically growing ESCC cells were seeded at a density of 5 × 103 cells per well in 96-well plates containing serum and cultured for 12, 24, 36, and 48 h. Subsequently, 20 μL of CCK-8 solution (Beyotime Institute of Biotechnology) was added to each well, and the cells were incubated for an additional 4 h. The WST-8 in the CCK-8 solution is reduced by dehydrogenases in living cells to produce a soluble orange formazan product. The absorbance at 450 nm, which reflects cell proliferation, was measured using a microplate reader (Bio-Rad Laboratories, Inc.).
2.4 Transwell assay
The migration and invasion of ESCC cells were determined via Transwell assay. In detail, the Transwell chamber coated with or without Matrigel (Corning) was applied in invasion or migration assay. 5 × 104 cells within 200 μL non-serum medium was added to the upper chamber and the complete serum (600 μL) was added to the corresponding lower chamber. Following the incubation for 48 h, migrated and invaded cells were fixed in 4% fixative (Beyotime Institute of Biotechnology) for 15 min and dyed using 0.1% crystal violet (Beyotime Institute of Biotechnology). Images of cells were taken from a light microscope (Olympus).
2.5 Reverse-transcription quantitative PCR
The total RNA extraction from cells was carried out via TriZol reagent (Invitrogen) and the PrimeScript™ RT Reagent Kit (Takara Bio, Inc.) was used to reverse-transcribe the extracted RNA into complementary DNA. Hereafter, Fast SYBR™ Green Master Mix (ThermoFisher Scientific) was applied for the PCR assay. The relative expression of genes was finally quantified via 2−ΔΔCT method with GAPDH as the normalization control [28, 29]. See Table 1 for the primers used.
2.6 Western blot
Radioimmunoprecipitation assay lysis buffer (P0013B, Beyotime Institute of Biotechnology) was applied to lyse the total protein from cells. Subsequently, following the determination on the concentration via BCA method, the protein sample (30 μg) was separated by SDS-PAGE and moved onto the PVDF membrane. TBST containing 5% defatted milk was used for blocking non-specific binding during 1-h incubation at room temperature. The relevant primary and secondary antibodies were applied for further incubation at 4 ℃ overnight (with primary antibodies) and at room temperature for 1 h (with secondary antibodies). The signals were developed via ECL chemiluminescence assay kit (P0018S, Beyotime Institute of Biotechnology) and the results were evaluated via Quantity One 4.6.6 (Bio-Rad Laboratories, Inc.). Antibodies used were listed in Table 2.
2.7 Statistical analyses
All data from independent triplicates were shown in the form of as mean ± standard deviation and analyzed in GraphPad Prism 8.0.2. Independent samples t test were performed for two-group data comparison. The correlation between CCR7 and metastasis-related proteins was explored via Pearson’s correlation test. The data were deemed as statistically significant when the P-value was lower than 0.05.
3 Results
3.1 Higher expression of CCR7 in ESCC
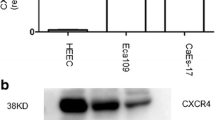
With the aim to determine the involvement of CCR7 in ESCA, 4 ESCA cells lines were collected and the levels of CCR7 in these cells were quantified, where an increased CCR7 expression in 4 ESCC cells lines was seen (Fig. 1A-B, P < 0.001). Of note, the expression of CCR7 was relatively higher in KYSE410 and KYSE520 cells; Thus, these two cells were applied for subsequent assays. This is mainly due to the fact that stable expression of such genes enables subsequent findings to be easily detected and analyzed.

Higher expression of CCR7 in ESCC. A, B CCR7 expression level in ESCA cells (KYSE410, KYSE510, KYSE520 and KYSE150) and normal esophageal epithelial cells (HEEC). The results from independent triplicates were expressed as mean ± standard deviation and the data with P < 0.05 were deemed to be statistically significant. ns non-significant; ***P < 0.001, ****P < 0.0001
3.2 Silencing CCR7 represses ESCA cell viability, migration and invasion
To be able to explore the potential effects of CCR7 on ESCC development and progression, we validated the changes in ESCC cells after knocking down CCR7 expression. Hereafter, CCR7 level was forced to be knocked down using the relevant siRNAs and the decreased CCR7 level in these two aforementioned ESCA cells confirmed the successful intervention. In other words, knockdown of CCR7 expression was followed by significant changes in CCR7 in ESCC cells (Fig. 2A–D, P < 0.001). In addition, the results of CCK-8 assay showed that the silencing of CCR7 could repress the viability of ESCC cells KYSE410 and KYSE520, as reflected by the reduced optical density (OD) value in these two cells (Fig. 2E, F, P < 0.001).

Silencing CCR7 represses the viability of ESCC cells. A-D Validation on the knockdown efficiency of si-CCR7 #1 and si-CCR7#2 in ESCC cells KYSE410 (A, B) and KYSE520 (C, D). E, F CCK-8 assay results showing the effects of CCR7 silencing on the viability of KYSE410 (E) and KYSE520 (F) cells. The results from independent tripli7cates were expressed as mean ± standard deviation and the data with P < 0.05 were deemed to be statistically significant. ***P < 0.001, ****P < 0.0001
In the meantime, the data from Transwell assay showed that silencing CCR7 reduced the quantity of invaded and migrated cells at 48 h (Fig. 3A-D, P < 0.001). Further exploration on the molecular mechanisms was then initiated. We focused on E-cadherin (E-cad), Vimentin (Vim) and MMP-9, all of which have been addressed to be the metastasis-related proteins in cancers. In ESCC cells and HEEC, we found higher expressions of Vim and MMP-9 yet lower expression of (CDH1) in ESCC cells (Fig. 4A-B, P < 0.0001). The subsequent Pearson’s correlation analysis in ESCA cells has additionally proven the negative correlation between CCR7 and CDH1 (Fig. 4C) yet the positive correlation between CCR7 and Vim as well as CCR7 and MMP9 (Fig. 4D, E). These results further emphasize the ability of CCR7 to enhance migration and invasion of ESCC cells, which may be accomplished through the regulation of E-cad, Vim, and MMP-9.

Silencing CCR7 inhibits the migration and invasion of ESCC cells. A–D Transwell assay results demonstrating the migration (A, C) and invasion (B, D) of ESCC cells KYSE410 and KYSE520. Scale bar: 50 μm. The results from independent triplicates were expressed as mean ± standard deviation and the data with P < 0.05 were deemed to be statistically significant. ***P < 0.001, ****P < 0.0001

Exploration on the mechanisms underlying the effects of CCR7 in ESCC. A, B Quantification on the levels of E-cadherin (E-cad, CDH1), Vimentin (Vim) and matrix metalloproteinase-9 (MMP-9) in ESCC cells KYSE410 and KYSE520 and HEEC. C–E Analysis on the correlation between CCR7 and CDH1 (C), Vim (D) and MMP-9 in ESCC cells (E). F, G Expression levels of E-cad, Vim and MMP-9 in ESCA cells KYSE410 (F) and KYSE520 (G). The results from independent triplicates were expressed as mean ± standard deviation and the data with P < 0.05 were deemed to be statistically significant. ns non-significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Following the silencing of CCR7, the expression levels of these proteins in ESCC cells were further quantified, an increased CDH1 mRNA level and decreased levels of Vim and MMP-9 were additionally seen in both KYSE410 and KYSE520 cells transfected with CCR7-specific siRNAs (Fig. 4F, G, P < 0.05). Altogether, it could be concluded that CCR7 silencing could repress ESCA cell proliferation, migration and invasion in vitro.
3.3 Determination on JAK2/STAT3 pathway and its relationship with CCR7 in ESCC
Finally, we shifted our attention to JAK2/STAT3 pathway, since its involvement in ESCC has been already addressed. Increased phosphorylation of STAT3 and JAK2 in ESCC cells KYSE410 and KYSE520 has been witnessed (Fig. 5A–D, P < 0.05), hinting the activation status of JAK2/STAT3 pathway in ESCC.

Determination on JAK2/STAT3 pathway and its relationship with CCR7 in ESCC. A–D Quantification on the phosphorylation of JAK2/STAT3 in ESCC cells KYSE410 (A, C) and KYSE520 (B, D). E–H Effects of CCR7 silencing on the phosphorylation of JAK2/STAT3 in ESCC cells KYSE410 (E, G) and KYSE520 (F, H). The results from independent triplicates were expressed as mean ± standard deviation and the data with P < 0.05 were deemed to be statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
With the purpose of exploring the association between CCR7 and JAK2/STAT3 pathway in ESCC, the phosphorylation levels of JAK2/STAT3 in ESCC cells KYSE410 and KYSE520 were additionally quantified, unveiling a sharp reduction on the phosphorylation of JAK2 and STAT3 (Fig. 5E-H, P < 0.001). Therefore, it could be hinted that CCR7 could target JAK2/STAT3 pathway in ESCA cells.
4 Discussion
Cancer is one of the deadliest diseases and its cure is far from being discovered and only a few patients respond to the treatments [30, 31]. Esophageal cancer is a common malignant disease of esophageal epithelium, which can be categorized into two primary histological subtypes including ESCC [32, 33]. A review has addressed the efficacy of CCR7 as a biomarker in ESCC, whilst the detailed mechanisms have not been expounded [18]. Here, we validated the exploration of the potential mechanism of action of CCR7 on cancer progression based on an in vitro ESCC cell model. We first evaluated the expression of CCR7 in esophageal squamous cell carcinoma cells and its effects on cell proliferation, migration and invasion by Western blot, CCK-8 and transwell assays. Subsequently, the specific mechanisms by which CCR7 regulates the JAK2/STAT3 signaling pathway in these processes were explored by correlation analysis and signaling pathway studies.
CCR7 was known as a GPCR containing 7 transmembrane domains expressed on a variety of cells like central memory T cells, natural killer cells, naïve T/B cells, regulatory T cells, immature/mature dendritic cells, and even a minority of tumor cells [34]. Aberrant expression of CCR7 in some tumor types has already been detected and was linked to pro-survival and invasive pathways [35]. The involvement of CCR7 in some tumors has been addressed already, like TSCC [15], colorectal cancer [16] and glioblastoma [17]. When examining the relevant molecular mechanisms underlying the pro-survival effects of CCR7, it has been shown that CCR7 could promote the proliferation and strengthen the migration and invasion of prostate cancer cells T24 via increasing the expression of MMP-9 (an extracellular matrix (ECM) protein widely discovered to the pathology of cancers) [36, 37]. Another study on breast cancer has demonstrated the cancer-promoting effects of CCR7, together with its repressive effects on E-cad (a calcium-dependent adhesion molecule and one of the most crucial molecules for the metastasis of tumor cells) [38, 39]. Lung cancer cell migration and invasion could be initiated via epithelial-to-mesenchymal transition (EMT, of which Vim is a key biomarker and a type III intermediate filament that has an upregulated expression during cancer metastasis) [40, 41].
Considering the dim molecular mechanisms of CCR7 in ESCC, we, in addition to the observation and determination on the effects of CCR7 on the proliferation, migration and invasion of ESCC cells via the relevant assays like CCK-8 and Transwell, further quantified the levels of these three proteins (E-cad, MMP-9 and Vim) of interest. It was observable in the results of this study that E-cad was lowly expressed yet MMP-9 and Vim were highly expressed in ESCC cells and the silencing of CCR7 increased the expression of E-cad (which was negatively correlated with CCR7) and decreased the levels of Vim and MMP-9 (which was positively correlated with CCR7) in ESCC cells. After conducting a thorough analysis of bioinformatics data, Zhang and colleagues discovered that MMP-9 was overexpressed in the majority of malignant tumors. Additionally, they observed a significant correlation between MMP-9 expression and the clinical outcomes of cancer patients. This extensive examination across various cancer types highlights the potential utility of MMP-9 as a valuable biomarker for targeted therapy [42]. Specifically, research has shown that activation of CCR7 by its particular ligand, external chemokine ligand 19 (CCL19), was linked to a notable linear rise. The RT-qPCR and Western blot revealed that CCL19/CCR7 markedly increased MMP9 expression, a gene associated with metastasis [43]. In addition, Guo et al. revealed that CCR7 favors chemotaxis and migration of squamous cell carcinoma of the head and neck cell line, upregulates MMP-9 protein, stimulates MMP-9 protein activity, and induces reorganization of the actin cytoskeleton [44]. These findings collectively indicate that MMP-9 plays a crucial role in mediating the pro-tumorigenic effects of CCR7, particularly in enhancing tumor metastasis and invasion. Of note, there exist some other proteins that may be accountable for the pro-ESCC effects of CCR7, like N-cadherin [45], MMP-2 [46]. Whether CCR7 could exert its cancer-promoting effects via positively modulating the expressions of these proteins, despite not being addressed here, will be covered in our future study.
The JAK protein was originally known as “Just another kinase” is a family of non-membrane receptor tyrosine kinases (TYKs) which possess catalytic domains, while the STAT proteins are those playing crucial roles in signal transduction and gene expression activation [47]. As a downstream of JAK/STAT pathway, signals from the extracellular could be transduced to the intercellular domain of specific cell surface receptors by JAK2/STAT3 pathway, which has multiple biological functions including cell apoptosis, proliferation, and differentiation [48, 49]. The abnormal activation status and the promoting effects of JAK2/STAT3 in ESCC have been addressed [24,25,26]. So far as we are concerned, the interplay between C–C chemokine receptor family and JAK2/STAT3 signaling pathway has been addressed in some other diseases like subarachnoid hemorrhage [50, 51]. To our surprise, CCR7, the interested C–C chemokine receptor family member, can promote the ETM and stemness of oral squamous cell carcinoma via JAK2/STAT3 signaling pathway, as reflected by the evidently increased phosphorylation of JAK2 and STAT3 [52]. Similar results were also seen in our current study, where the phosphorylation of JAK2 and STAT3 was proven to be increased in ESCC cells, whilst CCR7 knockdown led to the decreased level of phosphorylation. This further emphasizes the impact of CCR7 on ESCC and reveals the mechanism of CCR7 in influencing ESCC progression through the JAK2/STAT3 signaling pathway.
To consolidate such result of our study, this requires a comprehensive understanding of the limitations that exist in the study. First, the small variety of cell lines used in this study is not fully representative of the biology of all ESCAs. Therefore, more ESCC cell lines from different sources and with different characteristics should be added in subsequent studies to improve the breadth and representativeness of the results. Second, future studies will also need to add in vivo experiments, such as using mouse xenograft models to validate the role of CCR7 in vivo. Finally, this study focused on the JAK2/STAT3 signaling pathway, but tumor progression is usually the result of multiple signaling pathways acting together. Studying only one signaling pathway may not be able to fully reveal the mechanism of CCR7's role in ESCA. Future studies should expand the scope of the study to explore other signaling pathways that may be regulated by CCR7 and perform multi-pathway cross-analysis to fully understand the function of CCR7.
5 Conclusion
Collectively, the results showed the abnormally higher levels of CCR7 and JAK2/STAT3 pathway in ESCC and it could be concluded from the current study that CCR7 silencing could diminish the phosphorylation of JAK2/STAT3 and the malignant behaviors of ESCA cells.
Data availability
All data included in this study are available upon request by contact with the corresponding authors. Available at Weian Zeng: zengweian_sysucc@163.com.
Abbreviations
- ESCC:
-
Esophageal squamous cell carcinoma
- EAC:
-
Esophageal adenocarcinoma
- GPCR:
-
G-protein coupled receptor
- CCR7:
-
C-C chemokine receptor type 7
- TSCC:
-
Tongue squamous cell carcinoma
- EGFR:
-
Epidermal Growth Factor receptor
- JAK2/STAT3:
-
Janus kinase 2/signal transduction and transcription activation 3
- CCL18:
-
Chemokine C–C motif ligand 18
- SOX12:
-
Sex‑determining region Y box 12
- ECM:
-
Extracellular matrix
- E-cad:
-
E-cadherin
- Vim:
-
Vimentin
- TYKs:
-
Tyrosine kinases
References
Codipilly DC, Wang KK. Squamous cell carcinoma of the esophagus. Gastroenterol Clin N Am. 2022;51(3):457–84.
Rogers JE, et al. Esophageal cancer: emerging therapeutics. Expert Opin Ther Targets. 2022;26(2):107–17.
Smyth EC, et al. Oesophageal cancer. Nat Rev Dis Primers. 2017;3:17048.
Morgan E, et al. The global landscape of esophageal squamous cell carcinoma and esophageal adenocarcinoma incidence and mortality in 2020 and projections to 2040: new estimates from GLOBOCAN 2020. Gastroenterology. 2022;163(3):649-658.e2.
Zhang R, et al. Endoscopic diagnosis and treatment of esophageal squamous cell carcinoma. Methods Mol Biol. 2020;2129:47–62.
Wu C, et al. Joint analysis of three genome-wide association studies of esophageal squamous cell carcinoma in Chinese populations. Nat Genet. 2014;46(9):1001–6.
Zhang B, et al. TSPAN15 interacts with BTRC to promote oesophageal squamous cell carcinoma metastasis via activating NF-κB signaling. Nat Commun. 2018;9(1):1423.
Hao JJ, et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat Genet. 2016;48(12):1500–7.
Cui Y, et al. Whole-genome sequencing of 508 patients identifies key molecular features associated with poor prognosis in esophageal squamous cell carcinoma. Cell Res. 2020;30(10):902–13.
Cao W, et al. Multi-faceted epigenetic dysregulation of gene expression promotes esophageal squamous cell carcinoma. Nat Commun. 2020;11(1):3675.
Xu WW, et al. Genome-wide identification of key regulatory lncRNAs in esophageal cancer metastasis. Signal Transduct Target Ther. 2021;6(1):88.
Talukdar FR, et al. Genome-wide DNA methylation profiling of esophageal squamous cell carcinoma from global high-incidence regions identifies crucial genes and potential cancer markers. Cancer Res. 2021;81(10):2612–24.
Salem A, et al. CCR7 as a therapeutic target in cancer. Biochim Biophys Acta Rev Cancer. 2021;1875(1): 188499.
Mishan MA, Ahmadiankia N, Bahrami AR. CXCR4 and CCR7: two eligible targets in targeted cancer therapy. Cell Biol Int. 2016;40(9):955–67.
Wang Q, et al. CCR7-CCL21 axis promotes the cervical lymph node metastasis of tongue squamous cell carcinoma by up-regulating MUC1. J Craniomaxillofac Surg. 2021;49(7):562–9.
Gao L, et al. CCR7 high expression leads to cetuximab resistance by cross-talking with EGFR pathway in PI3K/AKT signals in colorectal cancer. Am J Cancer Res. 2019;9(11):2531–43.
Geraldo LH, et al. CCL21-CCR7 signaling promotes microglia/macrophage recruitment and chemotherapy resistance in glioblastoma. Cell Mol Life Sci. 2023;80(7):179.
Goto M, Liu M. Chemokines and their receptors as biomarkers in esophageal cancer. Esophagus. 2020;17(2):113–21.
Kohal R, et al. Targeting JAK2/STAT3 for the treatment of cancer: a review on recent advancements in molecular development using structural analysis and SAR investigations. Bioorg Chem. 2024;143: 107095.
Owen KL, Brockwell NK, Parker BS. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers (Basel). 2019. https://doi.org/10.3390/cancers11122002.
Kim HS, et al. STAT1 deficiency redirects IFN signalling toward suppression of TLR response through a feedback activation of STAT3. Sci Rep. 2015;5:13414.
Jiang S, et al. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. 2010;70(8):3119–27.
Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282(28):20059–63.
Sui X, et al. Integrative analysis of bulk and single-cell gene expression profiles to identify tumor-associated macrophage-derived CCL18 as a therapeutic target of esophageal squamous cell carcinoma. J Exp Clin Cancer Res. 2023;42(1):51.
Li C, et al. SOX12 contributes to the activation of the JAK2/STAT3 pathway and malignant transformation of esophageal squamous cell carcinoma. Oncol Rep. 2021;45(1):129–38.
Chen X, et al. B7–H4 facilitates proliferation of esophageal squamous cell carcinoma cells through promoting interleukin-6/signal transducer and activator of transcription 3 pathway activation. Cancer Sci. 2016;107(7):944–54.
Liu FY, et al. CCR7 regulates cell migration and invasion through JAK2/STAT3 in metastatic squamous cell carcinoma of the head and neck. Biomed Res Int. 2014;2014: 415375.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8.
Sindhuja S, Amuthalakshmi S, Nalini CN. A review on PCR and POC-PCR - a boon in the diagnosis of COVID-19. Curr Pharm Anal. 2022;18(8):745–64. https://doi.org/10.2174/1573412918666220509032754.
Roque N, et al. The interface of cancer, their microenvironment and nanotechnology. Oncologie. 2022;24(3):371–411.
Lu S, et al. A novel biological nano confinement inhibits cancer metastasis. Oncologie. 2022;24(3):591–7.
Ai H, Wang Y, Gu H. Effect of cluster nursing mode combined with blood pressure regulation on surgical tolerance of patients with esophageal cancer and hypertension. Oncologie. 2021;23(2):185–93.
Alsina M, Moehler M, Lorenzen S. Immunotherapy of esophageal cancer: current status, many trials and innovative strategies. Oncol Res Treat. 2018;41(5):266–71.
Ma X, et al. Leveraging a disulfidptosis/ferroptosis-based signature to predict the prognosis of lung adenocarcinoma. Cancer Cell Int. 2023;23(1):267.
Mburu YK, et al. CCR7 mediates inflammation-associated tumor progression. Immunol Res. 2006;36(1–3):61–72.
Mo M, et al. CCL21/CCR7 enhances the proliferation, migration, and invasion of human bladder cancer T24 cells. PLoS ONE. 2015;10(3): e0119506.
Huang H. Matrix metalloproteinase-9 (MMP-9) as a cancer biomarker and MMP-9 biosensors: recent advances. Sensors (Basel). 2018. https://doi.org/10.3390/s18103249.
Xu B, et al. CCR7 mediates human breast cancer cell invasion, migration by inducing epithelial-mesenchymal transition and suppressing apoptosis through AKT pathway. Cancer Med. 2017;6(5):1062–71.
Petrova YI, Schecterson L, Gumbiner BM. Roles for E-cadherin cell surface regulation in cancer. Mol Biol Cell. 2016;27(21):3233–44.
Zhong G, et al. Chemokine (C-C motif) ligand 21/C-C chemokine receptor type 7 triggers migration and invasion of human lung cancer cells by epithelial-mesenchymal transition via the extracellular signal-regulated kinase signaling pathway. Mol Med Rep. 2017;15(6):4100–8.
Usman S, et al. Vimentin is at the heart of epithelial mesenchymal transition (EMT) mediated metastasis. Cancers (Basel). 2021. https://doi.org/10.3390/cancers13194985.
Zhang J, et al. Pan-cancer analysis of the prognostic and immunological role of matrix metalloproteinase 9. Medicine (Baltimore). 2023;102(30): e34499.
Zhang W, et al. Matrix metalloproteinase-9 is up-regulated by CCL19/CCR7 interaction via PI3K/Akt pathway and is involved in CCL19-driven BMSCs migration. Biochem Biophys Res Commun. 2014;451(2):222–8.
Guo N, et al. Chemokine receptor 7 enhances cell chemotaxis and migration of metastatic squamous cell carcinoma of head and neck through activation of matrix metalloproteinase-9. Oncol Rep. 2014;32(2):794–800.
Mrozik KM, et al. N-cadherin in cancer metastasis, its emerging role in haematological malignancies and potential as a therapeutic target in cancer. BMC Cancer. 2018;18(1):939.
Kwon Y, et al. Multi-layered proteogenomic analysis unravels cancer metastasis directed by MMP-2 and focal adhesion kinase signaling. Sci Rep. 2021;11(1):17130.
Mengie Ayele T, et al. Role of JAK2/STAT3 signaling pathway in the tumorigenesis, chemotherapy resistance, and treatment of solid tumors: a systemic review. J Inflamm Res. 2022;15:1349–64.
Kiu H, Nicholson SE. Biology and significance of the JAK/STAT signalling pathways. Growth Factors. 2012;30(2):88–106.
Park SY, et al. The JAK2/STAT3/CCND2 axis promotes colorectal cancer stem cell persistence and radioresistance. J Exp Clin Cancer Res. 2019;38(1):399.
Tian Q, et al. Inhibition of CCR1 attenuates neuroinflammation via the JAK2/STAT3 signaling pathway after subarachnoid hemorrhage. Int Immunopharmacol. 2023;125(Pt A): 111106.
Lin J, et al. CCL5/CCR5-mediated peripheral inflammation exacerbates blood–brain barrier disruption after intracerebral hemorrhage in mice. J Transl Med. 2023;21(1):196.
Chen Y, et al. CCL21/CCR7 interaction promotes EMT and enhances the stemness of OSCC via a JAK2/STAT3 signaling pathway. J Cell Physiol. 2020;235(9):5995–6009.
Acknowledgements
None.
Funding
The authors declare that they have received no funding.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by [XWZ] and [YJA]. The first draft of the manuscript was written by [DMM], [WAZ] and [WH], and the revised version was conducted by [XWZ] and [YJA]. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The study required neither patient nor informed consent for the review of patients’ images and medical records.
Competing interests
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, X., An, Y., Mai, D. et al. Modulation of esophageal squamous cell carcinoma progression: the impact of CCR7 on JAK2/STAT3 signaling pathway. Discov Onc 15, 421 (2024). https://doi.org/10.1007/s12672-024-01289-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-024-01289-2