
Abstract
We propose a new two-dimensional metallic carbon allotrope named QHOD-net using first-principles calculations, the structure of which includes five carbon rings; quadrangular, pentagonal, hexagonal, octagonal, and decagonal. This metastable phase metallic carbon material displays anisotropic mechanical properties, and its smallest and largest in-plane stiffness have been calculated to be Ca = 261 GPa nm and Cb = 240 GPa nm, respectively, both much lower than for graphene. The Poisson's ratio is as low as 0.29, which has good toughness. The DFT indicates that QHOD-net is metallic with no bandgap in the entire BZ region and one band crosses the Fermi level. At the Fermi level, the electron density of states per atom is much higher, reaching ~ 0.297 eV/states/per atom. In addition, we have performed the 3D stacked structure of the two-dimensional structure QHOD-net, and the results of our study indicate that the stacked structure is a super-hard 3D carbon material (74.8 GPa nm). The two-dimensional structure QHOD-net contains a large number of tetragonal, pentagonal, octagonal, and decagonal carbon rings than the perfect hexagonal shape of ideal graphene. The disorder of the material is increased compared to that of graphene. It is this disorder that triggers these interesting findings, and in addition we provide a new strategy for the design of 2D structures with multiple carbon rings.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Due to the rich electronic hybridization configurations of sp, sp2, and sp3 orbitals, carbon is the most versatile element and able to form many allotropes, of which zero-dimensional fullerenes [1], one-dimensional carbon nanotubes [2], and two-dimensional (2D) graphene [3] are the three most typical examples. Among them, the unique 2D inorganic honeycomb structure and the fascinating physical properties of graphene have greatly motivated researchers to explore new 2D carbon materials [4,5,6,7]. Many allotropes of carbon with peculiar properties have been discovered, such as penta-graphene [8], which has a negative Poisson’s ratio and ultra-high ideal strength. Furthermore, 2D carbon allotropes such as QPHT-graphene [9], h567 [10], net-τ [11], Kust-I [12], Thgraphene [13], and Bp-sheet 5 [14] have been predicted. These new carbon allotropes exhibit various electronic properties ranging from metals to semimetals and semiconductors [15].
The biphenylene network (BPN) [16], a fully sp2-hybridized carbon allotrope with a planar structure, has recently been successfully synthesized in an experiment. In addition, theoretical predictions of the 2D M-C crystals [17] can also be made using the already synthesized structure γ-graphyne [18]. This has greatly promoted theoretical prediction studies of 2D carbon allotropes. Based on previous research, researchers have been able to achieve the construction of 2D graphene allotropes in two ways. The first approach is to obtain 2D carbon allotropes by exfoliating them from known materials, such as penta-graphene from T12 carbon [19]. The second method is theoretical design without changing the periodicity and symmetry of crystals, such as QPHT-graphene, net-τ, and Kust-I. Although many 2D carbon allotropes have been proposed, considering the unparalleled advantages of elemental carbon, the search for new 2D carbon-based materials with outstanding properties is far from complete.
In the present work, a new 2D carbon allotrope named QHOD-net, composed of tetragonal, pentagonal, hexagonal, octagonal, and decagonal carbon rings, was predicted using first-principles calculations. The energetic, dynamic, thermal, and mechanical stability were assessed. Interestingly, analysis of the electronic structures showed that QHOD-net is a metallic carbon allotrope possessing anisotropic mechanical properties. Significantly, we have also studied a three-dimensional (3D) extended QHOD-net stacked structure.
2 Computational Methods
First-principles calculations were performed using the CASTEP code based on density function theory [20]. Ultrasoft pseudopotentials were used to study ion–electron interactions [21], and GGA and PBE functions were used to model the exchange–correlation potential of electrons [22]. To avoid the influence of adjacent layers, a vacuum space of 15 Å was set along the C-axis, based on the Monkhorst–Pack method [23, 24] with a cutoff energy of 500 eV and K-mesh of 2π × 0.03 Å−1. To ensure better structural optimization, the total energy converged to 1.0 × 10−6 eV/atom and the interatomic forces converged to 10−3 eV/Å. To calculate an accurate electronic band structure, the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional was used [25]. The phonon spectrum was verified for kinetic stability using the density functional perturbation theory [26]. This method uses first-principles molecular dynamics simulation to study the thermal stability of QHOD-net under canonical ensemble [27].
3 Results and Discussion
3.1 Structure and Stability
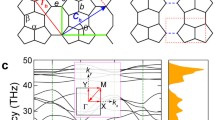
The optimized structure of QHOD-net is shown in Fig. 1, which can be viewed as a rectangular primitive cell with 22 atoms, including seven chemically non-equivalent carbon atoms, labelled as C1–C7 and colored differently. The lattice constants of QHOD-net are a = 6.246 Å, b = 10.085 Å, respectively. Wyckoff position coordinates are 2j(0.886, 1.0, 0.5), 4z(0.810, 0.865, 0.5), 4z(0.619, 0.795, 0.5), 4z(0.619, 0.652, 0.5), 4z(0.798, 0.572, 0.5), 2n(1.0, 0.643, 0.5), and 2n(1.0, 0.782, 0.5). The bond lengths were plotted as d1–d11. The maximum bond length (1.485 Å) has been found to be rather large compared with other bonds (1.383–1.481 Å). In particular, the individual carbon bond lengths of the four-membered rings are longer than those of any other carbon ring (d3 = 1.485 Å, d4 = 1.445 Å, d5 = 1.481 Å). All of these bond lengths are comparable to graphene, and they exhibit similar sp2 hybrid bonding properties to those of graphene, but there are differences from the physical and electronic properties of graphene [28]. In addition, QHOD-net has numerous 8- and 10-carbon rings, which makes the atomic density of QHOD-net much lower than that of graphene. This result strongly suggests that the total energy of QHOD-net is likely to be higher than that of graphene.

Atomic configuration of QHOD-net
To confirm the stability of this structure, the energy of QHOD-net and other 2D graphene allotropes was investigated. The results show that the relative energy of QHOD-net is higher than that of graphene (Fig. 2a), which may be due to a change in bond length leading to an increase in total energy, indicating lower stability for QHOD-net. It is worth noting that the energy of QHOD-net is lower than that of net-C, BPC, and graphene, suggesting that QHOD-net is energetically more stable than these structures.

a Total energy per atom as a function of the 2D crystal area per atom for QHOD-net in comparison with some other 2D carbon allotropes. b Phonon band structure of QHOD-net. c Total potential energy fluctuation of QHOD-net during AIMD simulation at 300 K. d Strain energy of QHOD-net under different kinds of in-plane strain in the harmonic region
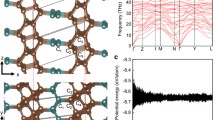
To verify whether it is dynamically stable, we calculated the phonon spectrum of QHOD-net (Fig. 2b). No imaginary frequencies have been measured in the Brillouin zone, indicating that QHOD-net is dynamically stable [29].
MD simulation results for QHOD-net are shown in Fig. 2c. The geometry of QHOD-net is found to be well retained at temperatures of 300 K for 3 ps with a time step of 1 fs. Furthermore, the total potential energies of the QHOD-net supercell only vary around a constant value [30]. The dynamic stability of QHOD-net is further confirmed by these calculations.
In addition, to ensure the mechanical stability of a 2D material, the strain energy of QHOD-net was also calculated for different stress planes (Fig. 2d). The elastic strain energy U(ε) for a 2D material per unit area was calculated using standard Voigt notation as [31]:
where \(\varepsilon_{xx}\) and \(\varepsilon_{yy}\) are uniaxial strains along the a and b directions, and \(\varepsilon_{xy}\) is the equi-biaxial strain. C11, C22, C12, and C44 are components of the independent elastic stiffness tensor. By fitting the strain energy curves associated with uniaxial, biaxial and shear strains, we calculated elastic stiffness tensor C11, C22, C12, and C44 to be 289.93, 265.83, 86.45, and 24.81 GPa nm, respectively. The calculated independent elastic constants must satisfy the Born–Huang criteria [32] (C11C22–C122 > 0 and C44 > 0). It is clear that they satisfy the Born–Huang criteria, indicating QHOD-net is mechanically stable.
3.2 Mechanical Properties
The mechanical properties of QHOD-net can be described by material independent intrinsic constants, namely Young’s modulus and Poisson’s ratio. From the elastic constants, we can derive the Young’s modulus (E) and Poisson’s ratio (v) for any angle, expressed as:
For isotropic (anisotropic) materials, the polar plots of Young's modulus and Poisson's ratio are generally circular (non-circular). We may conclude that QHOD-net is anisotropic based on the plots of Young’s modulus and Poisson's ratio (Fig. 3). In addition, as shown in Table 1, the Young's modulus values along the a- and b-axes are 261 and 240 GPa nm, respectively, which are lower than graphene, h567, and net-τ, but higher than Kust-I. This means that QHOD-net is softer than graphene, h567, and net-τ, and a little stiffer than Kust-I. Hence, we can conclude that QHOD-net has good ductility. Additionally, Poisson’s ratio shows a tendency to increase and then decrease from the a to b direction. The maximum value of Poisson’s ratio of 0.75 occurs in the 45° direction with the a direction and the minimum value of 0.29 in the b direction. QHOD-net is a ductile carbon material (Poisson’s ratio > 0.25) compared to graphite, h567, and net. The high Poisson’s ratio indicates that QHOD-net has excellent ductility. In addition, we found that the Poisson’s ratio of QHOD-net is smaller than that of Kust-I, which may be related to the fact that Kust-I has a large number of 10-carbon rings.

Polar diagrams for a plane Young’s modulus E and b Poisson’s ratio υ for QHOD-net
3.3 Electronic Properties
To understand the electronic structure and properties of QHOD-net, we calculated the energy band diagram and density of states of QHOD-net, respectively. The electronic band calculation results for HSE06 and GGA-PBE are shown in Fig. 4a. The DFT indicates that QHOD-net is metallic since there is no bandgap in the entire BZ zone and one band crosses the Fermi level. The HSE06 hybrid functional calculated result agrees with the GGA- PBE. The density of states (DOS) is shown in Fig. 4b, and the DOS value of 0.297 states/eV per atom implies a large density of states in the metallic carbon structure of QHOD-net. This is twice that of QPHT-graphene (0.15 states/eV per atom), four times that of 5.3 Å diameter (4, 4) carbon nanotubes (0.07 states/eV per atom) [33], and three times that of planar T-graphene (0.11 states/eV per atom) [34].

Electrical properties of QHOD-net. a Band structure of QHOD-net. b Total density of states (TDOS) of QHOD-net. c Projected density of states (PDOS) of seven unequal carbon atoms (C1-C7) calculated using the PBE function
To understand the origin of QHOD-net metallicity is very important, so we calculated the projected density of states (PDOS) of seven non-equivalent carbon atoms by using the PBE function (Fig. 4c). PDOS analysis reveals that the electronic states near the Fermi level are mainly from pz orbitals, and pz orbitals occupy the bonding and antibonding states simultaneously, and generating a delocalized π-network in a–b plane.
In particular, to compare the contribution of non-equivalent carbon atoms to the charge density. We visualize the charge distribution around the Fermi energy level by calculating the decomposition charge density of the occupied bands (bands 44 and 45) in Fig. 5a. The results show relatively high electron accumulation around C3, C4 and C7 atoms, and relatively low electron accumulation around C2 and C5 atoms, which is consistent with the size of the electronic states on the Fermi energy level of PDOS.

a decomposed charge density of occupied band (No. 44, 45 bands). b Electron localization function (ELF)
Furthermore, to probe the local electronic character around non-equivalent carbon atoms, the electronic localized function (ELF) was also calculated [35]. The value of electron localization ranges from 0.0 to 1.0, and the slice is clearly seen to be uniformly distributed (Fig. 5b). Moving 1 Å up from the plane forms an off-domain π-network (ELF = 0.5) [36], and the metallicity of QHOD-net improves the charge transfer efficiency. As can be seen from ELF = 0.75 in Fig. 5b, the equivalents that are inside the four-membered rings move outward toward the rings, which decreases the strength of the related bonds, so these bonds become longer, and this electron distribution also weakens the structural stability, leading to a higher energy of QHOD-net than graphene.
We also studied a three-dimensional (3D) extended QHOD-net stacked structure, as shown in Fig. 6a. It is composed of 44 carbon atoms. It consists of 44 carbon atoms with sp3 hybridization. In this structure, the orange-red atoms are separated by layer gaps with a minimum of 1.62 Å and a maximum of 1.83 Å, and the blue atoms are separated by a distance of 2.85 Å.

3D buckled QHOD-net. a The structure of 3D buckled QHOD-net. b Electronic band structure of 3D buckled QHOD-net. c Young's modulus diagram of 3D buckled QHOD-net. d Hardness diagram of QHOD-net
In order to investigate the electronic properties of the 3D stacked structure, its electronic energy band diagram was calculated as shown in Fig. 6b. The results show that the 3D stacked structure has a band gap of 2.791 eV, and the valence band top and conduction band bottom are the high-symmetry points of S and Γ, respectively, and that the 3D stacked structure is a semiconductor with an indirect band gap. The large band gap range can also extend the range of absorbable light, which also indicates that the 3D stacked structure has potential applications in photocatalysis.
The 3D stacked structure of QHOD-net is an orthorhombic lattice with 9 independent elastic constants C11, C22, C33, C44, C55, C66, C12, C13, and C23 calculated as 953, 933, 590, 257, 404, 303, 224, 119, and 60 GPanm, respectively. The stability conditions for the stiffness matrix of an orthorhombic crystal need to satisfy the following necessary and sufficient Born criteria [37]: C11 > 0, C44 > 0, C55 > 0, C66 > 0, C11C22C33 + 2C12C13C23 − C11C2 23 − C22C2 13—C33C2 12 > 0, C11C22 – C2 12 > 0. Therefore, the 3D stacked structure of QHOD-net is mechanically stable. The Young's modulus of the 3D stacked structure is not circular in three directions, as shown in Fig. 6c. The 3D stacked structure is anisotropic. The Young's modulus has a maximum value of 890 GPa nm and a minimum value of 567 GPa nm. According to the definition of hardness \(H_{v} = 2\left[ {\left( {G/B} \right)^{2} G} \right]^{0.585} - 3.0\), the hardness of the 3D stacked structure has different values in the three directions as shown in Fig. 6d. The maximum and minimum values are 74.8 and 40 GPa nm, respectively. In the H-xz direction, part of the area is larger than the T-carbon (61.1 GPa nm) [38].
4 Conclusions
Herein, we systematically studied the structural and electronic properties of QHOD-net using first-principles simulation. The structure is composed of quadrangular, pentagonal, hexagonal, octagonal, and decagonal carbon rings. The energetic results show that QHOD-net is metastable compared to graphene. The dynamic, mechanical, and thermal stabilities of QHOD-net were assessed by phonon spectroscopy, Born–Huang criteria, and molecular dynamics calculations, respectively. The elastic constant calculations showed that QHOD-net displays anisotropic mechanical properties, and its smallest and largest in-plane stiffness were calculated to be Ca = 261 GPa nm and Cb = 240 GPa nm, respectively, which are lower than graphene, h567, and net-τ, but higher than Kust-I. This means that QHOD-net is softer than graphene, h567, and net-τ, and a little stiffer than Kust-I. Interestingly, the electronic structure of QHOD-net was further explored, and QHOD-net was found to be metallic with a high electronic state at the Fermi energy level of ~ 0.297 eV/states per atom. The 3D stacked structure of QHOD-net is of greater hardness (74.8 GPa nm) than that of T-carbon in the H-xz direction in some regions. The findings further expand our understanding of the structural and electronic properties of carbon allotropes.
References
H.W. Kroto, J.R. Heath, S.C. O’Brien, R.F. Curl, R.E., Nature 318 (1985) 162.
S. Iijima, Nature 354 (1991) 56.
K.S. Novoselov, A.K. Geim, S.V. Morozov, D. Jiang, Y. Zhang, S.V. Dubonos, I.V. Grigorieva, A.A. Firsov, Science 306 (2004) 666.
C. Lee, X. Wei, J.W. Kysar, J. Hone, Science 321 (2008) 385.
R.H. Baughman, H. Eckhardt, M. Kertesz, J. Chem. Phys. 87 (1987) 6687.
Y. Zhang, T.T. Tang, C. Girit, Z. Hao, M.C. Martin, A. Zettl, M.F. Crommie, Y.R. Shen, F. Wang, Nature 459 (2009) 820.
K.S. Novoselov, A.K. Geim, S.V. Morozov, D. Jiang, M.I. Katsnelson, I.V. Grigorieva, S.V. Dubonos, A.A. Firsov, Nature 438 (2005) 197.
S. Zhang, J. Zhou, Q. Wang, X. Chen, Y. Kawazoe, P. Jena, P. Natl. Acad. Sci. USA 112 (2015) 2372.
X. Wang, J. Rong, Y. Song, X. Yu, Z. Zhan, J. Deng, Phys. Lett. A 381 (2017) 2845.
S. Thomas, H. Jung, S. Kim, B. Jun, C. H. Lee, S. U. Lee, Carbon 148 (2019) 344.
X. Wang, Z. Feng, J. Rong, Y. Zhang, Y. Zhong, J. Feng, X. Yu, Z. Zhan, Carbon 142 (2019) 438.
X. Yu, J. Hou, H. Wu, J. Rong, X. Wang, K. Xu, J. Feng, J. Mater. Chem. A 9 (2021) 21158.
W. Wang, J. Meng, Y. Hu, J. Wang, Q. Li, J. Yang, J. Mater. Chem. A 10 (2022) 9848.
N. N. Karaush, G. V. Baryshnikov, B. F. Minaev, Chem. Phys. Lett. 612 (2014) 229.
Y. Aierken, O. Leenaerts, F.M. Peeters, Phys. Rev. B 94 (2016) 155410.
Q. Fan, L. Yan, M.W. Tripp, O. Krejčí, S. Dimosthenous, S.R. Kachel, M. Chen, A.S. Foster, U. Koert, P. Liljeroth, Science 372 (2021) 852.
S. Li, K.M. Yam, N. Guo, Y. Zhao, C. Zhang, NPJ 2D Mater. Appl. 5 (2021) 52.
Q. Li, C. Yang, L. Wu, H. Wang, X. Cui, J. Mater. Chem. A 7 (2019) 5981.
Z. Zhao, F. Tian, X. Dong, Q. Li, Q. Wang, H. Wang, X. Zhong, B. Xu, D. Yu, J. He, H.T. Wang, Y. Ma, Y. Tian, J. Am. Chem. Soc. 134 (2012) 12362.
M. Segall, P.J. Lindan, M.a. Probert, C.J. Pickard, P.J. Hasnip, S. Clark, M. Payne, J. Phys-Condens. Mat. 14 (2002) 2717.
G. Kresse, D. Joubert, Phys. Rev. B 59 (1999) 1758.
A.E. Mattsson, R. Armiento, P.A. Schultz, T.R. Mattsson, Phys. Rev. B 73 (2006) 195123.
P. Wisesa, K.A. McGill, T. Mueller, Phys. Rev. B 93 (2016) 155109.
R. Evarestov, V. Smirnov, Phys. Rev. B 70 (2004) 233101.
J. Heyd, G.E. Scuseria, M. Ernzerhof, J. Chem. Phys. 118 (2003) 8207.
S. Baroni, S.D. Gironcoli, A.D. Corso, P. Giannozzi, Rev. Mod. Phys. 73 (2001) 515.
P. Rosales-Pelaez, I. Sanchez-Burgos, C. Valeriani, C. Vega, E. Sanz, Phys. Rev. E 101 (2020) 022611.
F. Tuinstra, J.L. Koenig, Raman spectrum of graphite, J. Chem. Phys. 53 (1970) 1126.
Y. Yao, J.S. Tse, J. Sun, D. Klug, R. Martoňák, T. Iitaka, Phys. Rev. Lett. 102 (2009) 229601.
F. Liu, P. Ming, J. Li, Phys. Rev. B 76 (2007) 064120.
E. Cadelano, P.L. Palla, S. Giordano, L. Colombo, Phys. Rev. B 82 (2010) 235414.
Y. Ding, Y. Wang, J. Phys. Chem. C 117 (2013) 18266.
R. Saito, M. Fujita, G. Dresselhaus, M.S. Dresselhaus, Appl. Phys. Lett. 60 (1992) 2204.
X. Zhang, L. Jin, X. Dai, G. Chen, G. Liu, Appl. Surf. Sci. 527 (2020) 146849.
B. Silvi, A. Savin, Nature 371 (1994) 683.
J.T. Wang, C. Chen, H.D. Li, H. Mizuseki, Y. Kawazoe, Sci. Rep. 6 (2016) 1.
F. Mouhat, F.X. Coudert, Phys. Rev. B 90 (2014) 224104.
X. L. Sheng, Q.B. Yan, F. Ye, Q.R. Zheng, G. Su, Phys. Rev. Lett. 106 (2011) 155703.
Acknowledgements
This work was supported financially by the Scientific Research Fund of Yunnan Education Department (Grant Nos. 2020J0416 and 2019J0039); Key projects of basic research plan of Yunnan Science and Technology Department (Grant Nos. 202001AS070048, 202201AU070085).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kong, D., Zhong, L., Wu, C. et al. QHOD-Net: A New Highly Metallic Two-Dimensional Carbon Allotrope Material. Trans Indian Inst Met 76, 829–835 (2023). https://doi.org/10.1007/s12666-022-02779-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12666-022-02779-z