
Abstract
In recent years, modeling and simulation techniques have been pioneered in the field of prediction existence or ability to synthesize new structures and to study physical and chemical properties. One of these methods is first-principles computations, which are based on the Kohn–Sham density functional theory (DFT). In this work, we predict a new kind of two-dimensional (2D) carbon allotrope by a tiny size building block with interesting properties. A systematic study of the structural and electronic properties on a non-hexagonal flat carbon allotrope has been performed in two different phases that consist of pentagonal (P\(\equiv \{C_5\}\)) and heptagonal (H\(\equiv \{C_7\}\)) rings, as well as a carbon nanotube (CNT), by using the DFT computational method. Hence, we obtain optimized lattice structures, bond lengths, density of states (DOS), band structure, the isosurface, and the difference charge density for these both novel two-dimension (2D) materials. The results show that regardless of the type of structure, the nanostructures are electrically metallic. It is anticipated that the results of the present work can be useful in the experimental synthesis of these materials and their potential applications in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Recently, carbon-based nanostructures have received much attention due to their potential for application improvement in electronics and optoelectronics with novel properties [1]. In the periodic table of elements, the carbon atom is the sixth element that makes up its electron is \(1s^22s^22p^2\). The two 2s and 2p orbitals have very little energy difference compared to the energy released in chemical bonds, so the first-type orbital may be combined with a number of the second-type orbitals to create the equivalent hybrid orbitals. Hybridization of carbon element in combination can appear different as sp, \(sp^2\) and \(sp^3\). This causes a variety of compounds including carbon amorphous, diamond and graphite (3D), graphene and its family (2D), carbon nanotubes (CNT) and nanoribbons (1D), and also fullerenes or \(\hbox {C}_{{60}}\) (0D), which are made by this element. The discovery of new carbon allotropes including the fullerene by Smalley et al. [2], CNT by Iijima in 1991 [3], and the graphene by Novoselov et al. in 2004 [4] has created a new research area to investigate the physical and chemical properties of different types of carbon-based materials.
Among these allotropes, graphene is a 2D sheet with one atom thick, and flat which carbon atoms are arranged in a hexagonal lattice [4]. Because of its unusual structure, electronic and magnetic properties, this 2D sheet structure is known as a revolutionary material and promising with a high potential for thermal management applications in nanoelectronic circuits [5] and thermal interface materials [6]. Moreover, it has attracted extreme attention due to its application in circuits and electronic and optoelectronic devices [7,8,9], photonic devices, clean energy and sensors [8, 10, 11], energy and thin film applications [12], and solar cell technologies [13].
The most common types of CNT can be created by rolling into a hexagonal honeycomb lattice in a particular direction. Accordingly, there are three types zigzag, armchair, and chiral category of CNT, which are stable due to the symmetry of the atomic lattice. In recent years, numerous applications of CNT have attracted much attention in various fields including hydrogen storage [14], supercapacitor electrodes [15, 16], actuators and electromagnetic shielding lightweight [12], optical elements type cathode-ray tube and fluorescence display panels-vacuum [17], field-effect transistors, single-electron and rectifier diodes [18], and also nanostructured electrodes [19].
Moreover, boron nitride and aluminum nitride hexagonal layers are other 2D topological materials whose structures are similar to graphene. A boron nitride crystal forms with a periodic of boron and nitrogen atoms similar to carbon allotropes, with a lattice spacing similar carbon-based nanostructure. Unlike graphene, the h-BN sheet behaves like an insulator, electrically. This sheet-like can be combined with different 2D materials to make a heterostructure device via the van der Waals interaction. With these properties, impressive quantum structures such as tunneling double barriers and multiple quantum well structures based on heterostructures of graphene and BN can be constructed. Also, there are the quasi-2D materials that are composed mainly of atoms of transition metal chalcogenides and as an essential loss graphene structure [20]. Other 2D structures of carbon atoms can also imagine that they, unlike graphene lattice, are constructed from non-hexagonal rings.
In recent years, inspired by the abundant physics studies in the field of finding ways to manipulate new materials is controlled to improve the physical and chemical properties for various applications. One of these approaches functionality of a carbon lattice, for example through the partial replacement of carbon atoms with boron atoms of various doping especially nitrogen, dislocations, and vacancies [21,22,23,24]. In all mentioned structures, a hexagonal lattice is used in building a nanotube, graphene, and graphene-like material as a base. Moreover, there are other species of carbon allotropes with the construction of non-hexagonal rings [25,26,27]. In another work, the electronic and structural properties of the graphene-like boron nitride have been investigated within tight-binding calculations using the density functional theory (DFT) [28].
It is necessary to point out the combined pentagonal, and heptagonal rings in graphene, graphite, and CNTs have been extensively studied and are commonly known as a Stone–Wales defect. The Stone–Wales defect in these crystalline structures affects on their physical and chemical. The formation of this defect in a graphene sheet, four of the six-sides in deformation to two five-sided, and two seven-side is converted. For this reason, two carbon–carbon covalent bonds are rotated by 90-degree concerning the mid-point of their bond. Because for this process, an energy of about a few electronvolts (eV) is required; this defect can be seen in the low graphene structure [29]. These topological defects in \(sp^2\)-bonded carbon structures [30] play an important role in the transformation and formation of carbon-based nanostructures [31, 32]. Besides graphene, other flat 2D structures of carbon atoms and the corresponding nanotubes can imagine with \(sp^2\) hybridization. Moreover, there are other species of carbon allotropes with the construction of non-hexagonal rings (see Refs. [26, 27, 33,34,35]). Here, we look into the structural and electronic properties of the novel materials based on carbon allotropes (graphene and nanotube) with a structure consisting of pentagonal (P\(\equiv \{C_5\}\)) and heptagonal (H\(\equiv \{C_7\}\)) cycles instead of hexagonal-like lattice [36]. The calculation has been carried out using DFT simulation, as implemented in the Vienna ab initio simulation package (VASP) [37, 38].
The outline of this article is organized as follows: In Sect. 2, we clarify the computational details of the method to determine the structure and electronic properties of the graphene-like and CNT-like and its family as mentioned earlier. In Sect. 3, some numerical results on these properties in both structures consisting of the P and H cycles. The conclusion of our findings is presented in the last section.
2 Description of DFT calculations
In this work, we have calculated the atomic and electronic structure properties of the considered PH-graphene and PH-CNTs in the projector augmented wave (PAW) approximation [39, 40] as implemented within the VASP code [37, 38]. The PAW method is described as the electron–ion interactions in the structures mentioned above. This computational code is based on first-principle DFT calculations and the plane-wave pseudopotential approach, which solves Kohn–Sham equations using a self-consistent procedure. In order to consider the electron–electron interaction via the exchange-correlation energy, we used the Perdew–Wang (PW91) method of the generalized gradient approximation (GGA) [41]. Here, the related electrons to \(2s^2 2p^2\) are created, such as the valence electrons for the carbon atoms. As an initial assumption in these structures, the whole of bond lengths, such as the C-C bond length in graphene, are assumed as 1.42 Å[42].
2.1 Computational details of PH-graphene sheet structures
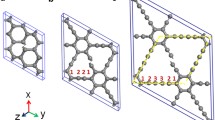
Figure 1 shows a 2D non-hexagonal lattice of an allotrope of carbon atoms with an \(sp^2\)-bonded hybridization in two \(\alpha \) and \(\beta \) phases, which are formed from the rings with the covalent bonds within the P and H cycles. In the \(\alpha \) and \(\beta \) phases, the unit cell consists of 8 and 16 carbon atoms in diamond shapes that can be seen in Figs. 1a, b, respectively. In the lattice structure of two phases, a conventional rectangular cell is introduced with two \(\mathbf{a }_\mathbf{1} \) and \(\mathbf{a }_\mathbf{2} \) primitive vectors. Each cell contains 16 carbon atoms that are bonded in four pentagon rings and four heptagon rings. For the electron wave function, a plane wave basis set was considered with a cutoff energy of 450 eV. Using Monkhorst–Pack mesh method, the above structures fully were relaxed with the \(k-\)points mesh of \(4\times 5\times 1\) and \(4\times 8\times 1\) for sampling the \(\alpha \) and \(\beta \) phases, respectively [43]. All atomic positions were fully relaxed by the conjugate gradient (CG) algorithm [37] and their structural geometry via coordinates were optimized with the converging tolerance 0.01 eV \(\hbox {nm}^{-1}\) for the force on all atoms in the structure. In order to refrain the interlayer interaction between adjacent layers, we have considered a vacuum separation with a length of 12 Åalong the perpendicular to the plane of sheets. After the relaxation of the structures, in order to calculate the density of states (DOS), the mesh of k space was increased to \(21\times 31\times 1\) and \(15\times 29\times 1\) for two \(\alpha \) and \(\beta \) phases, respectively [43]. The band structure calculation is done in the path with high-symmetry for 54 points of the grid inversion.

The balls and sticks model for flat and 2D structure consisting of rings pentagonal and heptagonal carbon construction rings: a \(\alpha \) phase, and b \(\beta \) phase. \(\mathbf{a}_1\) and \(\mathbf{a}_2\) are the primitive lattice vectors, respectively
2.2 Computational details of PH-CNT structures
In Fig. 2, the formation of non-hexagonal CNTs is displayed from the spreadsheet structure. The CNT is created with rolling the spreadsheet, which circumferential of the tube is described with a chiral vector \(\mathbf{C}\). This vector is defined using the primitive vectors of the spreadsheet as \(\mathbf{C} = m\mathbf{a}_1 + n\mathbf{a}_2\). Also, the primitive vector of the tube is expressed by \(\mathbf{T}\) and described according to primitive vectors of the sheet, which is the shortest lattice vector perpendicular to the chiral vector, \(\mathbf{C}\). Here, some different chirality of nanotubes, (0, 3), (0, 4), (2, 0), (3, 0), and (4, 0) are studied, which have 48, 64, 32, 48, and 64 atoms, respectively. The plane-wave energy cutoff used for all calculations in the PH-CNT structures is 425 eV. To avoid the interactions between the adjacent tubes, we consider a vacuum separation of 6.2 Å. Like before, here, the positions of all atoms are fully relaxed using a CG algorithm [39]. Also, their structural geometries are optimized so that the force convergence is less than 0.01 eV \(\hbox {nm}^{-1}\) on each atom. Using the Monkhorst–Pack method, the structures as mentioned above fully relaxed with the \(k-\)points mesh of \(1\times 1\times 9\) (including \(\Gamma \) point) [43]. Moreover, the mesh of k space is increased to \(1\times 1\times 31\) in the density of states calculations. The band structure calculation is done in \(\Gamma -Z\) path with high-symmetry for 10 points of the grid inversion.

Top (upper figures) and side (lower figures) views of two type PH-graphene and PH-CNT structure as a spreadsheet (2, 0) with the construction of pentagonal, and heptagonal rings
3 Numerical results and discussion
3.1 Structural properties of PH-graphene sheet structures
At first, we perform the self-consistent calculations to relax the structural and the perfect geometric optimization for the aforementioned 2D carbon lattices in two \(\alpha \) and \(\beta \) phases. Then, the lattice vector lengths for the PH-graphene were obtained as 5.08 Å and 7.38 Å for the \(\alpha \) phase, and 4.71 Å and 9.08 Å for the \(\beta \) phase, respectively. The length of carbon–carbon bond is in the range of 1.38 Å and 1.47 Å, for both phases, which its length average is about 1.42 Å. This is compatible with the length of graphene lattice [42]. Also, in another work, Zhao et al. obtained the C-C bond lengths in a C-57 carbon (a two-dimensional, flat carbon allotrope consisting of 5-sided and 7-sided rings) between 1.361 Åand 1.534 Å[44]. They have theoretically investigated the structural stability and electronic properties of a C-57 carbon using the first-principles calculations. Moreover, Wang et al. calculated the bond lengths of carbon atoms in other allotropes of carbon, including 4-sided, hexagonal, and octagonal rings (in two different phases C and W), between 1.389 and 1.477 [45].
In order to compare the stability between two allotropes of \(\alpha -\)graphene and \(\beta -\)graphene, we have calculated the difference total energy \(\delta E\), for each carbon atom between the phase \(E_{\mathrm{total}}^{\mathrm{allotrope}}\) and the pristine graphene, \(E_{\mathrm{total}}^{\mathrm{graphene}}\). These values are obtained as 0.298 eV and 0.319 eV for the \(\alpha \) and \(\beta \) phases, respectively. The result displays that these allotropes, despite having stability less than the pristine graphene, are stable, relatively, so they can be synthesized in a standard environment. Also, we have shown surface electrical charge density distribution of both the phases in Fig. 3. It is seen that the charge density has been distributed uniformly along with C-C carbon bonds. The electrons are localized environs the carbon atoms, and hence, the charge density decreases inside the porous cycles in the radial direction.

2D charge density of PH-graphene for a \(\alpha \) phase, and b \(\beta \) phase. Colors in a rainbow of blue for the lowest to the highest charge density charge density are displayed in red
The band structure and DOS curves of both \(\alpha -\)graphene and \(\beta -\)graphene phases are plotted in Figs. 4a, b, respectively. The Fermi energy is shifted to zero in all panels of the figure. It is seen that the DOS has been administrated around of the Fermi level due to the partial occupation of p band and their overlap with s band of carbon atom. The curves in the panels of Fig. 4 show that two phases of graphene are metallic in comparison with the pristine graphene. Such behavior has been observed in another 2D carbon allotrope [46]. The schematic profile of direct and inverse lattice vectors, as well as a symbolic first Brillouin zone, is shown in Fig. 5. The first Brillouin zone is a rectangle. The area has four non-equivalent points \(\Gamma \), X, Y, and S, with high symmetry, which we have plotted the band structure curve according to the symmetry of the first Brillouin zone in this path.

Band structure diagrams (left panels) and DOS diagrams (right panels) of PH-graphene for a \(\alpha \) phase, and b \(\beta \) phase. The horizontal dotted line shows the position of the Fermi energy which is shifted to zero

The schematic profile of direct and inverse lattice vectors as well as a symbolic first Brillouin zone with four non-equivalent high symmetry points \(\Gamma , X, Y\), and S, which the band structure curve is generally plotted according to these path

The partial DOS of s and p orbitals for the identified atom in the a \(\alpha \) phase and b \(\beta \) phase
In order to look into the role of atomic orbitals of s and p in the electronic properties, we consider the partial density of state (PDOS) diagrams presented in Fig. 6. It is seen that upper energy orbitals of s share more in the low-energy, but especially around the Fermi energy in the p orbitals plays a more critical role. As shown in Fig. 6, the 2s and 2p orbitals of a single carbon atom take part in the valence band, and the 2p orbitals play a better pattern in the conduction band. Thus, it may be mentioned that the carbon atoms lose an electron in the s orbital and receive an electron in the p orbital. By comparing the extracted PDOS curves, we notice that the 2p orbitals of the considered atom C in the left panels of Fig. 6a, b take part in the occupied states close the Fermi energy that displays the transfer of an electron from its atom to \(\alpha \)-graphene and \(\beta \)-graphene, respectively.
3.2 Structural properties of PH-CNT structures
Figure 7 shows the side view of the introduced relaxation nanotube structures in the previous section. In order to introduce the relaxed bond lengths, we have numbered the carbon atoms in a unique area in the figure. These values are reported in Table 1 in the angstrom unit. As can be seen, the carbon–carbon bond lengths are in the range 1.38–1.47 Å. These values fit very well with the carbon bonds in the conventional hexagonal CNT, which are in the range of 1.40 Å and 1.45 Å [47]. The shortest length (or most durable bond) is related to linked bonds parallel to the nanotube axis, which is owned by the intersection of two heptagonal ring atoms. On the other hand, the most extended length (or weakest bond) generally belongs to the \(\hbox {C}^2\)–\(\hbox {C}^3\) linked bonds, which shares a pentagonal ring and a heptagonal ring.

The relaxed structures of (3, 0), (4, 0), (0, 2), (0, 3), and (0, 4) CNTs with the construction of the pentagonal and heptagonal rings
In order to better understand the PH-CNT structures, we have some of the structural and electrical properties of the nanotube structures with types of (3,0), (4,0), (0,2), (0,3), and (0,4) after performing structural relaxation calculations in Table 2. The numerical values of the total energy of the ground state for the PH-CNT systems, the energy of ground state per atom, the minimum and maximum diameters, and length of the nanotubes are reported in the table. As you can see, the range of change in nanotube diameter (m, 0) to nanotubes diameter (0, n) is less effective, resulting in more uniform diameters. The average length of the nanotubes environment vector obtained as \(|\mathbf{C}| = \pi d\), where d is the diameter of the nanotubes.
Figure 8 shows the electronic density of states and the dispersion function of the PH-CNT systems for (3,0), (4,0), (0, 2), (0, 3), and (0, 4) types in the left and right of each panel, respectively. In all panels, the Fermi energy is shifted to zero. As shown in Fig. 8, the value of the DOS at \(E_{\mathrm{F}}\) takes a non-constant, since the p orbital is partially occupied and, so, the Fermi level in this material intersects not only the s orbital but also the p orbital. Also, we can observe that the conduction and valence bands around the Fermi energy perfectly intersect and there is no energy gap for them. So, unlike the usual CNT with a structure consisting of the hexagonal lattice, all these structures show metallic behavior [46]. The results are consistent with the findings of Zhao et al for the C-57 nanotube [44], which the band structural calculations and the charge density distribution in the C-57 nanotube indicate that it is a metal nanotube. Also, the charge density distribution around carbon atoms is non-uniform. As a result, it is seen that the presence of these topological shapes creates an asymmetry in the DOS spectrums.

Band structure diagrams (left panels) and DOS diagrams (right panels) of the PH-CNTs. The horizontal dotted line shows the position of the Fermi energy which is shifted to zero
Based on the optimized geometrical structure, the electric charge density for each PH-CNT is computed. According to Fig. 9, due to one type of atoms, the charge density of the PH-CNTs evenly repartitioned all over nanotube surrounding carbon atoms and also along with the C-C bonds. A change in the form of a rainbow of blue to red for the least and most amount of strength of charge density is shown in Fig. 9b.

a Electron density isosurfaces and b two-dimensional charge density of the studied PH-NTs (see Fig. 7)
4 Concluding remarks
Using first-principles calculations, we display two new 2D carbon allotrope constructed by the pentagonal and heptagonal building rings in graphene and CNT. The structural and electronic properties of them have been investigated using computational methods based on DFT. The main conclusions can be summarized as follow: The relaxed lattice vector lengths for PH-graphene were obtained as 5.08 Å and 7.38 Å for the \(\alpha \) phase, and 4.71 Å and 9.08 Å for the \(\beta \) phase. It was also observed that the length of the C-C bond in two phases of the graphene sheet was computed in the range of 1.38–1.47 Å. These values are compatible with the average length of carbon–carbon bond in pristine graphene [42]. The total energy difference of 0.298 eV and 0.319 eV per atom were achieved between pristine graphene and its \(\alpha \) and \(\beta \) phases, respectively. Despite of less stable of the structures than graphene, they remain stable and have the ability to synthesize in a standard situation. Also, the band structure and DOS curves showed that the PH-graphene is metallic in both phases.
Moreover, the numerical results on the PH-CNT showed that the C–C bond lengths are in the range of 1.38–1.47 Å. The shortest length is related to linked bonds parallel to the nanotube axis, which is owned by the intersection of two heptagonal ring atoms. On the other hand, the longest length belongs to the \(\hbox {C}^2\)–\(\hbox {C}^3\) linked bonds, which share a pentagonal ring and a heptagonal ring. Also, from the band structure of the aforementioned PH-CNTs, we found that the systems are metallic as well as their counterparts of the DOS around the Fermi energy, which the energy gap is not observed. These 2D carbon allotropes may be useful for developing highly efficient photocatalysts, [48] and optoelectronic devices [49].
Data Availability Statement
This manuscript has associated data in a data repository. [Authors’ comment: All data included in this manuscript are contacting with the corresponding author].
References
R. Saito, G. Dresselhaus, M.S. Dresselhaus et al., Physical Properties of Carbon Nanotubes, vol. 35 (World Scientific, Singapore, 1998)
H.W. Kroto, J.R. Heath, S.C. ÓBrien, R.F. Curl, R.E. Smalley, Nature 318(6042), 162–163 (1985)
S. Iijima et al., Nature 354(6348), 56–58 (1991)
K.S. Novoselov, A.K. Geim, S.V. Morozov, D. Jiang, Y. Zhang, S.V. Dubonos, I.V. Grigorieva, A. Firsov, Science 306(5696), 666–669 (2004)
S. Ghosh, I. Calizo, D. Teweldebrhan, E. Pokatilov, D. Nika, A. Balandin, W. Bao, F. Miao, C.N. Lau, Appl. Phys. Lett. 92(15), 151911 (2008)
K.M. Shahil, A.A. Balandin, Solid State Commun. 152(15), 1331–1340 (2012)
W. Hua-Qiang, L. Chang-Yang, L. Hong-Ming, Q. He, Chin. Phys. B 22(9), 098106 (2013)
X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey, H. Zhang, Small 7(14), 1876–1902 (2011)
A. Vahedi, M. Sadr Lahidjani, Eur. Phys. J. Plus 132(10), 420 (2017)
W. Chen, F. Li, P.C. Ooi, Y. Ye, T.W. Kim, T. Guo, Sens. Actuators B Chem. 222, 763–768 (2016)
G. Jiang, M. Goledzinowski, F. J. Comeau, H. Zarrin, G. Lui, J. Lenos, A. Veileux, G. Liu, J. Zhang, S. Hemmati et al. Adv. Funct. Mater. (2016)
S. Guo, S. Dong, Chem. Soc. Rev. 40(5), 2644–2672 (2011)
L. Lancellotti, T. Polichetti, F. Ricciardella, O. Tari, S. Gnanapragasam, S. Daliento, G. Di Francia, Thin Solid Films 522, 390–394 (2012)
R.H. Baughman, A.A. Zakhidov, W. de Heer, Science 297(5582), 787–792 (2002)
D.N. Futaba, K. Hata, T. Yamada, T. Hiraoka, Y. Hayamizu, Y. Kakudate, O. Tanaike, H. Hatori, M. Yumura, S. Iijima, Nat. Mater. 5(12), 987–994 (2006)
M.F. De Volder, S.H. Tawfick, R.H. Baughman, A. Hart, J. Sci. 339(6119), 535–539 (2013)
Y. Saito, S. Uemura, Carbon 38(2), 169–182 (2000)
V.N. Popov, Mater. Sci. Eng. R Rep. 43(3), 61–102 (2004)
J.J. Gooding, Electrochim. Acta 50(15), 3049–3060 (2005)
Y. Ding, Y. Wang, J. Ni, L. Shi, S. Shi, W. Tang, Phys. B Condens. Matter 406(11), 2254–2260 (2011)
D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang, G. Yu, Nano Letters 9(5), 1752–1758 (2009)
M.T. Lusk, L.D. Carr, Phys. Rev. Lett. 100(17), 175503 (2008)
E. Beheshti, A. Nojeh, P. Servati, Carbon 49(5), 1561–1567 (2011)
A. Shokri, E.K. Safari, Indian J. Phys. 89(1), 23–29 (2015)
Fan, L.J. Xinyu, G. Chen, RSC Adv. 7(1), 17417 (2017)
A. Balaban, C.C. Rentia, E. Ciupitu, Revue Romaine De Chimie 13(2), 231 (1968)
A.N. Enyashin, A.L. Ivanovskii, Phys. Status solidi (b) 248(8), 1879–1883 (2011)
A. Enyashin, A. Ivanovskii, Chem. Phys. Lett. 509(4), 143–147 (2011)
E. Brayfindley, E. Irace, C. Castro, W. Karney, J. Org. Chem. 80(8), 3825–3831 (2015)
A. Stone, D. Wales, Chem. Phys. Lett. 128(5), 501–503 (1986)
B. Eggen, M. Heggie, G. Jungnickel, C. Latham, R. Jones, Science 272, 87–90 (1996)
J. Ma, D. Alfè, A. Michaelides, E. Wang, Phys. Rev. B 80(3), 033407 (2009)
H. Bao, L. Wang, C. Li, J. Luo, ACS Appl. Mater. Interfaces 11(3), 2717–2729 (2019)
G. Brunetto, P. Autreto, L. Machado, B. Santos, R. dos Santos, D. Galvão, J. Phys. Chem. C 116(23), 12810–12813 (2012)
V. Gaikwad, ACS Omega 4(3), 5002–5011 (2019)
Z. Wang, Z. Kang, J. Phys. Chem. 100(45), 17725–17731 (1996)
G. Kresse, J. Furthmüller, Comput. Mater. Sci. 6(1), 15–50 (1996)
G. Kresse, D. Joubert, Phys. Rev. B 59(3), 1758 (1999)
G. Kresse, J. Furthmüller, J. Hafner, Phys. Rev. B 50(18), 13181 (1994)
P.E. Blöchl, Phys. Rev. B 50(24), 17953 (1994)
J.P. Perdew, Y. Wang, Phys. Rev. B 46(20), 12947 (1992)
H. Şahin, S. Cahangirov, M. Topsakal, E. Bekaroglu, E. Akturk, R.T. Senger, S. Ciraci, Phys. Rev. B 80(15), 155453 (2009)
H. Monkhorst, J. Pack, Phys. Rev. B 13(12), 5188 (1976)
C-X. Zhao, et al., Comput. Mater. Sci. 160(1), 115–119 (2019)
X-Q. Wang, et al., Phys. Chem. Chem. Phys. 15(6), 2024–2030 (2013)
X. Li, Q. Wang, J. P, J. Phys. Chem. Lett. 8(14), 3234–3241 (2017)
H. Liu, C. Chan, Phys. Rev. B 66(11), 115416 (2002)
S. Guo, Y. Jiang, F. Wu, P. Yu, Liu, Y. Hand Li, L. Mao, ACS Appl. Mater. Interfaces 11(3), 2684–2691 (2019)
T. Lin, J. Wang, ACS Appl. Mater. Interfaces 11(3), 2638–2646 (2019)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shokri, A., Safari, E.K. Theoretical studies on electronic properties of a new carbon allotrope with paring of pentagonal and heptagonal rings. Eur. Phys. J. Plus 135, 771 (2020). https://doi.org/10.1140/epjp/s13360-020-00708-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1140/epjp/s13360-020-00708-x