
Abstract
This paper investigates cyclic hot corrosion of plasma sprayed CoCrAlY + Al2O3 + YSZ (C1) and CoCrAlY + CeO2 (C2) composite coatings on MDN 321 and Superni 76 substrates in molten salt (Na2SO4-60%V2O5) environment exposed to 700 °C. Weight change technique is used to evaluate the corrosion performance. Both C1 and C2 coatings showed better corrosion resistance than uncoated alloy. Both the coatings showed linear weight gain during the initial cycles and parabolic weight gain nature with subsequent hot corrosion cycles. The parabolic rate constant (Kp) of C1 and C2 coating was observed to be in the range 0.29–0.32 × 10−10 g2 cm−4 s−1 and 1.0–1.13 × 10−10 g2 cm−4 s−1 respectively. In C1 coating, the globular and continuously packed structure on the corroded surface having CoO, Cr2O3, CoCr2O4 and CoAl2O4 spinel oxides provided superior hot corrosion resistance. While in case of C2 coating, the outward growth of CeVO4 irregular crystals as a corrosion product of CeO2 and V2O5 salt deteriorated the oxide scales resulting in higher corrosion rate.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The unique combination of MCrAlY (M = Ni, Co and Fe) is prominent for its oxidation and corrosion resistance which can be used as both overlay and bond coat. These coatings are desirable for many parts exposed to high temperatures like gas turbines, aero engines, land based gas turbines and naval diesel engine [1]. James and Rajagopalan [1] have reported that components such as combustor and transition duct of gas turbine operate at temperature of nearly 800 °C. Turbine exit blades, vanes and disks operate between the temperatures of 500–600 °C [2]. These components also work typically under higher mechanical and fatigue stress, and in oxidising, corrosive and wear environment [3,4,5]. In order to extend the life of such components, surface coatings are necessary. Thermal spray surface coating is one of the promising techniques to protect against such degradations [6]. The induced irregularities in substrates are filled with melted or partially melted powder by high impact pressure, which undergoes rapid solidification resulting in their interlocking contributing to the coating thickness and its properties. Particularly the plasma spray technique is used for bond coat and is also the most common method for oxide coatings. Plasma sprayed ceramic coatings are widely used for structural materials and machine parts to improve tribological, oxidation and corrosion resistance [7, 8].
Generally, MCrAlY coatings with Cr content of 18–22% and Al of 8–12% is known for its good oxidation and corrosion resistance behaviour at higher temperatures [9,10,11,12,13]. MCrAlY coatings applied for gas turbines experience degradation under aggressive conditions due to the salts formed potentially by fuel contaminants particularly if the fuel is of low quality (low cost), e.g., sodium, potassium, sulphur, chlorine, etc. The molten salts on the surface usually induce two types of hot corrosion: (1) high temperature type I (850–1000 °C) and (2) low temperature type II (below 800 °C). Both corrosion types will lead to the depletion of beneficial constituents, internal sulfidation and oxidation, and more severe failure of MCrAlY coating system [9]. Nicholls, et al. [11] have reported that NiCrAlY and CoNiCrAlY performs better than Co-based system under oxidising environment at temperatures above 900 °C. At lower temperatures (650–800 °C), type II hot corrosion is predominant and corrosion rates of CoCrAlY are lower than the NiCrAlY and NiCoCrAlY coatings [9]. However, Bolelli et al. [12] have reported that pure MCrAlYs, are unsuitable for tribological applications as their low hardness leads to severe wear under sliding, rotating and oscillating test conditions. In order to improve the mechanical properties with coupled oxidation and hot corrosion resistance of MCrAlY coating, a lot of experimental investigations have been undertaken, such as adding modifying elements (Re, Zr, Hf, Ta, Si, etc.) and oxide reinforcement (Cr2O3, YSZ, Al2O3 etc.) to MCrAlY coating system. The composite coating appears as a feasible solution to couple the oxidation resistance of metal matrix and mechanical/chemical stability of the ceramics [12,13,14,15,16,17,18,19].
Plasma sprayed Al2O3 reinforced NiCrAlY composite coating shows incremental improvement in hardness and nominal improvement in high temperature wear resistance by forming dense glaze layer [12]. Ayyapan et al. [14] have suggested that 20–40% addition of reinforcements to the coating is preferable for improving the coating properties. Further increase in the reinforcement content can deteriorate the properties, due to the increase in porosity and pore size that generate high stress concentration as reported by Kim et al. [15]. Hence in case of CoCrAlY/28%Al2O3/2%YSZ coating, the total reinforcements added is 30%. The addition of Ce to bond coat can effectively resist crack propagation of the thermal barrier coating up to 1000 °C [16]. Zhou and Ouyang [17] have studied that electro deposited Ni–CeO2 nanocrystalline coating has better interfacial adhesion and dense structure of oxides as compared to pure Ni coating. Wang et al. [18] have reported that CeO2 reinforcement should be 2%. The NiAl coating having 2% CeO2 shows less porosity, higher hardness, and higher elastic modulus than the NiAl coatings having 0, 4 and 8% CeO2. Hence, in case of CoCrAlY + CeO2 coating, only 2% CeO2 reinforcement is added to the coating. Many studies investigating on plasma sprayed FeAl matrix composite coating reinforced with hard phases like Al2O3, Cr2O3, CeO2, ZrO2, Y2O3 have reported increase in mechanical and tribological properties as compared to FeAl [19].
The major focus in studies on hard phase reinforced MCrAlY based composite coatings includes phase evolution and tribological properties [12,13,14,15,16,17,18,19]. There is a need and great demand to evaluate the performance of these composite coatings in oxidising and corrosive environment. However, there is not much published literature presenting detailed investigations of the corrosion behaviour of these wear resistant composite coatings.
In the present work, attempt has been made to develop CoCrAlY + Al2O3 + YSZ and CoCrAlY + CeO2 composite coating using plasma spray technique. The high temperature cyclic hot corrosion behaviour of these composite coatings in molten salt (Na2SO4-60%V2O5) environment at 700 °C has been investigated.
2 Experimentation
2.1 Coating Powder and Deposition
The austenitic steel (MDN321) with chemical composition of 18.13Cr-10.36Ni-Mn1.46-Ti0.62-Si0.55-0.10C-bal Fe and Ni-based superalloy (Superni 76) with 21.87Cr-20Fe-9Mo-1.39Co-0.52WC-0.44Mn-0.3Si-0.14Ti-0.08C-bal Ni (wt%) were used as substrate materials procured from M/s MIDHANI, India. These alloys were cut to the dimensions of 25 × 25 × 4 mm and 25 × 25 × 3 mm and the surface of the cut sample was grit blasted using alumina particle of 150 µm to create irregularities/roughens for better mechanical interlocking. NiCrAlY with composition Ni-22Cr-9Al-1Y (wt%) was coated on substrate as bond coat to provide better adhesion with the top coat and to take care of the mismatch by thermal expansion between substrate and top coat.
The Co based composite powder of CoCrAlY (23Cr-13Al-0.65Y-bal Co) was reinforced with Al2O3 + YSZ (28% + 2%) and CeO2 (2%) individually to produce feedstock. The composite powders were developed by using mechanical mixer with the rotation speed of 150 rpm and mixing time of 24 h. The two composite feedstock powders namely CoCrAlY + 28%Al2O3 + 2%YSZ and CoCrAlY + 2%CeO2 were deposited individually over the bond coat by plasma spray technique. The coding for CoCrAlY + Al2O3 + YSZ and CoCrAlY + CeO2 coatings on MDN321 and Superni substrates are referred in Table 1.
The spraying parameters employed during plasma spray deposition are specified by the feedstock powder manufacturer. All the process parameters, including the spray distance were kept constant throughout coating process. The parameters mentioned for CoCrAlY powder was chosen. Also similar spray parameters were reported by [20, 21] for MCrAlY coating. The coating was carried out by plasma spray technique using METCO USA 3 MB equipment with the powder feed rate of 60 g/min; Ar and H2 flow rate of 40 and 7 L/min; current of 490 A; voltage of 60 V. The reinforcements were in weight fraction and the feedstock particle size ranged between − 45 + 15 µm. The morphology and microstructure of the feedstock powders and as-sprayed coatings were observed using scanning electron microscope (SEM, JOEL-JSM-6380LA) equipped with energy-dispersive spectroscopy (EDS) analysis system. The constituents and phases of the powders and coatings were characterized by X-ray diffraction (XRD).
2.2 Hot Corrosion Studies
Hot corrosion studies were performed in static air and molten salt (Na2SO4-60%V2O5) environment at 700 °C under cyclic conditions. The eutectic composition of Na2SO4-60%V2O5 was selected based on the phase diagram for Na2SO4–V2O5 System [22]. The salt mixture was dissolved in distilled water and was coated on the warm samples with the help of camel hair brush. The amount of the salt on the samples varied from 3.0 to 5.0 mg/cm2. All the six faces of the samples were coated by salt. The salt coated specimen was dried in the oven for 3–4 h at 100 °C and weighed before being exposed to hot corrosion tests. The uncoated and coated alloys were subjected to cyclic hot corrosion for 1 h furnace heating at 700 °C in air followed by 20 min cooling at the ambient temperature for 50 cycles. Then the weight change was measured using an electronic balance with sensitivity of 0.1 mg. In order to create severe testing conditions, cyclic loading of sample was considered, since spallation and cracks of protective scale occurred at cyclic loading due to thermal stress. The 50 h of testing time was considered to allow for steady-state oxidation [23, 24]. The weight change was measured after each cycle to understand the kinetics of oxidation and corrosion. The scanning electron microscope (SEM) and X-ray diffractometer (XRD) were used to characterise the corrosion products formed during the exposure.
3 Results and Discussion
3.1 Feed Stock and Coating Structure
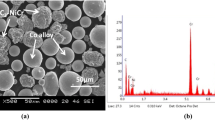
The distribution of hard phase reinforcement in C1 and C2 composite coating powders and particle morphology is shown in Fig. 1a, b. Co-alloy powders appear in typical spherical shape in both the composite powders. This is a good characteristic for spray coating. Distribution of angular Al2O3 and agglomerated YSZ is observed in Fig. 1a. CeO2 particles appear in cluster at the Co particle interspaces (Fig. 1b). The desired coating thickness is achieved by deposition of layers of melted/partially melted flattened particles called splats. Coating consists of two layers; bond coat and top coat (Fig. 1c, d). The average coating thickness as measured by SEM image taken along the coating cross section is 270–290 µm with top coat ranging between 145 and 165 µm. The bond coat has been used for better adhesion between top coat and substrate. The magnified image of coatings C1 and C2 in Fig. 1e, f show the completely and partially melted particle spalts. The completely melted particle has resulted in laterally elongated compact splats called as lamellar structure. The average porosity of C1 and C2 coating are measured using image analyser and are found to be 7.5 and 6% respectively.

SEM morphology of powder and as-coated cross section of C1 (a, c) and C2 (b, d) coatings. Magnified images of C1 (e) and C2 (f) coatings
3.2 Phase Analysis of Powder and Coatings
The XRD pattern of powders and as-sprayed C1and C2 coatings is shown in Fig. 2a, b. Cr and Al2O3 phases are indexed as major peaks with Co and AlCo indexed as intermediate peaks in C1 coating. In case of C2 coating, Cr and Co are indexed as major peaks with AlCo as intermediate peaks. In both the cases, no phase change is observed between the powder and as-sprayed coatings as the splats are formed by rapid solidification. The AlCo phase is indexed to the intermediate peaks in both the coated samples. AlCo phase shows its presence in wide range of temperatures ranging from 200 to 1600 °C and this phase is observed to be present in Co rich regions [25]. The coating structure and phases observed are similar on both the substrates. However the phenomenon of splat morphology on substrate is same when the substrate roughness and temperature remains unchanged [26].

XRD patterns of a C1 and b C2 powders and as-sprayed coatings
3.3 Thermo Cyclic Hot Corrosion Kinetics of Coatings and Substrate
The corrosion kinetics of uncoated alloys and coatings are represented as plot of weight change with respect to the number of cycles as shown in Fig. 3. The results of mass change of corroded samples have been normalized with respect to unit area (Fig. 3a) and squared to analyse the corrosion kinetics [24, 27]. The plot of square of weight gain versus time of both coatings and uncoated alloys at 700 °C in molten salt environment is shown in Fig. 3b. Uncoated alloys in the molten salt environment shows significant increase in weight exhibiting linear weight gain curve without any steady state conditions. Superni 76 alloy experiences 16% lesser weight gain as compared to MDN 321 with overall weight gain of about 8 mg/cm2.

Weight change of coatings versus number of hot corrosion cycles a weight gain per unit area and b square of weight gain per unit area
The initial weight gain of the coated sample is linear in nature and after first 10 cycles, the weight gain rate gradually reduces and reaches steady state. The reduction in weight gain rate or steady state is due to development of protective oxide layer on the coating surface. The steady state weight change is considered as parabolic for the purpose of comparing the hot corrosion performance of the coatings by calculating the parabolic rate constant (Kp) value.
The C1 coating shows weight gain to be parabolic in nature, while C2 coating shows slightly higher weight gain in the initial stages as compared to C1. Oxidation at the pores and splat boundaries result in drastic weight gain during initial cycles and formats stable/protective oxide layer thus resulting in steady state weight gain with the subsequent hot corrosion cycles. This is generally observed characteristics of corrosion resistant coatings. From the observation made, it can be inferred that the formation of protective oxide scale on the coating surface has taken place after the initial few cycles. There difference in weight gain curve of coatings with respect to MDN 321 and Superni 76 substrates do not show much difference. The overall weight gain of coating C1 is about 2.8 mg/cm2 and coating C2 is about 4.7 mg/cm2. Whereas, weight gain of MDN321 is 9.5 mg/cm2 and superni 76 is 8.1 mg/cm2.
The parabolic rate constant (Kp in 10−10 g2 cm−4 s−1) value of uncoated MDN 321 and Superni 76 are found to be 4.65 and 3.32 respectively. The Kp values of C1M and C1S coatings are 0.41 and 0.32 respectively, whereas the Kp values of C2M and C2S coatings are 1.10 and 1.16 respectively.
XRD patterns of hot corroded C1 and C2 coatings exposed to salt environment at 700 °C are shown in Fig. 4. The major peaks in C1 coatings correspond to CoO, Cr2O3, CoCr2O4 and CoAl2O4, while the intermediate peaks are indexed as αAl2O3, AlCo and CoCr2O4. In C2 coating the major peaks correspond to CoO, Cr2O3 and CoCr2O4, while the intermediate peaks are indexed as CeO2, θAl2O3, V2O5 and CeVO4. The major phases observed in the coatings of both the alloys are similar and major elements of substrate such as Fe or Ni are not observed in XRD analysis.

XRD patterns of a C1 and b C2 coated alloys subjected to hot corrosion in molten salt environment at 700 °C
3.3.1 Coating C1 Corrosion Kinetics and Mechanism
The total weight gain of C1 coating is 41% lesser as compared to C2 coating. The parabolic rate constant (Kp in 10−10 g2 cm−4 s−1) of C1 coating is approximately 2 times lesser than parabolic rate constant value of C2 coating. The EDS analysis of the corroded surface and cross section are shown in Fig. 5. The continuous closely packed globular structure is observed throughout the corroded surface. EDS analysis of region ‘A’ and ‘B’ in Fig. 5a shows the dominant presence of Co, Cr and O with traces of Na, S and V salts. The region ‘A’ in Fig. 5b shows Co, Al and O as a major constituents and the region ‘B’ in Fig. 5b is rich in Cr and oxygen. Thus from EDS results, it can be inferred that the corroded surface indicate the presence of CoO, Cr2O3, Al2O3 and their spinels. The presence of these phases is also evident from the XRD analysis and it is represented in Fig. 4a.

Hot corrosion of C1 coating showing a, b surface morphology and c, d cross section and point analysis
The cross section of corroded coating (Fig. 5c) has thin, dense and continuous oxide layer of thickness 12–15 µm. The elemental composition at various locations of the cross section is analysed by EDS analysis (Fig. 5d) and reported in Table 2. EDS of upper oxide scale at region 1 is dominant in Co, Cr, Al and O with V and S salts. The elemental analysis along the cross section elucidates that external surface of the coating is severely oxidized forming surface oxide scale. The salts such as V, S and Na are observed only at the upper most region of coating surface. The minor amount of oxygen is observed at point 3 and 4 which may be due to the intersplat oxidation.
The elemental X-ray mapping along the corroded coating cross section of C1 coating is shown in Fig. 6. The mapping of corroded cross section of C1 coating shows the distribution of considerable amount O, Co, Cr and salts on the surface oxide layer, while Al concentration is not continuous throughout the oxide scale. The distribution of Al2O3 and YSZ reinforcements appears as white spots in Al and Zr mapping. The distribution of oxygen observed throughout the coating cross section is probably due to the intersplat oxidation leading to the formation of oxide stringers.

BSEI and elemental X-ray mapping along the cross-section of the C1 coating subjected to hot corrosion
3.3.2 Coating C2 Corrosion Kinetics and Mechanism
The surface morphology and cross section of corroded C2 coatings are shown in Fig. 7. The corroded coating surface shows continuous closely packed globular structure and dark irregular crystals. EDS analysis of region ‘A’ in Fig. 7a shows the dominant presence of O, Co and Cr with traces of salts and small amount of Al. Thin rod like structure represents the V2O5 salt. The dark irregular crystals appearing on the corroded surface referred to as region ‘B’ has major percentage of oxides of Ce and with traces of Al. This appears to indicate the presence of CeVO4 phase which is also evident from the surface XRD analysis. The growth of irregular crystals on the corroded coating surface is clearly observed in Fig. 7b. The superficial cracks are observed on the coating surface (Fig. 7c).

Hot corrosion of C2 coating showing (a, b, c) surface morphology and (d) cross section and (e) point EDS analysis
Fig. 7d shows thick, delaminated scale ranging between 37 and 43 µm and many cracks are observed in the oxide scale. The elemental composition at various location of the cross section is analysed by EDS point analysis (Fig. 7e) and are reported in Table 3. EDS of upper oxide scale at point 1 is dominant in Co, Cr and O with traces of Na and V salts. However, at point 2, higher amount of O and V as compared to point 1 indicate the entry of corrosion products inside the coating surface.
The elemental X-ray mapping along the corroded coating cross section of C2 coating is shown in Fig. 8. The presence of oxygen throughout the coating along the splats is observed in the corroded coating. The upper scale is predominant in Co, whereas the O and V are observed through the coating surface. This shows the entry of corrosion products inside the coating. Al is not prominent at the coating surface which indicates the absence of Al2O3. Also the presence of Ce and V is prominent at the outer surface and this might have resulted in the formation of CeVO4 with subsequent corrosion cycles. This corrosion product will mainly be responsible for coating degradation.

BSEI and elemental X-ray mapping along the cross-section of the C2 coating subjected to hot corrosion
3.4 Discussion
It is revealed from the XRD analysis on the surface of the corroded C1 coating (Fig. 4a) that the CoO, Cr2O3, α-Al2O3 and there spinels are formed during high temperature exposure. α-Al2O3 is the thermodynamically stable phase showing slow-scale growth kinetics during the oxidation [28]. The Cr2O3 and Al2O3 react with CoO to form CoCr2O4 and CoAl2O4 spinel oxides respectively. The presence of phases CoO, Cr2O3, CoCr2O4 and CoAl2O4 revealed by XRD and presence oxygen rich Co, Cr, and Al elements by EDS are in accordance with the studies of Singh [28]. The protection shown by this coating by steady state weight gain may be due to the formation of Cr2O3, α-Al2O3 and their spinels. These spinel oxides will have lower diffusion coefficient than the parent oxide [28]. Luthra [29] has proposed that the formation of spinels may stop the diffusion activities through the CoO, which in turn suppress the further formation of this oxide. The increase in the growth of CoCr2O4 and Cr2O3 in competition with CoO formation increases the corrosion resistance of alloys. Cr2O3, CoCr2O4 and CoAl2O4 are protective oxides, providing the necessary protection against penetration of corrosive species into the coating. Similar observations are reported by Zhang et al. [30] and Somasundaram et al. [24] during the hot corrosion studies of Co-based coatings.
Afrasiabi et al. [31] have described that the formation of YVO4 crystal is responsible for the degradation of YSZ + 40%Al2O3 coating during hot corrosion. The higher percentage of YSZ is more prone to the formation of YVO4 due to the fluxing action of Y-oxide with V salt. However, in case of C1 coating, very small amount of YSZ and finely dispersed Al2O3 in Co-matrix will restrict the direct contact of V2O5 salt with YSZ. This, further restricts the formation of YVO4 crystals which is a severe corrosion product.
In case of C2 coating, the elemental analysis (EDS and XRD) of the oxide scale reveals the presence of major oxides of Co and Cr. The reaction between Na2So4 and V2O5 result in the formation of NaVO3 during high temperature exposure (Eq. 1). It is well known that the melting point of NaVO3 is 610 °C [29]. The CeO2 in the coating reacts with molten NaVO3 eutectic salt to form CeVO4 irregular crystals (Fig. 7b). On the other hand, Jones et al. [32] and Nejati et al. [33] have reported that the destabilization of CSZ and pure CeO2 by molten NaVO3 or V2O5 to form reaction corrosion product CeVO4 is a mineralization effect. The possible reaction is in the Eq. (2)
The reaction between the coating elements and molten salts will result in the formation of corrosion products [33]. The corrosion product CeVO4 is accompanied by a destructive volume expansion resulting in outward growth in the form of irregular crystals. This leads to the stresses on surrounding splats/oxide scale and the XRD analysis indicates the presence of CeVO4. The cross section of corroded C2 coating (Fig. 7c) shows thick, cracked and delaminated layer and the mapping evidence the presence of vanadium salt inside the coating. Similar observation as is reported by Nejati et al. [33] and Ahmadi-Pidani et al. [34] show that the growth of CeVO4 and YVO4 irregular crystals as corrosion products is responsible for the development of crack on Ce and Y stabilized thermal barrier coatings. Afrasiab and Kobayashi [35] reported these hot corrosion products as irregular crystals exhibiting compressive stresses on the surrounding splats leading to crack resulting in accelerated corrosion. Later, the molten salt infiltrates into the coating through the cracks and enhances the corrosion rate. This further leads to delamination of outer oxide layer and degradation of coating material. The reaction of active elements of the coating with the salt is not observed in XRD analysis, hence the stress developed by other hot corrosion products is not considered.
The major steps involved in the failure of C2 coating are:
-
1.
Reaction between molten eutectic salt NaVO3 and NaVO3–V2O5 mixture with reinforcement CeO2 resulting in the formation of CeVO4 irregular crystals.
-
2.
Growth of hot corrosion product CeVO4 (irregular crystals) causes additional stresses and superficial cracks on the coating.
-
3.
Infiltration of molten salt into the coating through the cracks and delamination of protective oxide scale.
-
4.
Progress of superficial cracks in the cyclic hot corrosion condition due to additional thermal stresses.
The observation from the previous research shows that, CeO2 (2 wt%) reinforced CoCrAlY coating provides superior elevated temperature erosion resistance than Al2O3 (30%) reinforced coating. This is due to the effect of CeO2 for providing better intersplat adhesion and toughness [36]. Whereas, in the present study, during hot corrosion condition, the molten salt makes an attempt to enter into the coating through splat boundaries. The CeO2 particles get distributed along the splat boundaries due to their surface active nature [36]. This results in substantial reaction between molten salt and CeO2 leading to the formation of severe corrosion product which diminishes the protective oxide scale and progresses the corrosion rate. From the above discussion, it can be inferred that the addition of 2 wt% CeO2 rare earth oxide to Co based coating may provide superior corrosion resistance at high temperature [36], but it is not beneficial to resist hot corrosion degradation.
4 Conclusion
-
Co-based cermet coatings with reinforcements Al2O3 and CeO2 were deposited by plasma spray technique. The coating formed showed layers of melted/partially melted splats and reinforcements homogeneously distributed within the coating matrix.
-
Uncoated alloys exhibited linear weight gain curve, while coatings show parabolic nature of weight gain. The coatings showed better hot corrosion resistance as compared to the substrates. Coating C1 showed better hot corrosion resistance than coating C2.
-
In C1 coating, the presence of Cr2O3, CoAl2O4, CoCr2O4 spinel oxides and thermodynamically stable α-Al2O3 was beneficial for resisting against hot corrosion.
-
The CoAl2O4 and CoCr2O4 spinel oxides formed in both the coatings resisted the corrosion degradation. This was due to lower diffusion coefficients of cations and anions in the spinel oxides than the parent oxides.
-
The superficial cracks observed in the C2 coating were due to the outward growth of CeVO4 irregular crystals as a reaction product of molten eutectic NaVO3 and CeO2. Infiltration of molten salt through the cracks led to degradation of the coating and further delamination of the protective oxide scale.
References
James W, and Rajagopalan S, Structural Alloys for Power Plants, (ed) Shirzadi A, and Jackson S, Woodhead Publishing Series in Energy (2014), p 3.
Becker W T, and Shipley R J, ASM Metals Handbook: Vol. 11. Failure analysis and prevention, ASM Publication (2002), p. 1533.
Gurrappa I, Surf. Coat. Technol. 139 (2001) 272.
Doolabi D S, Rahimipour M R, Alizadeh M, Pouladi S, Hadavi S M, and Vaezi M R, Vacuum 135 (2017) 22.
Ramesh M R, Prakash S, Nath S K, Sapra P K, and Venkataraman B, Wear 269 (2010) 197.
Lu J, Zhu S, Wang F, Surf. Coat. Technol. 205 (2011) 5053.
Cai J, Yang S Z, Ji L, Guan Q F, Wang Z P, and Han Z Y, Surf. Coat. Technol. 251 (2014) 2017.
Utu I D, Marginean G, Hulka I, Serban V A, Cristea D, Int. J. Refract. Met. Hard Mater. 51 (2015) 118.
Eliaz N, Shemesh G, and Latanision R M, Eng. Fail. Anal. 9 (2002) 31.
Baiamonte L, Marra F, Gazzola S, Giovanetto P, Bartuli C, Valente T, and Pulci G, Surf. Coat. Technol. 295 (2016) 78.
Nicholls J. R, Simms N J, Chan W Y and Evans H E, Surf. Coat. Technol. 149 (2002) 236.
Bolelli G, Candeli A, Lusvarghi L, Ravaux A, Cazes K, Denoirjean A, and Valette S, Wear 344 (2015) 69.
Cabral Miramontes J A, Gaona Tiburcio C, Almeraya Calderón F, Estupiñan Lopez F H, Pedraza Basulto G K, and Poblano Salas C A, Int. J. Corr (2014) 8.
Praveen A S, Sarangan J, Suresh S, and Subramanian J S, Int. J. Refract. Met. Hard Mater. 52 (2015) 209.
Kim H J, Hwang S Y, Lee C H, and Juvanon P, Surf. Coat. Technol.172 (2003) 262.
Ogawa K, Ito K, Shoji T, Seo D W, Tezuka H, and Kato H, J. Therm. Spray Technol. 15 (2006) 640.
Zhou X, and Ouyang C, Surf. Coat. Technol. 315 (2017) 67.
Wang Y, Yang Y, and Yan M F, Wear 263 (2007) 371.
Song B, Dong S, Liao H, and Coddet C, Surf. Coat. Technol. 268 (2015) 24.
He K, Chen J, Weng W, Li C, and Li, Q, Vacuum 151 (2018) 209.
Xiao J K, Zhang W, Liu L. M, Gan X P, Zhou K C, and Zhang C, Surf. Coat. Technol. 337 (2018) 159.
Otero E, Merino M C, Pardo A, Biezma M V, and Buitrago G, Proceedings of 10th ICMC 4 (1987) 3583.
Ramesh M R, Prakash S, Nath S K, Sapra P K and Krishnamurthy N, J. Therm. Spray Technol. 20 (2011) 992.
Somasundaram B, Kadoli R, and Ramesh M R, J. Therm. Spray Technol. 23 (2014) 1000.
McAlister A J, Alloy Phase Diagram, (ed) Hugh B, United States of America (1989), p 288.
Sampath S, Jiang X Y, Matejicek J, Leger A C, and Vardelle A, Mater. Sci. Eng. A 272 (1999) 181.
Zhu L, Zhu S, and Wang F, Appl. Surf. Sci. 268 (2013) 103.
Singh H, Kaur M, and Prakash S., J. Therm. Spray Technol. 6 (2016) 1192.
Luthra K L, J. Electrochem. Soc. 132 (1985) 1293
Zhang T, Huang C, Lan H, Du L, and Zhang W, J. Therm. Spray Technol. 6 (2016) 1208.
Afrasiabi A, Saremi M, and Kobayashi A, Mater. Sci. Eng. A 478 (2008) 264.
Jones R L, Williams C E and Jones A J, J. Electrochem. Soc. 133 (1986) 227.
Nejati M, Rahimipour M R, and Mobasherpou I, Ceram. Int. 40 (2014) 4579.
Ahmadi-Pidani R, Shoja-Razavi R, Mozafarinia R, and Jamali H, Mater. Des. 57 (2014) 336.
Afrasiab A, and Kobayashi A, Vacuum 88 (2013) 103.
Nithin H S, Desai V, and Ramesh M R, J. Mater. Eng. Perform. 26 (2017) 5251.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Nithin, H.S., Desai, V. & Ramesh, M.R. Hot Corrosion Behaviour of Refractory and Rare Earth Oxide Reinforced CoCrAlY APS Coatings at 700 °C. Trans Indian Inst Met 71, 2403–2413 (2018). https://doi.org/10.1007/s12666-018-1371-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12666-018-1371-3