
Abstract
There has been increasing evidence that pseudohypoxia—a phenomenon that we refer to as “gasping for air”––along with mitochondrial enzyme dysregulation play a crucial role in tumorigenesis, particularly in several hereditary pheochromocytomas (PHEOs) and paragangliomas (PGLs). Alterations in key tricarboxylic acids (TCA) cycle enzymes (SDH, FH, MDH2) have been shown to induce pseudohypoxia via activation of the hypoxia-inducible transcription factor (HIF) signaling pathway that is involved in tumorigenesis, invasiveness, and metastatic spread, including an association with resistance to various cancer therapies and worse prognosis. This review outlines the ongoing story of the pathogenesis of hereditary PHEOs/PGLs, showing the unique and most updated evidence of TCA cycle dysregulation that is tightly linked to hypoxia signaling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, substantial progress has been accomplished in the field of genetics and pathogenesis of pheochromocytoma/paraganglioma (PHEO/PGL) research. Advances in genetics and recognition of a high prevalence of PHEO/PGL in certain familial syndromes is now making it mandatory for routine screening of the tumor in patients with identified mutations, even in the absence of normally considered clinical signs and symptoms. Accumulating data also indicates that many more PHEOs/PGLs are caused by germline mutations than previously recognized, raising the importance of considering an underlying hereditary condition even when there is no obvious familial condition.
The number of PHEO/PGL susceptibility genes was recently increased to 21, a group that includes the von Hippel-Lindau (VHL) tumor suppressor gene [1], the rearranged during transfection (RET) proto-oncogene [2], the neurofibromatosis type 1 (NF1) tumor suppressor gene [3], the genes encoding the four succinate dehydrogenase complex (SDH) subunits (SDHA, -B, -C, -D) [4–7], and the gene encoding the enzyme responsible for flavination of the SDHA subunit (SDHAF2) [8]. Additionally, new susceptibility genes, transmembrane protein 127 (TMEM127) [9, 10], MYC-associated factor X (MAX) [11], and hypoxia-inducible factor 2α (HIF2A) [12, 13], have been identified. The kinesin family member 1B, transcript variant β (KIF1Bβ) [14, 15], prolyl hydroxylase 1 and 2 (PHD1/EGLN2 and PHD2/EGLN1) [16, 17], Harvey rat sarcoma viral oncogene (H-RAS) [18], Kirsten rat sarcoma viral oncogene (K-RAS) [19], isocitrate dehydrogenase 1 (IDH1) [20], fumarate hydratase (FH) [21, 22], and BRCA1-associated protein-1 (BAP1) [23] genes are also anecdotally reported. This year, germline mutations in malate dehydrogenase 2 (MDH2) [24] and somatic mutations in alpha thalassemia/mental retardation syndrome X-linked (ATRX) genes [25] were identified in PHEOs/PGLs.
Currently, there is a large effort to determine the similarities between PHEOs/PGLs resulting from different genetic mutations and to find the common signaling pathways and mechanisms involved in their pathogenesis. Microarray expression profile studies in hereditary PHEOs/PGLs showed two different signatures resulting from specific gene mutations thus dividing the neuroendocrine tumors into two clusters: cluster 1—pseudohypoxic (SDH, VHL, and lately FH and HIF2A mutations)—includes tumors presenting with impaired degradation and accumulation of HIF-1α/HIF-2α, which lead to changes in cell metabolism by pseudohypoxia (an aberrant activation of hypoxia response pathways in the absence of true oxygen deficiency [26]), angiogenesis, heightened reactive oxygen species (ROS) production, and diminished oxidation response. Cluster 2 gene mutations (RET, NF1, TMEM127, MAX, and KIF1Bβ) are connected by kinase signaling and protein translation pathway activation [27–33]. Although these two clusters seem to show distinct cell signaling and new susceptibility genes were recently described, the available data suggests that the majority of PHEO/PGL susceptibility genes mutations are associated with dysregulation of several metabolic pathways, subsequently leading to defects in the hypoxia signaling pathway and adaptive responses to it [32, 34]. Mutations in genes encoding metabolic enzymes, such as SDHx subunits, IDH1, FH, or MDH2 (all were identified in PHEOs/PGLs as well as in a variety of other tumors, including acute myelogenous leukemia, gliomas, chondrosarcomas, and kidney cancer), disrupt the tricarboxylic acid (TCA) cycle and increase dependence on oxidative mitochondrial metabolism [24, 35–40].
Recent evidence shows that pseudohypoxia and mitochondrial enzymes disruption may have direct oncogenic or tumor-suppressive effects by regulating and controlling diverse cellular processes [41–43]. Alterations in cell metabolism have been shown to be associated with tumorigenesis and resistance of cancers to therapy in the past. Presence of pseudohypoxia has been found in a majority of cancers, and nowadays, it is considered to play an important role in the cancer pathogenesis. This present review outlines the ongoing story of PHEO/PGL pathogenesis, which shows an important role of the TCA cycle and hypoxia signaling in this process.
TCA Cycle Overview and Tumorigenesis
The TCA cycle, also known as the Krebs cycle, is a key metabolic pathway that unifies carbohydrate, lipid, and protein metabolism [44]. This cycle takes place in mitochondria and is the most important cellular metabolic network for oxidation of various energy sources, such as glucose, glutamine, and lipids [45]. Simply put, the TCA cycle is a cyclic route consisting of the oxidation of acetyl-coenzyme A (acetyl-CoA), deriving from glycolysis through pyruvate dehydrogenase (PDH) and from lipid β-oxidation to CO2, with the concomitant production of NADH and FADH2, whose electrons fuel the electron transport chain (ETC) for ATP generation. Besides being a central pathway for energetic metabolism, the TCA cycle provides metabolic intermediates for biosynthetic reactions leading to the de novo synthesis of proteins, lipids, and nucleic acids [46]. That means that most of the metabolic pathways in cells are directly or indirectly linked to mitochondria.
In proliferating cells, the TCA cycle operates in a different manner that is characterized by the exit of intermediates from the cycle to supply various biosynthetic pathways. Under these conditions, oxaloacetate (OAA) would become a limiting factor unless it was produced by another pathway that did not flow from mitochondrial citrate. OAA producing pathways, so-called anaplerosis, enable the TCA cycle to function as a biosynthetic pathway in addition to energy generation [47, 48].
For the TCA cycle to function properly, sufficient amounts of its substrates are needed and particular enzymes: citrate synthase (CS), aconitase (ACO), IDH, α-ketoglutarate dehydrogenase (α-KGDH), succinyl-CoA synthetase (SUCLG), SDH, FH, and MDH2. The most important source of carbon for energy-generating pathways that provide acetyl-CoA for oxidative metabolism in mitochondria is glucose [48]. The second most abundant nutrient is glutamine, which serves as a shuttle of carbon and nitrogen between organs. Glutamine is a major source of nitrogen for nonessential amino acids, nucleotides, and hexosamines [49]. Moreover, glucose and glutamine are versatile and in some cases can compensate each other to maintain TCA cycle function [48].
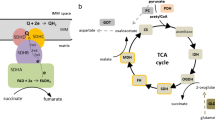
The TCA cycle (Fig. 1) begins with the condensation of acetyl-CoA with OAA to produce citrate, catalyzed by CS. Citrate can be then exported to the cytoplasm to be used as a precursor for lipid biosynthesis (via conversion by ATP-citrate lyase; ACLY) or stays in the mitochondria and is converted to isocitrate by ACO. Isocitrate is subsequently decarboxylated to α-ketoglutarate (α-KG) by IDH. α-KG is then converted to succinyl-CoA by α-KGDH complex or can exit mitochondria and serve as a precursor for amino acid biosynthesis. Succinyl-CoA is transformed to succinate in the reaction catalyzed by SUCLG or it can be utilized for porphyrin biosynthesis. Succinate is then oxidized to fumarate by SDH, which also represents complex II of the ETC. Fumarate is hydrated to malate by FH and, finally, malate is oxidized by MDH to restore OAA. The TCA cycle can be divided into two stages: (1) decarboxylating, in which citrate is converted to succinyl-CoA and releases two CO2 molecules and (2) reductive, which comprises the successive oxidations of succinate to OAA [46, 50]. A defect in any of the TCA cycle enzymes as well as depletion or abundance of its substrates leads to malfunction of the cycle and activation of adaptive mechanisms to assure cell survival. Many of these mechanisms/pathways are related to processes linked with tumorigenesis.

TCA cycle. Acetyl-CoA, the source of energy for TCA cycle, is formed from pyruvate through oxidative decarboxylation by PDH enzyme complex. Acetyl-CoA then enters and fuels TCA cycle as described in the text. α-KG alpha-ketoglutarate, α-KGDH alpha-ketoglutarate dehydrogenase, acetyl-CoA acetyl coenzyme A, ACLY ATP-citrate lyase, ACO aconitase, ADP adenosine diphosphate, ATP adenosine triphosphate, CO 2 carbon dioxide, CS citrate synthase, FAD flavin adenine dinucleotide, FADH 2 reduced FAD, FH fumarate hydratase, GDP guanosine diphosphate, GTP guanosine triphosphate, H 2 O water, IDH isocitrate dehydrogenase, MDH2 malate dehydrogenase 2, NAD + nicotinamide adenine dinucleotide (oxidized), NADH reduced form of NAD, PDH pyruvate dehydrogenase, SDH succinate dehydrogenase complex, succinyl-CoA succinyl coenzyme A, SUCLG succinyl-CoA synthetase
Mitochondrial Substrates in Tumorigenesis
Genetic defects––recessive mutations in genes of TCA cycle enzymes have been known to be associated with multisystem disorders and severe neurodegenerative diseases for many years [51–53]. In recent years, dominant mutations associated with tumorigenesis were also described in TCA cycle enzymes, namely in SDH, FH, IDH, and MDH [7, 24, 54, 55]. These mutations usually lead to accumulation of TCA cycle substrates that, via the feedback loop or epigenetic changes, can promote tumorigenesis.
Pyruvate
Pyruvate is the metabolic intermediate and glycolytic end product that plays a crucial role in the metabolic switch between aerobic and anaerobic metabolism and is an important precursor for glucose, amino acid, and lipid synthesis. It can be utilized both in the cytosol and in mitochondria; under normoxic conditions, mitochondria mostly utilize pyruvate. Pyruvate transfer to mitochondria is mediated by the mitochondrial pyruvate carrier (MPC), a process that links the mitochondrial TCA cycle with the cytosolic glycolytic pathway [45]. Within the mitochondrial matrix, conversion of pyruvate to acetyl-CoA, NADH, and CO2 is catalyzed by PDH. In mitochondria, pyruvate is oxidized to fuel the TCA cycle and oxidative phosphorylation ATP production (OXPHOS) [45]. In the cytosol, pyruvate is reduced to l-lactate by LDH, and during this reaction, one molecule of ATP is produced and NAD+ is regenerated [56].
The PDH complex is the gatekeeper enzyme that links glycolysis to the TCA cycle and lipogenic pathways [57–59]. Pseudohypoxia is one of the mechanisms involved in pyruvate deregulation. In cancer cells, metabolic changes to aerobic glycolysis may be due, in part, to inhibition of the PDH complex that is caused by increased expression of pyruvate dehydrogenase kinase (PDK) 1, 2, and 4 isoforms, leading to mitochondrial dysfunction. The activity of PDK is stimulated by ATP, NADH, and acetyl-CoA; inhibitory effects are exerted by ADP, NAD+, CoA-SH, and pyruvate [60]. Moreover, HIF-α activation also mediates the change of pyruvate distribution in the cell. HIF-1α was found to induce transcriptional upregulation of some of the proteins participating in pyruvate metabolism; for instance, PDK1 and LDH-A [61, 62], reviewed in [45]. PDK1 phosphorylates the E1 subunit of PDH complex that leads to its inactivation and consequently prevents the entry of pyruvate into mitochondria and, thus, its accumulation in the cytosol [59, 63]. In the metabolic environment of cancer cells, the allosteric modifications of PDK activity results in downstream mitochondrial dysfunctions, such as defects in the TCA cycle and/or the electron transport chain and, thus, are responsible for changes in the metabolic phenotype [60]. Besides the TCA cycle, the PDH complex is closely connected to the ETC. In the situation when PDH complex activity (and as a consequence the TCA cycle) is impaired, there is decrease in generation of the reducing equivalents NADH and FADH2, which donate electrons to the respiratory chain to complete the OXPHOS by generation of ATP. As a result of a defective PDH complex, TCA cycle, or respiratory chain, cells develop common features such as lactic acidosis, alteration in redox status, and deregulation of the ADP/ATP ratios and free CoA/acetyl-CoA [64].
The other player in disruption of pyruvate metabolism is pyruvate kinase isoenzyme M2 (PKM2), overexpressed in many cancers [56, 65]. Its knockdown was shown to decrease glycolysis and inhibit cell proliferation in cancer cells [66]. Upregulation of PKM2 is controlled by the pohosphoinositide-3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway and is transcriptionally mediated by HIF-1α and by c-Myc-heterogenous nuclear ribonucleoprotein-dependent gene splicing. PKM2 is a direct transcriptional target of HIF-1α and simultaneously promotes HIF-1α-mediated transactivation and reprogramming of glucose metabolism. Hydroxylation of PKM2 by PHD3 facilitates direct interaction with HIF-1α and promotion of transcriptional transactivation of HIF-1α target genes. Both PKM2 and PHD3 are targets of HIF-1α, meaning that the positive feedback loop between them sustains the expression of glycolytic genes and, thus, facilitates the Warburg effect. Moreover, PKM2 acts as a transcriptional co-activator not only for HIF-1α but also for Oct-4, β-catenin, and STAT3 (reviewed in [45]).
Succinate
Succinate, as mentioned above, is oxidized to fumarate by complex II of ETC, also known as SDH or succinate:ubiquinone oxidoreductase (SQR). ETC consists of four multimeric protein complexes that are anchored to the inner mitochondrial membrane. Together, they catalyze the oxidation reducing equivalents (NADH), using O2 as a terminal acceptor of electrons [67, 68]. Complex II is a unique structure that consists of four functionally different subunits broken up into two groups: the SDH portion consists of SDHA and SDHB whereas SDHC and SDHD make up the SQR component [69, 70]. Its function is to couple the oxidation of succinate to fumarate in the mitochondrial matrix (as part of the TCA cycle) with the reduction of ubiquitin (UQ) in the membrane (as part of the ETC) [7, 32]. Two SDH accessory factors, SDH complex assembly factors (SDHAFs) 1 and 2, were identified lately and they are important for SDH complex assembly and flavination of the SDH catalytic subunit, respectively [8, 71]. The SDH portion of complex II catalyzes succinate to fumarate oxidation and the simultaneous production of FADH2 while the SQR pathway mediates the transfer of electrons generated during oxidation of succinate to reduce ubiquinone to ubiquinol and, subsequently, to respiratory complex III. Electron transfer within the ETC is coupled at specific points to the extrusion of protons into the mitochondrial intermembrane space. This fuels the ATP synthase complex (complex V) for ATP generation. Coupling between ETC and ATP synthesis is called OXPHOS [68].
Impaired function of complex II was shown to be associated with tumorigenesis via stabilization and activation of HIF-α and increased ROS production. Recent works have described complex II as an important site of ROS production in the form of superoxide [72–74] and have also linked it to ROS-mediated execution of apoptotic cell death [75]. Albayrak et al. [76] demonstrated that cell death induction by SDHC expression is associated with a transient inhibition of complex II and ROS generation. Moreover, cells deficient in SDHC are resistant to several proapoptotic cytostatic agents. It seems that complex II acts as a cell death sensor responding to acidification upon toxic stimuli by its disassembly and thereby further facilitating ROS-mediated apoptosis [68, 77]. The other oncogenic mechanism is mediated through SDH complex subunits dysfunction resulting from gene mutations. In SDHx-mutated PHEOs/PGLs, high succinate accumulation has been detected [78, 79] and the succinate:fumarate ratio has been found to be higher in SDHB-mutated and metastatic PHEOs/PGLs in contrast to SDHC/D-related tumors or tumors without metastases [79, 80]. Succinate has a structure similar to α-KG and exerts its ability to modulate the activity of α-KG-dependent dioxygenases––the enzyme family with diverse functions, including epigenetic regulation, oxygen sensing, collagen biosynthesis, fatty acid metabolism, and translation regulation [35, 81, 82]. The α-KG-dependent dioxygenase family also includes HIF prolyl hydroxylases (PHDs), which are needed for HIF-α hydroxylation and its further recognition by pVHL and eventual proteasome degradation [83–85]. Succinate acts as a competitive inhibitor of PHDs and thus promotes the activation of the HIF-α signaling pathway and expression of HIF target genes (Fig. 2) ([86–88]; reviewed in [32]). Additionally, several studies have demonstrated succinate’s ability to remodel the epigenome and alter gene expression. Succinate accumulation leads to DNA hypermethylation by inhibition of ten-eleven-translocation methylcytosine dioxygenase (TET) in PHEOs/PGLs and gastrointestinal stromal tumors [89, 90]. These findings emphasize the interconnection between the TCA cycle and epigenomic changes in cells. Moreover, succinate was shown to act as a signal for inflammation; during the inflammatory process, it accumulates in immune cells, leading to HIF-α activation [91]. These findings are very interesting, particularly because inflammation and tumorigenesis seem to be closely related.

Some of the metabolic pathways involved in HIF-α activation in PHEOs/PGLs. α-KG alpha-ketoglutarate, α-KGDH alpha-ketoglutarate dehydrogenase, acetyl-CoA acetyl coenzyme A, ACO aconitase, Akt RAC-alpha serine/threonine-protein kinase, CS citrate synthase, c-Myc Myc proto oncogene, eIF-4E eukaryotic translation initiation factor 4E, ERK mitogen-activated protein kinase 2, FH fumarate hydratase, HIF-α hypoxia-inducible factor α, IDH isocitrate dehydrogenase, MAX myc-associated factor X, mTORC1 mammalian target of rapamycin complex 1, mTORC2 mammalian target of rapamycin complex 2, MDH2 malate dehydrogenase 2, NF1 neurofibromin 1, PDH pyruvate dehydrogenase, PHD prolyl hydroxylase domain protein, PI3K phosphoinositide 3-kinase, pVHL von Hippel-Lindau protein, Raptor regulatory associated protein of mTOR, RAS rat sarcoma oncogene, RET rearranged during transfection proto-oncogene, Rheb RAS homolog enriched in brain, S6K S6 kinase, SDH succinate dehydrogenase complex, succinyl-CoA succinyl coenzyme A, SUCLG succinyl-CoA synthetase, TMEM127 transmembrane protein 127, TSC1/2 tuberous sclerosis complex 1/2, UQ ubiquitin
Fumarate
Fumarate is a TCA cycle metabolite that is converted to malate by FH. Inactivation of FH, e.g., due to its mutations, leads to fumarate accumulation. In high levels, fumarate can act as an oncometabolite through several mechanisms. Similarly to succinate, it acts as an α-KG-dependent dioxygenase inhibitor and consequently leads to HIF-α activation by PHD inhibition (Fig. 2) [35, 82, 86]. Recent findings suggest that there are also other HIF-independent mechanisms of oncogenesis. Fumarate is a moderately reactive α,β-unsaturated electrophilic metabolite that can covalently bind to cysteine residues of proteins in a process called succination and is a feature of FH-deficient tumors [92, 93]. Succination is a non-enzymatic and irreversible reaction [94]. Succinated proteins have been detected in a variety of animal and cellular models, and some of these proteins were identified to be associated with tumorigenesis [82, 93]. Furthermore, FH-mutated PHEOs/PGLs displayed the same epigenetic changes as SDH-mutated tumors involving the alterations in DNA methylation [21, 89].
Fumarate accumulation is probably also associated with mutations in another TCA cycle enzyme, MDH2. MDH2-mutated tumors have a transcriptional profile similar to SDH-mutated tumors. Like succinate and fumarate, malate also inhibits PHDs [95, 96] and, thus, HIF-α prolyl hydroxylation. Nevertheless, a high fumarate:succinate ratio was detected in MDH2-mutated tumors, suggesting fumarate accumulation, which is in contrast to SDHx-mutated tumors [24].
Citrate
Citrate occupies a central position in cellular metabolism as a substrate of the TCA cycle and as an important switch between metabolic pathways [45, 97]. It serves as a metabolic sensor and is involved in many biological processes, including inflammation, cancer, insulin secretion, and histone acetylation. After its synthesis in mitochondria, citrate becomes a substrate in the TCA cycle and, after oxidation, serves as a major source of cellular ATP production. Citrate inhibits and induces important strategic enzymes involved in glycolysis, the TCA cycle, gluconeogenesis, and fatty acids synthesis [98]. Under physiological conditions when citrate is sufficiently produced by TCA cycle, it represses the TCA cycle by inhibition of PDH and SDH enzymes and, when transported outside the mitochondria by the citrate carrier (CIC), acts as a glycolytic inhibitor (reviewed in [45, 56]). This means that it slows down or arrests both the TCA cycle and glycolysis and stimulates gluconeogenesis and lipid synthesis. In normal cells, together with ATP, citrate inhibits enzymatic activity of phosphofructokinase (PFK) 1 (a rate-limiting enzyme in glycolysis) and PFK2 [45, 56]. PFK2 produces fructose 2,6-bisphosphate, which is a powerful allosteric activator of PFK1 in cancer cells. Increased levels of PFK2 enable to overcome citrate and ATP inhibition of PFK1 when glucose uptake is high. Moreover, overexpression of the nuclear isoform of PFK2 (PFK3B) sustains a high rate of glycolysis and stimulates proliferation. Inhibition of PFK2 suppresses cell proliferation [99–101]; reviewed in [45, 56]. Citrate also indirectly inhibits pyruvate kinase (PK) by inhibition of PFK1, leading to decreased levels of fructose 1,6-bisphosphate, a strong allosteric activator of PK [102]. Moreover, cytosolic citrate is cleaved by ACLY to OAA and acetyl-CoA, which are used for pyruvate, fatty acid, and sterol synthesis, respectively [98]. ACLY has been shown to be involved in metabolic modulation mediated by PI3K/AKT signaling pathway. Activation of the PI3K/AKT pathway induces glucose uptake and promotes glucose carbon flux into biosynthetic pathways but its central role in oncogenesis is to relay the reprogamming of citrate metabolism [103]. In PHEOs/PGLs arising from SDHx and VHL mutations, lower levels of citrate, isocitrate, and ACO have been observed compared to those harboring RET, NF1, or TMEM127 mutations, demonstrating decreased OXPHOS and presence of pseudohypoxic state in those tumors [79].
The citrate level in the cytosol depends on the mitochondrial export rate, which can be modulated by the level of CIC expression; this is regulated by several transcription factors, including Sp1 and NF-κB [104]. CIC was shown to be overexpressed in cancer cells with its inhibition having antitumor activity, albeit no toxicity on adult normal tissues [105]. Citrate was also suggested to be used in cancer treatment since it was shown to induce cancer cell death directly or facilitate toxic effects of conventional anticancer drugs; reviewed in [45].
Glutamine, α-KG, and D2HG
Glutamine plays a crucial role in the metabolism of cancer cells since it is involved in cell bioenergetics and is a source of nitrogen for the production of molecules essential for cell growth [106–108]. Glutamine is the second most important carbon-based source of energy after glucose [45]. It serves a number of anabolic processes in cancer cells, such as protein and nucleotide synthesis and conversion to glutamate. Glutamate is used for amino acid synthesis or is transformed into α-KG that enters the TCA cycle [109]. Besides that, glutamine regulates the activity of mammalian target of rapamycin complex 1 (mTORC1). mTORC1 is a master regulator of cell growth, activates protein translation, and inhibits macroautophagy in response to abundance of amino acids and growth factor signaling [110, 111]. Glutamine conversion to glutamate is mediated by glutaminases, GLS and GLS2, which are under control of transcription factor c-Myc [98, 112]. The role of GLS in cancer is isoform-specific: GLS was proposed to work as an oncogene whereas GLS2 seems to exert antitumor activity [107, 108, 113–115]. Oncogenic expression of GLS was demonstrated to be dependent on the c-Myc transcription factor mediated by suppression of miR-23a/b [116] and c-Myc has also been found to stimulate glutamine catabolism and cell proliferation via the upregulation of glutamine importer SLC1A. GLS expression can also be activated by ErbB2 signaling through activation of NF-κB [45, 117].
The product of GLS activity, glutamate, is an essential precursor for antioxidant glutathione synthesis and also serves as a donor of amino groups for non-essential amino acids. Glutamate is transformed to α-KG in a reaction catalyzed either by transaminases for production of non-essential amino acids or by glutamate dehydrogenase (GDH). Under conditions of limited glucose supply, GDH-mediated conversion is prevalent. α-KG is then utilized in the TCA cycle, either for succinate (via α-KGDH) or for citrate (via IDH) production. Both of them are important substrates—succinate for the TCA cycle and citrate for fatty acid synthesis. The reductive pathway of citrate production is favored in cells under (pseudo)hypoxic conditions to compensate the drop in glucose-derived citrate production [45]. Lately, it has been described how low levels of intracellular citrate and high levels of α-KG are associated with HIF-dependent reductive carboxylation of glutamine [118–120]. According to Sun and Denko [121], hypoxia causes a decrease in glucose-derived citrate because of decreased PDH activity and also increases α-KG levels as a result of decreased αKGDH activity. These changes in substrate concentrations also drive the reverse reaction of IDH. This metabolic switch keeps maintenance of lipid synthesis that is required for the growth of cells in hypoxia.
D2HG is an oncometabolite that is synthesized in place of α-KG in IDH1/2-mutated cells [122]. D2HG is structurally very similar to α-KG and glutamate; this similarity results in the competitive inhibition of α-KG-dependent dioxygenases [123]. Unlike succinate and fumarate, the effect of D2HG on PHDs is not completely clear since there is evidence of both inhibition [123, 124] and activation of PHDs [125, 126]; reviewed in [127]. According to the work of Williams et al. [128], mutant IDH1 expression in gliomas was not sufficient to stabilize HIF-1α. But there is evidence that accumulation of D2HG is associated with epigenetic changes, disruption of defense mechanisms against ROS, and changes in redox homeostasis. Mechanisms of its involvement in tumorigenesis are not fully understood yet but are thought to be based on epigenetic modifications that result in differential expression of genes involved in cell proliferation, changes in HIF-α levels, as well as aberrant extracellular matrix structures induced by changes in collagen synthesis [39, 45, 127]. Further studies are needed to fully explore metabolic and physiological defects present in IDH-mutated tumors as well as their consequences and roles in tumorigenesis.
Current Views on Cancer Cell Metabolism Linked to Craving for Oxygen in the Pathogenesis of PHEO/PGL
Although in recent years scientists were able to reveal several crucial signaling pathways involved in tumor development, precise metabolic changes still remain hidden and further research is needed. It is currently known that cancer cells switch from OXPHOS-producing ATP to aerobic glycolysis-producing lactate and suppression of mitochondrial function. This is mostly mediated by changes in the fate of the end product of glycolysis––pyruvate. Under normal conditions in healthy cells, pyruvate is directed to mitochondria, metabolized by PDH to acetyl-CoA, and enters the TCA cycle to fuel OXPHOS. But in most cancer cells, PDH activity is suppressed, leading to reduced flow of pyruvate to the mitochondria and a decrease in OXPHOS. Under these conditions, pyruvate is converted to lactate by LDH [45]. This is the situation when the cell starts to “gasp” for the air even in the presence of oxygen. This feature, referred to as the “Warburg effect” or “aerobic glycolysis,” was first observed by Otto Warburg in 1924 and is considered a defect in mitochondrial respiration and/or ATP production [129–131]. Although glycolysis is much less efficient in the production of ATP compared to OXPHOS (2 vs. 36 molecules of ATP), it makes cancer cells more resistant to hypoxia (oxygen deprivation) as a result of excessive growth or their high metabolic activity and poor oxygen supply. Thus, aerobic glycolysis is present especially in the early avascular phase of tumorigenesis, where it provides an advantage in hypoxic conditions [132]. This metabolic shift also provides the cell with resources to sustain proliferation. In the past, it was anticipated that these metabolic changes are caused by defects in mitochondria but it is now clear that they emerge from specific metabolic regulatory signaling. Should we, therefore, refer to some cancers as a metabolic disease? Certainly, yes, as suggested for kidney cancer by Linehan et al. [40, 133]. Mutations in known kidney cancer susceptibility genes lead to dysregulation of at least one metabolic pathway involved in the sensing of oxygen, iron, or nutrients, and particularly in the TCA cycle enzymes. Special attention should also be given to PHEOs/PGLs as several hereditary PHEOs (related to mutations in VHL, SDHx, FH) are closely linked to the development of kidney cancer and can occur concurrently. Thus, for PHEOs/PGLs, as outlined above, several metabolic hits in the TCA cycle occur that lead to pseudohypoxia. Succinate or fumarate accumulation occurs in SDHx- or FH- and MDH2-mutated PHEOs/PGLs, respectively. In SDHx- and VHL-mutated PHEOs/PGLs, lower levels of citrate, isocitrate, and ACO were detected, which is associated with epigenetic and metabolic changes involved in tumorigenesis. All these metabolic changes result in a metabolic shift from OXPHOS to aerobic glycolysis with further “gasping for air” by TCA metabolite-specific activation of the HIF signaling pathway. HIF-α stabilization and inhibition of α-KG-dependent dioxygenases was described in VHL- and SDHx-mutated tumors [28, 134–139]. HIF enhances the glycolytic pathway by increasing expression of target genes involved in the glycolytic and anabolic processes (glucose transporters (GLUT) 1, 2, hexokinase (HK) 2, PKM2, LDH-A) [34, 140]. Although some studies have not found the GLUT1 overexpression of SDHx-related tumors [29, 135, 141], there was still increased expression of GLUT3 and HK2 as well as LDH-A [135, 141]. Functional analysis of PHEO/PGL tumor tissue from patients with somatic HIF2A and germline PHD1 and PHD2 mutations also showed increased activity of the HIF signaling pathway and its target gene expression, including GLUT1 and in PHD-mutated PHEOs/PGLs also LDH-A [12, 16]. Recently, Berkel et al. [142] reported high expression of HK2 in SDHx-related PHEOs/PGLs when compared to sporadic and RET-, NF1-, and MAX-related tumors. All PHEOs/PGLs in their study showed similar GLUT1 and GLUT3 expression.
HIF-α activation is becoming a significant detriment to PHEOs/PGLs but most likely, not solely, results in tumorigenesis acceleration, tumor growth, and metastasis development. Now, these tumors become truly hypoxic and are “gasping for air,” culminating in no escape from normal cell function collapse and ongoing tumorigenesis. Indeed, this is not the only oncogenic mechanism in PHEOs/PGLs but it seems to play an important role in their development.
Cells, in craving for oxygen, activate a number of adaptive responses, coordinated by various signaling pathways, most of them controlled by HIFs [143–145]. HIFs are transcription factors comprised of ubiquitously expressed three α subunit isoforms (oxygen-regulated, HIF-1α, HIF-2α, HIF-3α) and a constitutively expressed β subunit [144–146]. HIF-α regulates the expression of a large number of genes related to adaptation to hypoxia, such as those involved in angiogenesis, glycolysis, survival, differentiation, or proliferation. Under normoxic conditions, HIF-1α and HIF-2α are degraded via the ubiquitin-proteasome pathway, a process involving prolyl hydroxylation by PHDs (PHD1, PHD2, and PHD3) and the recognition of hydroxylated HIF-α subunits by pVHL. pVHL is a component of an E3 ubiquitin ligase complex that targets HIF-α for proteasomal degradation [145]. Under hypoxic conditions, HIF-α becomes stabilized, heterodimerizes with HIF-β, recruits co-activators, binds to the DNA at hypoxia-responsive elements (HREs), and activates transcription of the target genes (reviewed in [32]). There are also multiple other oxygen-independent oncogenic pathways involved in HIF-α regulation. Two main pathways that induce the pseudohypoxic state independently of oxygen tension are the PI3K and mTOR pathways. Their signaling leads to stabilization of HIF-1α, which causes a strong increase in glycolysis––the Warburg phenotype [45, 147, 148]. Full glucose oxidation in mitochondria is decreased, resulting in decreased ATP and citrate production. In turn, the feedback of these molecules on the main regulator enzyme for glycolysis, PFK1, is broken. This leads to acceleration of glycolysis and enhanced production of lactic acid [147]. HIF-α enhances glycolysis by tilting the balance between PDH and LDH in favor of LDH [63, 77, 149]. Occasionally, impairment of mitochondrial respiration can be observed, resulting in defective OXPHOS and contributing towards tumorigenesis [147]. Reduced activity of the respiratory chain results in reduced ROS generation and, thus, a decrease in apoptotic signals [77, 150]. Interestingly, HIF-α-driven pseudohypoxia might drive overproduction of TCA substrates, for instance D2HG [151], giving the bidirectional connection of TCA with metabolic pathways in cell.
SDHx mutations disrupt complex II activity, resulting in succinate accumulation and inhibition of PHD-catalyzed HIF-α hydroxylation required for HIF-α proteasomal degradation [86–88, 152], thus, mimicking hypoxia. A similar scenario happens in FH- and MDH2-deficient tumors, since fumarate has the ability to competitively inhibit PHDs (Fig. 2). Additionally, both metabolites inhibit other members of α-KG-dependent dioxygenases, including histone lysine demethylases (KDMs) and TET enzymes [89, 90, 153, 154]. Competitive inhibition of chromatin-modifying α-KG-dependent dioxygenases results in marked impairment of epigenetic regulation of gene expression and DNA hypermethylation phenotype [89, 90]. Abnormal fumarate and succinate accumulation also exerts tumorigenic effects by other mechanisms: overproduction of ROS may participate in oncogenesis and tumor progression by irreversible DNA modifications and protein oxidation. Moreover, protein succination may result in the constitutive activation of the NRF2-mediated antioxidant defense pathway that has the ability to promote tumorigenesis by enhancing ROS elimination as well as by generating a reductive environment that can facilitate cell proliferation and survival. The functional impairment of multiple proteins leads to subsequent dysregulation of cellular metabolism and activation of oncogenic pathways [151, 154–157].
In PHEOs/PGLs, besides the mutations in genes encoding TCA cycle enzymes, other mutations in genes that directly or indirectly influence the TCA cycle, HIF signaling, and other pathways involved in tumorigenesis were described. Mutations in VHL, PHD1/2, and HIF2A genes are associated with direct HIF-α pathway dysregulation (Fig. 2) [24, 32, 34]. In VHL mutations, oxygen sensing is impaired, resulting in HIF-α stabilization in different ranges of activation depending on the type of mutation. In VHL type 1 disease, mutations are usually nonsense or deletions and result in complete dysfunction of pVHL, whereas VHL type 2 disease mutations are commonly missense and cause mild changes in pVHL structure and compromise the activity of ubiquitin-ligase complex in variant extent [158–160]. Interestingly, VHL mutations associated with mild changes in pVHL function confer a higher risk for PHEO/PGL development than those associated with complete dysfunction of pVHL, suggesting that besides pseudohypoxia there are also other mechanisms involved in PHEO/PGL pathogenesis (reviewed in [161]). As mentioned above, HIF-α prolyl hydroxylation is a crucial step for its further proteasomal degradation. Germline loss-of-function PHD1 and PHD2 mutations described in patients with PHEO/PGL and polycythemia lead to stabilization and activation of HIF-α by the decreased or lost ability to hydroxylate HIF-α prolyl residues [16, 17, 144, 145]. The role of HIF-α, especially HIF-2α, in PHEO/PGL development is supported by the detection of somatic and germline gain-of-function mutations in the HIF2A gene, causing HIF-α stabilization. HIF2A mutations are associated with a specific clinical phenotype: multiple PHEOs/PGLs, polycythemia, and, often, duodenal somatostatinomas [12, 13, 162, 163].
Indirect activation of HIF-α can be found in cluster 2 PHEOs/PGLs arising from NF1, RET, TMEM127, K-RAS, H-RAS, and MAX mutations (Fig. 2). HIF-α activation in these tumors is not expressed to the same extent as in cluster 1 tumors. However, these clusters are most probably interconnected via mTOR and PI3K signaling pathways [32]. The PI3K signaling pathway is linked to growth control, glucose metabolism, and can also activate HIF [164]. Activation of PI3K and mTOR pathways leads to HIF-α accumulation and to induction of its downstream genes transcription. In RET, NF1, and K-/H-RAS mutations, HIF up-regulation is driven through activation of the RAS/RAF/MEK/ERK, or mitogen-activated protein kinase (MAPK), signaling pathway, resulting in activation of downstream pathways (PI3K and mTORC) involved in oncogenic processes (reviewed in [32]). mTOR-dependent translation of HIF-α is induced by the loss of tuberous sclerosis 1 and 2 (TSC1, TSC2) or serine/threonine kinase LKB1 tumor-suppressor genes and via the activation of the AKT signaling pathway (Fig. 2) [165–170]. Moreover, mutations in tumor suppressor MAX lead to deregulation of c-Myc signaling that enhances both the glycolytic pathway and OXPHOS. c-Myc is a very powerful gene transcription activator, labeled also as the “oncogene from hell” [171]. It regulates genes involved in the biogenesis of ribosomes and mitochondria, in the regulation of glucose and glutamine metabolism, and can also induce DNA damage, increase ROS production, and genome instability [151, 172–176]. c-Myc was shown to regulate glycolysis in cells under normoxic conditions via direct activation of LDH-A and other glycolytic genes [148]. Metabolic reprogramming in cancer cells can be mediated via crosstalk between HIF-α and c-Myc. c-Myc transcriptional activity is also directly regulated by sirtuin 1 (SIRT1), either by c-Myc deacetylation or by binding c-Myc and promoting its association with MAX [151, 177, 178].
In K-RAS-mutated tumors, in addition to activation MAPK signaling pathway and subsequent HIF-α activation, the strong link between RAS, mitochondria, and the Bcl-2 family proteins was described. Increased K-RAS expression is associated with mitochondrial dysfunction, mitochondrial ROS production, and, thus, with tumorigenesis [179, 180].
The KIF1β tumor suppressor gene is a downstream target of PHD3 necessary for neuronal apoptosis and seems to be connected to both the HIF and MAPK signaling pathways [14, 15].
Conclusions and Future Therapeutic Perspectives
There is increasing evidence that pseudohypoxia—gasping for the air—plays an important role in PHEO/PGL development and is a result of interplay between several metabolic signaling pathways. The role of mitochondria in tumorigenesis is indubitable, since most metabolic pathways in the cell are somehow interconnected with this organelle and with the TCA cycle. Mitochondrial dysfunction is quite common in cancers and results in changes of mitochondrial energy metabolism, specifically in the metabolic switch from OXPHOS to glycolysis. Pseudohypoxia and the metabolic switch to aerobic glycolysis are associated with a higher aggressiveness of tumors, resistance to systemic and radiation therapies, along with worse prognosis for patients [181–185].
This article shows how initial stages of “gasping for air” in PHEOs/PGLs are closely linked to metabolic and hypoxic (HIF signaling pathways directly) reprogramming of these tumors. In the past years, there is clear evidence of the direct role of TCA cycle enzymes and substrates in tumor development, including PHEOs/PGLs. Defects in SDH, FH, IDH, and MDH2 enzymes lead to accumulation of TCA metabolites, resulting in dysregulation of cell metabolism, generation of a pseudohypoxic state, alterations in epigenetic homeostasis, and other protumorigenic processes. The key role of TCA cycle enzymes in tumorigenesis designates them as very promising therapeutic targets. Based on actual understanding of the biochemical routes that redirect and reprogram cell metabolism in PHEOs/PGLs, two strategies in treatment could be used: one that directly focuses on inhibiting HIF pathway signaling and another aimed at fixing metabolic reprogramming in cancer cells.
Nowadays, several agents affecting HIF-1α signaling have been introduced, with varying results depending on a cancer’s HIF-α phenotype [43, 186–188]. They include antiangiogenic agents, such as inhibitors of vascular endothelial growth factor (VEGF), mTOR, and heat shock protein 90 inhibitors, or agents that restore or activate PHD enzyme activity. Drugs selectively targeting HIF-2α signaling are under development; for example, two cembrane diterpenes were found to selectively inhibit HIF-2α and modulate its downstream effectors [189–191]. In terms of inhibiting HIF signaling pathway, agents targeting both HIF-1α and HIF-2α subunits, such as PI3K inhibitors, dual PI3K/mTOR inhibitor, or JNK inhibitor are of great interest since they can activate different genes and, in many cancers, both are expressed in a certain balance [138, 192–194].
Dysregulated TCA cycle is another very promising therapeutical target. Restoring the activity of malfunctional enzymes or providing the depleted substrates could reverse energy production back to OXPHOS and, thus, restore mitochondrial functions. Several promising agents are under development, e.g., drugs restoring the functionality of mitochondrial enzymes, small molecule inhibitors of certain proteins, or glycolysis inhibitors. For example, proteostasis regulators, such as histone deacetylase inhibitors (HDACis), have the potential to increase the stability and, in effect, the total amount of mitochondrial SDHB protein in SDHB-deficient cells [195]. Another compound––redox-silent vitamin E analog α-tocopheryl succinate (α-TOS)––with known antiproliferative effects, has been found to target the UbQ binding site of complex II [196] and shown to be effective in several experimental cancers in vitro and in vivo with no toxicity to normal cells [197–200]. The other possibility of treatment is inhibition of glycolysis by small molecule inhibitors of GLUT1 [201, 202] or its reversion using dichloroacetate (DCA) [203, 204] or 3-bromopyruvate (3BP) [205–207]. Activation of mitochondrial respiration can also be induced by downregulation of LDH by small interfering ribonucleic acid (siRNA), which prevents conversion of pyruvate to lactate and, as a result, allows for the entry of pyruvate into the mitochondria in order to fuel the TCA cycle and OXPHOS [208]. Discovery of IDH1/2 mutations led to the development of small-molecule inhibitors that reduced D2HG levels in cancer cells and inhibited tumorigenesis; IDH1/2 inhibitors were proven to be successful in a glioma xenograft model and produced cytostatic rather than cytotoxic effects [209–211]. Since glutamine is highly utilized in cancer cells, several approaches aimed to influence different steps of glutamine metabolism are studied: suppressing cancer cell glutamine uptake, suppressing glutamine-dependent anaplerosis, inhibiting complex I, targeting glutamine-dependent mTOR activation, and enzymatic lowering of blood glutamine levels (reviewed in [111]). Therapies targeted against glutamine seem to be very promising, particularly in tumors displaying glutamine dependence. Another attractive target in PHEO/PGL and cancer treatment is citrate, levels of which are decreased in tumor cells and have the capability to induce apoptotic cell death or facilitate the toxic effects of conventional anticancer drugs [212–214].
The other direction in PHEO/PGL research is development of novel diagnostic methods based on biomarkers associated with altered metabolic pathways and pseudohypoxia that will help choose targeted therapy and, thus, personalize anticancer treatment.
References
Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML et al (1993) Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 260:1317–1320
Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E et al (1993) Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 363:458–460
Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK et al (1990) Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 62:187–192
Burnichon N, Briere JJ, Libe R, Vescovo L, Riviere J, Tissier F et al (2010) SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet 19:3011–3020
Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E et al (2001) Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 69:49–54
Niemann S, Muller U (2000) Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet 26:268–270
Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A et al (2000) Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287:848–851
Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H et al (2009) SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 325:1139–1142
Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES et al (2010) Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet 42:229–233
Yao L, Schiavi F, Cascon A, Qin Y, Inglada-Perez L, King EE et al (2010) Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. JAMA 304:2611–2619
Comino-Mendez I, Gracia-Aznarez FJ, Schiavi F, Landa I, Leandro-Garcia LJ, Leton R et al (2011) Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 43:663–667
Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E et al (2012) Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med 367:922–930
Toledo RA, Qin Y, Srikantan S, Morales NP, Li Q, Deng Y et al (2013) In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr Relat Cancer 20:349–359
Schlisio S, Kenchappa RS, Vredeveld LC, George RE, Stewart R, Greulich H et al (2008) The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev 22:884–893
Yeh IT, Lenci RE, Qin Y, Buddavarapu K, Ligon AH, Leteurtre E et al (2008) A germline mutation of the KIF1B beta gene on 1p36 in a family with neural and nonneural tumors. Hum Genet 124:279–285
Yang C, Zhuang Z, Fliedner SM, Shankavaram U, Sun MG, Bullova P et al (2015) Germ-line PHD1 and PHD2 mutations detected in patients with pheochromocytoma/paraganglioma-polycythemia. J Mol Med (Berl) 93:93–104
Ladroue C, Carcenac R, Leporrier M, Gad S, Le Hello C, Galateau-Salle F et al (2008) PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med 359:2685–2692
Crona J, Delgado Verdugo A, Maharjan R, Stalberg P, Granberg D, Hellman P et al (2013) Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J Clin Endocrinol Metab 98:E1266–1271
Hrascan R, Pecina-Slaus N, Martic TN, Colic JF, Gall-Troselj K, Pavelic K et al (2008) Analysis of selected genes in neuroendocrine tumours: insulinomas and phaeochromocytomas. J Neuroendocrinol 20:1015–1022
Gaal J, Burnichon N, Korpershoek E, Roncelin I, Bertherat J, Plouin PF et al (2010) Isocitrate dehydrogenase mutations are rare in pheochromocytomas and paragangliomas. J Clin Endocrinol Metab 95:1274–1278
Castro-Vega LJ, Buffet A, De Cubas AA, Cascon A, Menara M, Khalifa E et al (2014) Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet 23:2440–2446
Clark GR, Sciacovelli M, Gaude E, Walsh DM, Kirby G, Simpson MA et al (2014) Germline FH mutations presenting with pheochromocytoma. J Clin Endocrinol Metab 99:E2046–2050
Wadt K, Choi J, Chung JY, Kiilgaard J, Heegaard S, Drzewiecki KT et al (2012) A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma Res 25:815–818
Cascon A, Comino-Mendez I, Curras-Freixes M, de Cubas AA, Contreras L, Richter S et al (2015) Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J Natl Cancer Inst. doi:10.1093/jnci/djv053
Fishbein L, Khare S, Wubbenhorst B, DeSloover D, D'Andrea K, Merrill S et al (2015) Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun. doi:10.1038/ncomms7140
Sudarshan S, Sourbier C, Kong HS, Block K, Valera Romero VA, Yang Y et al (2009) Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol 29:4080–4090
Eisenhofer G, Huynh TT, Pacak K, Brouwers FM, Walther MM, Linehan WM et al (2004) Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome. Endocr Relat Cancer 11:897–911
Dahia PL, Ross KN, Wright ME, Hayashida CY, Santagata S, Barontini M et al (2005) A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet 1:72–80
Lopez-Jimenez E, Gomez-Lopez G, Leandro-Garcia LJ, Munoz I, Schiavi F, Montero-Conde C et al (2010) Research resource: transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol Endocrinol 24:2382–2391
Gimenez-Roqueplo AP, Dahia PL, Robledo M (2012) An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res 44:328–333
Vicha A, Musil Z, Pacak K (2013) Genetics of pheochromocytoma and paraganglioma syndromes: new advances and future treatment options. Curr Opin Endocrinol Diabetes Obes 20:186–191
Jochmanova I, Yang C, Zhuang Z, Pacak K (2013) Hypoxia-inducible factor signaling in pheochromocytoma: turning the rudder in the right direction. J Natl Cancer Inst 105:1270–1283
Favier J, Amar L, Gimenez-Roqueplo AP (2015) Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol 11:101–111
Vicha A, Taieb D, Pacak K (2014) Current views on cell metabolism in SDHx-related pheochromocytoma and paraganglioma. Endocr Relat Cancer 21:R261–277
Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL et al (2005) HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8:143–153
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K et al (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361:1058–1066
Arai M, Nobusawa S, Ikota H, Takemura S, Nakazato Y (2012) Frequent IDH1/2 mutations in intracranial chondrosarcoma: a possible diagnostic clue for its differentiation from chordoma. Brain Tumor Pathol 29:201–206
Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X et al (2014) IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res 74:3317–3331
Linehan WM, Ricketts CJ (2013) The metabolic basis of kidney cancer. Semin Cancer Biol 23:46–55
Bertout JA, Patel SA, Simon MC (2008) The impact of O2 availability on human cancer. Nat Rev Cancer 8:967–975
Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM et al (2011) Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A 108:19611–19616
Semenza GL (2010) Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29:625–634
Scheffler IE (2008) Mitochondria. 2nd Aufl.: Wiley
Kruspig B, Zhivotovsky B, Gogvadze V (2014) Mitochondrial substrates in cancer: drivers or passengers? Mitochondrion 19(Pt A):8–19
Desideri E, Vegliante R, Ciriolo MR (2015) Mitochondrial dysfunctions in cancer: genetic defects and oncogenic signaling impinging on TCA cycle activity. Cancer Lett 356:217–223
Owen OE, Kalhan SC, Hanson RW (2002) The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 277:30409–30412
Yang C, Ko B, Hensley CT, Jiang L, Wasti AT, Kim J et al (2014) Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell 56:414–424
Hensley CT, Wasti AT, DeBerardinis RJ (2013) Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest 123:3678–3684
Cardaci S, Ciriolo MR (2012) TCA cycle defects and cancer: when metabolism tunes redox state. Int J Cell Biol 2012:161837
Guffon N, Lopez-Mediavilla C, Dumoulin R, Mousson B, Godinot C, Carrier H et al (1993) 2-Ketoglutarate dehydrogenase deficiency, a rare cause of primary hyperlactataemia: report of a new case. J Inherit Metab Dis 16:821–830
Wallace DC, Fan W, Procaccio V (2010) Mitochondrial energetics and therapeutics. Annu Rev Pathol 5:297–348
Saini AG, Singhi P (2013) Infantile metabolic encephalopathy due to fumarase deficiency. J Child Neurol 28:535–537
Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D et al (2002) Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30:406–410
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773
Icard P, Poulain L, Lincet H (2012) Understanding the central role of citrate in the metabolism of cancer cells. Biochim Biophys Acta 1825:111–116
Patel MS, Roche TE (1990) Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J 4:3224–3233
Reed LJ (2001) A trail of research from lipoic acid to alpha-keto acid dehydrogenase complexes. J Biol Chem 276:38329–38336
Kato M, Wynn RM, Chuang JL, Tso SC, Machius M, Li J et al (2008) Structural basis for inactivation of the human pyruvate dehydrogenase complex by phosphorylation: role of disordered phosphorylation loops. Structure 16:1849–1859
Saunier E, Benelli C, Bortoli S (2015) The pyruvate dehydrogenase complex in cancer: an old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int J Cancer. doi:10.1002/ijc.29564
Denko NC (2008) Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 8:705–713
Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR (2011) High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 117:e207–217
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3:187–197
Adeva-Andany M, Lopez-Ojen M, Funcasta-Calderon R, Ameneiros-Rodriguez E, Donapetry-Garcia C, Vila-Altesor M et al (2014) Comprehensive review on lactate metabolism in human health. Mitochondrion 17:76–100
Wong N, Ojo D, Yan J, Tang D (2015) PKM2 contributes to cancer metabolism. Cancer Lett 356:184–191
Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R et al (2008) The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452:230–233
Lenaz G, Genova ML (2010) Structure and organization of mitochondrial respiratory complexes: a new understanding of an old subject. Antioxid Redox Signal 12:961–1008
Grimm S (2013) Respiratory chain complex II as general sensor for apoptosis. Biochim Biophys Acta 1827:565–572
Yankovskaya V, Horsefield R, Tornroth S, Luna-Chavez C, Miyoshi H, Leger C et al (2003) Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 299:700–704
Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D et al (2005) Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 121:1043–1057
Ghezzi D, Goffrini P, Uziel G, Horvath R, Klopstock T, Lochmuller H et al (2009) SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat Genet 41:654–656
Ishii T, Yasuda K, Akatsuka A, Hino O, Hartman PS, Ishii N (2005) A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res 65:203–209
Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD (2012) Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem 287:27255–27264
Moreno-Sanchez R, Hernandez-Esquivel L, Rivero-Segura NA, Marin-Hernandez A, Neuzil J, Ralph SJ et al (2013) Reactive oxygen species are generated by the respiratory complex II—evidence for lack of contribution of the reverse electron flow in complex I. FEBS J 280:927–938
Ricci JE, Gottlieb RA, Green DR (2003) Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J Cell Biol 160:65–75
Albayrak T, Scherhammer V, Schoenfeld N, Braziulis E, Mund T, Bauer MK et al (2003) The tumor suppressor cybL, a component of the respiratory chain, mediates apoptosis induction. Mol Biol Cell 14:3082–3096
Hwang MS, Rohlena J, Dong LF, Neuzil J, Grimm S (2014) Powerhouse down: complex II dissociation in the respiratory chain. Mitochondrion 19(Pt A):20–28
Rao JU, Engelke UF, Rodenburg RJ, Wevers RA, Pacak K, Eisenhofer G et al (2013) Genotype-specific abnormalities in mitochondrial function associate with distinct profiles of energy metabolism and catecholamine content in pheochromocytoma and paraganglioma. Clin Cancer Res 19:3787–3795
Richter S, Peitzsch M, Rapizzi E, Lenders JW, Qin N, de Cubas AA et al (2014) Krebs cycle metabolite profiling for identification and stratification of pheochromocytomas/paragangliomas due to succinate dehydrogenase deficiency. J Clin Endocrinol Metab 99:3903–3911
Lendvai N, Pawlosky R, Bullova P, Eisenhofer G, Patocs A, Veech RL et al (2014) Succinate-to-fumarate ratio as a new metabolic marker to detect the presence of SDHB/D-related paraganglioma: initial experimental and ex vivo findings. Endocrinology 155:27–32
Loenarz C, Schofield CJ (2008) Expanding chemical biology of 2-oxoglutarate oxygenases. Nat Chem Biol 4:152–156
Yang M, Ternette N, Su H, Dabiri R, Kessler BM, Adam J et al (2014) The succinated proteome of FH-mutant tumours. Metabolites 4:640–654
Bruick RK, McKnight SL (2001) A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294:1337–1340
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M et al (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292:464–468
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ et al (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292:468–472
Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC et al (2005) Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 14:2231–2239
Briere JJ, Favier J, Benit P, El Ghouzzi V, Lorenzato A, Rabier D et al (2005) Mitochondrial succinate is instrumental for HIF1alpha nuclear translocation in SDHA-mutant fibroblasts under normoxic conditions. Hum Mol Genet 14:3263–3269
Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD et al (2005) Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7:77–85
Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C et al (2013) SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23:739–752
Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M et al (2013) Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov 3:648–657
Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G et al (2013) Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496:238–242
Alderson NL, Wang Y, Blatnik M, Frizzell N, Walla MD, Lyons TJ et al (2006) S-(2-Succinyl)cysteine: a novel chemical modification of tissue proteins by a Krebs cycle intermediate. Arch Biochem Biophys 450:1–8
Zheng L, Cardaci S, Jerby L, MacKenzie ED, Sciacovelli M, Johnson TI et al (2015) Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat Commun 6:6001
Merkley ED, Metz TO, Smith RD, Baynes JW, Frizzell N (2014) The succinated proteome. Mass Spectrom Rev 33:98–109
Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, Giaccia AJ et al (2007) Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol 27:912–925
Philip B, Ito K, Moreno-Sanchez R, Ralph SJ (2013) HIF expression and the role of hypoxic microenvironments within primary tumours as protective sites driving cancer stem cell renewal and metastatic progression. Carcinogenesis 34:1699–1707
Costello LC, Franklin RB (2013) A review of the important central role of altered citrate metabolism during the process of stem cell differentiation. J Regen Med Tissue Eng 2:pii 1
Iacobazzi V, Infantino V (2014) Citrate—new functions for an old metabolite. Biol Chem 395:387–399
Chesney J (2006) 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase and tumor cell glycolysis. Curr Opin Clin Nutr Metab Care 9:535–539
Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL et al (2009) Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J Biol Chem 284:24223–24232
Yalcin A, Telang S, Clem B, Chesney J (2009) Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp Mol Pathol 86:174–179
Jurica MS, Mesecar A, Heath PJ, Shi W, Nowak T, Stoddard BL (1998) The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure 6:195–210
Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB (2005) ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24:6314–6322
Iacobazzi V, Infantino V, Palmieri F (2008) Epigenetic mechanisms and Sp1 regulate mitochondrial citrate carrier gene expression. Biochem Biophys Res Commun 376:15–20
Catalina-Rodriguez O, Kolukula VK, Tomita Y, Preet A, Palmieri F, Wellstein A et al (2012) The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 3:1220–1235
Mazurek S, Boschek CB, Hugo F, Eigenbrodt E (2005) Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol 15:300–308
Szeliga M, Albrecht J (2014) Opposing roles of glutaminase isoforms in determining glioblastoma cell phenotype. Neurochem Int. doi:10.1016/j.neuint.2014.11.004
Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM et al (2011) Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A 108:8674–8679
Kishton RJ, Rathmell JC (2015) Novel therapeutic targets of tumor metabolism. Cancer J 21:62–69
Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124:471–484
Wise DR, Thompson CB (2010) Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 35:427–433
Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK et al (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 105:18782–18787
Lobo C, Ruiz-Bellido MA, Aledo JC, Marquez J, Nunez De Castro I, Alonso FJ (2000) Inhibition of glutaminase expression by antisense mRNA decreases growth and tumourigenicity of tumour cells. Biochem J 348:257–261
Turner A, McGivan JD (2003) Glutaminase isoform expression in cell lines derived from human colorectal adenomas and carcinomas. Biochem J 370:403–408
Perez-Gomez C, Campos-Sandoval JA, Alonso FJ, Segura JA, Manzanares E, Ruiz-Sanchez P et al (2005) Co-expression of glutaminase K and L isoenzymes in human tumour cells. Biochem J 386:535–542
Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T et al (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458:762–765
Qie S, Chu C, Li W, Wang C, Sang N (2014) ErbB2 activation upregulates glutaminase 1 expression which promotes breast cancer cell proliferation. J Cell Biochem 115:498–509
Fendt SM, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM et al (2013) Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells. Nat Commun 4:2236
Gameiro PA, Laviolette LA, Kelleher JK, Iliopoulos O, Stephanopoulos G (2013) Cofactor balance by nicotinamide nucleotide transhydrogenase (NNT) coordinates reductive carboxylation and glucose catabolism in the tricarboxylic acid (TCA) cycle. J Biol Chem 288:12967–12977
Gameiro PA, Yang J, Metelo AM, Perez-Carro R, Baker R, Wang Z et al (2013) In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab 17:372–385
Sun RC, Denko NC (2014) Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab 19:285–292
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM et al (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462:739–744
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH et al (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19:17–30
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P et al (2009) Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 324:261–265
Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S et al (2012) Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483:484–488
Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C et al (2013) (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339:1621–1625
Parker SJ, Metallo CM (2015) Metabolic consequences of oncogenic IDH mutations. Pharmacol Ther. doi:10.1016/j.pharmthera.2015.05.003
Williams SC, Karajannis MA, Chiriboga L, Golfinos JG, von Deimling A, Zagzag D (2011) R132H-mutation of isocitrate dehydrogenase-1 is not sufficient for HIF-1alpha upregulation in adult glioma. Acta Neuropathol 121:279–281
Warburg O, Wind F, Negelein E (1927) The metabolism of tumors in the body. J Gen Physiol 8:519–530
Lu J, Tan M, Cai Q (2015) The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett 356:156–164
Koppenol WH, Bounds PL, Dang CV (2011) Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer 11:325–337
Gillies RJ, Gatenby RA (2007) Adaptive landscapes and emergent phenotypes: why do cancers have high glycolysis? J Bioenerg Biomembr 39:251–257
Linehan WM, Srinivasan R, Schmidt LS (2010) The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol 7:277–285
Qin Y, Buddavarapu K, Dahia PL (2009) Pheochromocytomas: from genetic diversity to new paradigms. Horm Metab Res 41:664–671
Favier J, Briere JJ, Burnichon N, Riviere J, Vescovo L, Benit P et al (2009) The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS One 4:e7094
Favier J, Gimenez-Roqueplo AP (2010) Pheochromocytomas: the (pseudo)-hypoxia hypothesis. Best Pract Res Clin Endocrinol Metab 24:957–968
Smith EH, Janknecht R, Maher LJ 3rd (2007) Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum Mol Genet 16:3136–3148
Keith B, Johnson RS, Simon MC (2012) HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 12:9–22
Her YF, Nelson-Holte M, Maher LJ 3rd (2015) Oxygen concentration controls epigenetic effects in models of familial paraganglioma. PLoS One 10:e0127471
Soga T (2013) Cancer metabolism: key players in metabolic reprogramming. Cancer Sci 104:275–281
Fliedner SM, Kaludercic N, Jiang XS, Hansikova H, Hajkova Z, Sladkova J et al (2012) Warburg effect’s manifestation in aggressive pheochromocytomas and paragangliomas: insights from a mouse cell model applied to human tumor tissue. PLoS One 7:e40949
van Berkel A, Rao JU, Kusters B, Demir T, Visser E, Mensenkamp AR et al (2014) Correlation between in vivo 18F-FDG PET and immunohistochemical markers of glucose uptake and metabolism in pheochromocytoma and paraganglioma. J Nucl Med 55:1253–1259
Semenza GL (2010) Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med 2:336–361
Schofield CJ, Ratcliffe PJ (2004) Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 5:343–354
Kaelin WG, Ratcliffe PJ (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30:393–402
Wang GL, Jiang BH, Rue EA, Semenza GL (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 92:5510–5514
Icard P, Lincet H (2012) A global view of the biochemical pathways involved in the regulation of the metabolism of cancer cells. Biochim Biophys Acta 1826:423–433
Jang M, Kim SS, Lee J (2013) Cancer cell metabolism: implications for therapeutic targets. Exp Mol Med 45:e45
Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P et al (1996) Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem 271:32529–32537
Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M et al (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334:1278–1283
Menendez JA, Alarcon T, Joven J (2014) Gerometabolites: the pseudohypoxic aging side of cancer oncometabolites. Cell Cycle 13:699–709
Briere JJ, Chretien D, Benit P, Rustin P (2004) Respiratory chain defects: what do we know for sure about their consequences in vivo? Biochim Biophys Acta 1659:172–177
Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H et al (2012) Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26:1326–1338
Yang M, Soga T, Pollard PJ, Adam J (2012) The emerging role of fumarate as an oncometabolite. Front Oncol 2:85
Frezza C, Pollard PJ, Gottlieb E (2011) Inborn and acquired metabolic defects in cancer. J Mol Med (Berl) 89:213–220
Adam J, Yang M, Soga T, Pollard PJ (2014) Rare insights into cancer biology. Oncogene 33:2547–2556
Sullivan LB, Chandel NS (2014) Mitochondrial metabolism in TCA cycle mutant cancer cells. Cell Cycle 13:347–348
Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M et al (1995) Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlation with phenotype. Hum Mutat 5:66–75
Knauth K, Bex C, Jemth P, Buchberger A (2006) Renal cell carcinoma risk in type 2 von Hippel-Lindau disease correlates with defects in pVHL stability and HIF-1alpha interactions. Oncogene 25:370–377
Yang C, Huntoon K, Ksendzovsky A, Zhuang Z, Lonser RR (2013) Proteostasis modulators prolong missense VHL protein activity and halt tumor progression. Cell Rep 3:52–59
Gossage L, Eisen T, Maher ER (2015) VHL, the story of a tumour suppressor gene. Nat Rev Cancer 15:55–64
Pacak K, Jochmanova I, Prodanov T, Yang C, Merino MJ, Fojo T et al (2013) New syndrome of paraganglioma and somatostatinoma associated with polycythemia. J Clin Oncol 31:1690–1698
Yang C, Sun MG, Matro J, Huynh TT, Rahimpour S, Prchal JT et al (2013) Novel HIF2A mutations disrupt oxygen sensing, leading to polycythemia, paragangliomas, and somatostatinomas. Blood 121:2563–2566
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Pore N, Jiang Z, Shu HK, Bernhard E, Kao GD, Maity A (2006) Akt1 activation can augment hypoxia-inducible factor-1alpha expression by increasing protein translation through a mammalian target of rapamycin-independent pathway. Mol Cancer Res 4:471–479
Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC (2006) Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell 21:521–531
Yuan G, Nanduri J, Khan S, Semenza GL, Prabhakar NR (2008) Induction of HIF-1alpha expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J Cell Physiol 217:674–685
Harada H, Itasaka S, Kizaka-Kondoh S, Shibuya K, Morinibu A, Shinomiya K et al (2009) The Akt/mTOR pathway assures the synthesis of HIF-1alpha protein in a glucose- and reoxygenation-dependent manner in irradiated tumors. J Biol Chem 284:5332–5342
Sudhagar S, Sathya S, Lakshmi BS (2011) Rapid non-genomic signalling by 17beta-oestradiol through c-Src involves mTOR-dependent expression of HIF-1alpha in breast cancer cells. Br J Cancer 105:953–960
Agani F, Jiang BH (2013) Oxygen-independent regulation of HIF-1: novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr Cancer Drug Targets 13:245–251
Soucek L, Evan G (2002) Myc—is this the oncogene from hell? Cancer Cell 1:406–408
Pelengaris S, Khan M, Evan G (2002) c-MYC: more than just a matter of life and death. Nat Rev Cancer 2:764–776
Patel JH, Loboda AP, Showe MK, Showe LC, McMahon SB (2004) Analysis of genomic targets reveals complex functions of MYC. Nat Rev Cancer 4:562–568
Dang CV, Kim JW, Gao P, Yustein J (2008) The interplay between MYC and HIF in cancer. Nat Rev Cancer 8:51–56
Meyer N, Penn LZ (2008) Reflecting on 25 years with MYC. Nat Rev Cancer 8:976–990
van Riggelen J, Yetil A, Felsher DW (2010) MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer 10:301–309
Yuan J, Minter-Dykhouse K, Lou Z (2009) A c-Myc-SIRT1 feedback loop regulates cell growth and transformation. J Cell Biol 185:203–211
Mao B, Zhao G, Lv X, Chen HZ, Xue Z, Yang B et al (2011) Sirt1 deacetylates c-Myc and promotes c-Myc/Max association. Int J Biochem Cell Biol 43:1573–1581
Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M et al (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 107:8788–8793
Neuzil J, Rohlena J, Dong LF (2012) K-Ras and mitochondria: dangerous liaisons. Cell Res 22:285–287
Aebersold DM, Burri P, Beer KT, Laissue J, Djonov V, Greiner RH et al (2001) Expression of hypoxia-inducible factor-1alpha: a novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res 61:2911–2916
Birner P, Schindl M, Obermair A, Breitenecker G, Oberhuber G (2001) Expression of hypoxia-inducible factor 1alpha in epithelial ovarian tumors: its impact on prognosis and on response to chemotherapy. Clin Cancer Res 7:1661–1668
Brahimi-Horn MC, Chiche J, Pouyssegur J (2007) Hypoxia and cancer. J Mol Med (Berl) 85:1301–1307
Verduzco D, Lloyd M, Xu L, Ibrahim-Hashim A, Balagurunathan Y, Gatenby RA et al (2015) Intermittent hypoxia selects for genotypes and phenotypes that increase survival, invasion, and therapy resistance. PLoS One 10:e0120958
Kim JI, Choi KU, Lee IS, Choi YJ, Kim WT, Shin DH et al (2015) Expression of hypoxic markers and their prognostic significance in soft tissue sarcoma. Oncol Lett 9:1699–1706
Liu XW, Cai TY, Zhu H, Cao J, Su Y, Hu YZ et al (2014) Q6, a novel hypoxia-targeted drug, regulates hypoxia-inducible factor signaling via an autophagy-dependent mechanism in hepatocellular carcinoma. Autophagy 10:111–122
Melillo G (2007) Hypoxia-inducible factor 1 inhibitors. Methods Enzymol 435:385–402
Melillo G (2007) Targeting hypoxia cell signaling for cancer therapy. Cancer Metastasis Rev 26:341–352
Grkovic T, Whitson EL, Rabe DC, Gardella RS, Bottaro DP, Linehan WM et al (2011) Identification and evaluation of soft coral diterpenes as inhibitors of HIF-2alpha induced gene expression. Bioorg Med Chem Lett 21:2113–2115
Scheuermann TH, Li Q, Ma HW, Key J, Zhang L, Chen R et al (2013) Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat Chem Biol 9:271–276
Rogers JL, Bayeh L, Scheuermann TH, Longgood J, Key J, Naidoo J et al (2013) Development of inhibitors of the PAS-B domain of the HIF-2alpha transcription factor. J Med Chem 56:1739–1747
Wang Y, Li Z, Zhang H, Jin H, Sun L, Dong H et al (2010) HIF-1alpha and HIF-2alpha correlate with migration and invasion in gastric cancer. Cancer Biol Ther 10:376–382
Joshi S, Singh AR, Zulcic M, Durden DL (2014) A macrophage-dominant PI3K isoform controls hypoxia-induced HIF1alpha and HIF2alpha stability and tumor growth, angiogenesis, and metastasis. Mol Cancer Res 12:1520–1531
Philips GK, Atkins MB (2014) New agents and new targets for renal cell carcinoma. Am Soc Clin Oncol Educ Book 2014:e222–227
Yang C, Matro JC, Huntoon KM, Ye DY, Huynh TT, Fliedner SM et al (2012) Missense mutations in the human SDHB gene increase protein degradation without altering intrinsic enzymatic function. FASEB J 26:4506–4516
Dong LF, Low P, Dyason JC, Wang XF, Prochazka L, Witting PK et al (2008) Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene 27:4324–4335
Prasad KN, Edwards-Prasad J (1982) Effects of tocopherol (vitamin E) acid succinate on morphological alterations and growth inhibition in melanoma cells in culture. Cancer Res 42:550–555
Neuzil J, Weber T, Schroder A, Lu M, Ostermann G, Gellert N et al (2001) Induction of cancer cell apoptosis by alpha-tocopheryl succinate: molecular pathways and structural requirements. FASEB J 15:403–415
Swettenham E, Witting PK, Salvatore BA, Neuzil J (2005) Alpha-tocopheryl succinate selectively induces apoptosis in neuroblastoma cells: potential therapy of malignancies of the nervous system? J Neurochem 94:1448–1456
Neuzil J, Weber T, Gellert N, Weber C (2001) Selective cancer cell killing by alpha-tocopheryl succinate. Br J Cancer 84:87–89
Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A et al (2011) Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med 3:94ra70
Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H et al (2012) A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther 11:1672–1682
Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99:989–994
Kumar A, Kant S, Singh SM (2012) Novel molecular mechanisms of antitumor action of dichloroacetate against T cell lymphoma: implication of altered glucose metabolism, pH homeostasis and cell survival regulation. Chem Biol Interact 199:29–37
Geschwind JF, Ko YH, Torbenson MS, Magee C, Pedersen PL (2002) Novel therapy for liver cancer: direct intraarterial injection of a potent inhibitor of ATP production. Cancer Res 62:3909–3913
Cardaci S, Desideri E, Ciriolo MR (2012) Targeting aerobic glycolysis: 3-bromopyruvate as a promising anticancer drug. J Bioenerg Biomembr 44:17–29
El Sayed SM, El-Magd RM, Shishido Y, Chung SP, Diem TH, Sakai T et al (2012) 3-Bromopyruvate antagonizes effects of lactate and pyruvate, synergizes with citrate and exerts novel anti-glioma effects. J Bioenerg Biomembr 44:61–79
Kim SH, Lee GM (2007) Down-regulation of lactate dehydrogenase-A by siRNAs for reduced lactic acid formation of Chinese hamster ovary cells producing thrombopoietin. Appl Microbiol Biotechnol 74:152–159
Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C et al (2013) An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340:626–630
Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E et al (2013) Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340:622–626
Kim J, DeBerardinis RJ (2013) Cancer. Silencing a metabolic oncogene. Science 340:558–559
Yousefi S, Owens JW, Cesario TC (2004) Citrate shows specific, dose-dependent lympholytic activity in neoplastic cell lines. Leuk Lymphoma 45:1657–1665
Lu Y, Zhang X, Zhang H, Lan J, Huang G, Varin E et al (2011) Citrate induces apoptotic cell death: a promising way to treat gastric carcinoma? Anticancer Res 31:797–805
Zhang X, Varin E, Allouche S, Lu Y, Poulain L, Icard P (2009) Effect of citrate on malignant pleural mesothelioma cells: a synergistic effect with cisplatin. Anticancer Res 29:1249–1254
Acknowledgments
This research was supported, in part, by the Intramural Research Program of the National Institutes of Health, NICHD.
Conflict of Interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jochmanová, I., Zhuang, Z. & Pacak, K. Pheochromocytoma: Gasping for Air. HORM CANC 6, 191–205 (2015). https://doi.org/10.1007/s12672-015-0231-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-015-0231-4