
Abstract
To evaluate the impact of mercury released into a river system by artisanal and small-scale gold mining (ASGM) activity in West Java, Indonesia, the temporal variations in elemental mercury, mercury ions, dissolved total mercury and particulate mercury concentrations in river water were observed in March 2015 (rainy season) and August 2015 (dry season) at a fixed observation point, and the migrated amount of mercury was estimated. Variations for other metals, total organic carbon (TOC), and total nitrogen (TN) contents were also measured. There have been few reports in which the amount of mercury transferred from ASGM activities is quantitatively estimated, and this study provides important data for evaluating the environmental impact of mercury released by ASGM activities. The concentration of filtered total mercury and the particulate mercury concentration observed in the dry season were 161 ± 167 μg L−1 and 142 ± 93 μg L−1, respectively, and these values were significantly higher than the values of 24.5 ± 23.9 μg L−1 and 53.7 ± 44 μg L−1 for filtered total mercury and particulate mercury, respectively, observed in the rainy season. On the other hand, the river flow in the rainy and dry seasons was 2.6 × 104 ± 5×103 m3 h−1 and 8.2 × 103 ± 8×102 m3 h−1, respectively, and an apparent increase in the amount of water in the rainy season was observed. The transfer amounts in the rainy and dry seasons obtained from these results were 6.2 × 102 ± 6.7 × 102 g h−1 and 1.6 × 103 ± 1.6 × 103 g h−1, respectively, for filtered total mercury and 1.3 × 103 ± 1.3 × 103 g h−1 and 1.1 × 103 ± 7×102 g h−1, respectively, for particulate mercury. The temporal variations in these values exhibited a wide range, indicating the effects of weather conditions and of the mining process.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The toxicity of mercury is well established; on 16 August 2017, the Minamata Convention on Mercury came into force. The Minamata Convention limits mercury mining, regulates trade, reduces the use of mercury in products and processes, decreases and eliminates the use of mercury in gold mining, controls mercury emissions into the air and water, and promotes sound waste disposal. However, artisanal and small-scale gold mining (ASGM) continues to be conducted illegally and is estimated to be responsible for approximately 37% of the mercury released into the atmosphere (UNEP 2013). Despite ASGM being a major source of mercury and given the fact that ASGM using mercury is illegal, an understanding of the entire scope of this activity is difficult to achieve. Moreover, since ASGM activities differ by region, detailed investigations for each region are indispensable for estimating the impact of the discharged mercury on ecosystems. High levels of mercury contamination in soil, atmosphere, rice, fish, and human tissue samples resulting from the discharge of mercury as mining waste and exhaust gases have been reported (Pfeiffer et al. 1989; Olivero and Solano 1998; Grandjean et al. 1999; Mol et al. 2001; Maurice-Bourgoin et al. 2003; Eisler 2004; Slowey et al. 2005; Ashe 2012; Tomiyasu et al. 2013; Wade 2013; Diringer et al. 2015; Tomiyasu et al. 2017). The results of many studies demonstrate that mercury contamination in river systems is caused by ASGM activity as a result of the discharge of mining wastes (Moreno-Brush et al. 2016; Appleton et al. 1999; Marrugo-Negrete et al. 2008; Roth et al. 2001; Niane et al. 2014; Reichelt-Brushett et al. 2017; Diringer et al. 2015; Pinedo-Hernández et al. 2015). These studies are important for debating the fate of discharged mercury and the impact it presents for ecosystems. The discussion of these studies is conducted with regard to the concentration levels of mercury and few studies have quantitatively reported the amount of mercury transported. However, to evaluate the impact of mercury and to understand the scale of pollution by ASGM activities, not only the concentration but also the absolute amount of mercury transferred by river flow must be better understood. In this study, the temporal variations in elemental mercury (Hg0); mercury ions (Hg2+); filtered total mercury (FT-Hg); and particulate mercury (P-Hg) concentrations in river water were investigated with respect to the variations of river flow in March (rainy season) and in August (dry season) in 2015 to estimate the transport amount of mercury by the river system. The study area in this investigation is in West Java, Indonesia, and ASGM operations here involve processing by adding metallic mercury during grinding of gold ore using a drum machine; the resulting mining waste is washed with water and directly discharged into the river system (Yustiawati et al. 2006; Hidayati et al. 2009). This type of activity in this area is illegal and the amount of mercury used in this area and the amount of mercury discharged into river systems is not well known. However, paddy fields are located downstream and the discharge of mercury may have a large influence on the health of the neighboring residents. This study offers important data for estimating the impact of mercury discharged into river systems and resulting from ASGM activities.
Materials and methods
Sampling point and ASGM activity in the study area
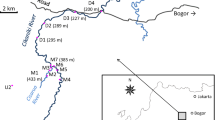
The study area is shown in Fig. S1. The mining work sites for the mining process are located in the villages in this area, where mercury is used in the extraction of gold from ore using traditional methods: Metallic mercury is added during the grinding process of the gold ore and the mining waste is washed with water, followed by discharge into the river system. The mercury remaining after the process is collected in a cloth. Excess mercury is squeezed out and the leftover gold amalgam remains in the cloth; heating with a burner is performed to evaporate the remaining mercury. Sampling for this study was conducted in March and August in 2015.
River water samples were carefully filled into 100-mL and 250-mL capacity Teflon bottles such that no air remained, and brought back to the laboratory. The temperature, pH, and redox potential of the water samples were measured at the sampling site using electrodes (pH meter, HM-20P having a range of measurement of pH 0.00–14.00, DKK-TOA Co., Ltd., Japan; ORP meter, RM-20P having a range of measurement of 0 to ± 1999 mV, DKK-TOA Co., Ltd., Japan).
The dissolved gaseous mercury (Hg0) was measured at the sampling site or measured as soon as possible after transport to the laboratory using samples collected in 250 mL capacity Teflon bottles using a Hg analyzer (EMP-2-Aqua system, Nippon Instruments Corporation Co., Ltd., Japan) without any pretreatment (see “The procedures for determining FT-Hg, Hg2+, and Hg0 concentrations in the water samples”). For filtered total mercury (FT-Hg) and mercury ion (Hg2+) analysis, the water samples collected in the 250-mL capacity were filtered through a membrane filter (with a pore size of 0.45 μm). The suspended particles on the filter were used for particulate mercury (p-Hg) analysis.
Water samples collected in the 100-mL capacity Teflon bottle were used for measurement of TOC, TN and other ions after filtration with a pore size of 0.45 μm membrane filter.
The time required for a float to drift 25 m downriver was used to estimate the flow rate of the river. Flow amount of the water (m3 hˉ1) was calculated using water levels, width of the river and the flow rate.
Analytical methods
The procedures for determining FT-Hg, Hg2+, and Hg0 concentrations in the water samples
The water samples collected in 250-mL Teflon bottle were filtered and the filtrates were used for Hg2+ and FT-Hg measurement. For Hg2+ measurements, the water sample was poured into a glass reaction vessel (< 20 mL) and 1 mL of 10% SnCl2 in 2 M HCl solution was added to reduce the Hg2+ to Hg0. The solution was bubbled with Hg-free air and the Hg0 swept from the water samples was measured by cold vapor atomic absorption spectrometry (CVAAS; EMP2-Aqua system, Nippon Instruments Corporation, Tokyo, Japan).
The method for determination of FT-Hg was based on acidification and oxidation with BrCl in combination with UV radiation (Logar et al. 2001). The BrCl solution was prepared by dissolving 1.1 g of analytical reagent grade KBrO3 and 1.5 g of analytical reagent grade KBr in 20 mL of pure water. Concentrated HCl (80 mL) was then slowly added to the solution with constant stirring (Bloom and Crecelius 1983). For FT-Hg measurements, the filtered water samples were poured into Teflon bottles and 0.5 mL of BrCl solution was added. After exposure to UV radiation overnight, 0.5 mL of 12% NH2OH·HCl was added to decompose any excess BrCl. The BrCl oxidation in combination with UV radiation decomposes mercury compounds contained in the water sample to Hg2+. Thus, the total mercury in the resulting solution was measured by CVAAS in the same manner as Hg2+. For the measurement of Hg0, unfiltered water samples poured into a glass reaction vessel (< 20 mL) were bubbled with Hg-free air without the addition of a SnCl2 solution. The Hg0 contained in the water sample was swept by the bubbling and was measured by CVAAS (EMP2-Aqua system, Nippon Instruments Corporation, Tokyo, Japan).
The measurements of the water samples were duplicated to verify their repeatability and variations of the duplicate measurements were 2.7 ± 1.9%; 4.0 ± 1.9%; and 14.9 ± 5.8%, for FT-Hg; Hg2+; and Hg0, respectively. The limit of detection of this method is 1.0 ng L−1.
Procedure for P-Hg
Suspended solids collected on the membrane filter (with a pore size of 0.45 μm) by the filtration of 250 mL river water samples were dried at room temperature for 3 days. The dried particle sample was weighed and placed with a filter in a 50-mL volumetric flask, to which 1 mL of pure water, 2 mL of a 1:1 nitric acid–perchloric acid solution, and 5 mL of concentrated sulfuric acid were added. After standing for a few minutes, the flask was heated on a hot plate at 230 °C for 20 min. After cooling, the volume of the digested sample was adjusted to 50 mL using water and a suitable aliquot of the resulting solution (< 10 mL) was analyzed for mercury by cold vapor atomic absorption spectrometry using a semiautomated mercury analyzer (Model Hg-3000, Sanso Seisakusho Co., Ltd., Japan). Separately, 1.0 mL of water and the standard mercury solution were transferred into three sample digestion flasks and treated using the procedure described above to obtain blank and standard test solutions for measuring the total mercury concentration (Committee for the Mercury Analysis Manual 2004). The absolute limit of detection was 0.02 ng, calculated on the basis of three standard deviations of the blank; when using 0.2 g of sample, the detection limit was 0.1 μg kg−1. The variation of duplicate measurements of sample solutions was 0.7 ± 0.5%.
The P-Hg concentration (μg L−1) was calculated by dividing the absolute amount of mercury by the volume of sample water used for the filtration.
To calculate the mercury concentration in SPM (Hg in SPM/mg kg−1), the absolute amount of mercury was divided by the weight of the SPM. The weight of SPM was obtained by subtracting the weight of an unused filter from the weight of the filter used for the filtration. P-Hg and Hg in SPM were calculated by the following equations:
P-Hg (μg L−1) = [the absolute amount of mercury in SPM (μg)]/[the volume of filtered river water sample (L)], and Hg in SPM (mg kg−1) = [the absolute amount of mercury in SPM (μg)]/[the weight of SPM on the filter (g)].
Determination of TOC, TN and other ions
Total nitrogen (TN), total carbon (TC), and inorganic carbon (IC) in filtered river water samples were determined by using a TOC/TN analyzer (model multi N/C 3100, Analytik Jena AG, Germany). For the IC determination, the 500 μL of sample injected into the devices was acidified by phosphoric acid. IC was released from the solution as CO2 and analyzed with an NDIR detector. For TC and TN determination, the sample injected into the device was subjected to high-temperature catalytic oxidation to form CO2 and NO. TC was analyzed with an NDIR detector and TN, with a chemiluminescent detector. Total organic carbon (TOC) was defined as the difference between total carbon (TC) and inorganic carbon (IC). Major anions (F−, Cl−, NO2−, Br−, NO3−, PO43−, SO42−) were measured simultaneously using ion chromatography (model ICS-900, Thermo Fisher Scientific Co-LTD, USA) incorporated with a separator column (AS12A: 4 × 200 mm), suppressor (AMMS 300 micro membrane suppressor), and electric conductivity detector at the eluent (0.3 mM NaHCO3–2.7 mM Na2CO3) flow rate of 1.65 mL min−1. Heavy metal ions were measured by simultaneous ICP atomic emission spectrometer (model ICPE-9000, Shimadzu Co-LTD, Japan. The ICP instrumental parameters are shown in Table S1). These measurements were conducted for water samples collected in March 2015.
Data analysis and statistics
A Student’s t test was used to evaluate if there were significant differences between the results from the two sampling seasons. Regression analysis was used to establish the relationships between the variables. The criterion for significance was set at < 0.05. The statistical analysis functions of Microsoft Excel 2010 (Microsoft Corporation, USA) were used for data analysis and to generate graphs.
Results and discussion
Variation of flow rate and water quality of river waters during the rainy season
Variation in flow rates of the river water is shown in Fig. S2. The flow rate of water in March was in the range of 2.1 × 104 to 3.5 × 104 m3 h−1 with an average of 2.6 × 104 m3 h−1. A rise in water level was observed after precipitation (14:05, 8th March). Redox potential (ORP), electrical conductivity (COND) and temperature were 117 ± 25 mV, 12.7 ± 1.3 mS m−1, and 23.5 ± 1.0 °C, respectively, and no large variation was observed during the 3 relevant days (Fig. S3). The pH value of the waters was 7.93 ± 0.90, but a significantly low value of 5.62 was observed on 8th March. On 8th March, rain occurred and the pH was then 5.2–5.4. Therefore, the decrease of pH in the river water may have been affected by the rain. The average river flow rate in August, 8.2 × 103 m3 h−1, was approximately one-third of what it was in March. No rain was observed in August and the variation of flow rate 7.1 × 103–9.6 × 103 m3 h−1 was smaller than in March.
Variation in the concentration of mercury species
In March, Hg0, Hg2+, Ft-Hg and P-Hg concentrations were 1.2 ± 0.8 μg L−1, 23.3 ± 22.6 μg L−1, 24.5 ± 23.9 μg L−1, and 53.7 ± 44.0 μg L−1, respectively. The predominant mercury species was P-Hg. Although the variation in water quality was not as large in March, marked concentration fluctuations were observed in the mercury species. In the dissolved mercury species (Fig. 1a–c), a high value was observed at 11 o’clock on the 7th (at the start of the survey), and the concentration declined from then into the evening. On the next day, the value varied with time and the highest value was observed at 10 o’clock. On the third day of the survey, the concentration increased from 6 to 10 o’clock. The fluctuation may mainly reflect the working step. On the other hand, P-Hg showed a different fluctuation from that of the dissolved Hg species (Fig. 1d). The value increased continuously from morning to evening on the second day, showing the highest value at 18 o’clock. SPM-Hg (mg kg−1) showed no significant variation with an average value of 227 ± 45 mg kg−1 and showed a tendency to decrease from morning to evening on the second day (Fig. 1e), when the P-Hg value increased. On 8th March (the second day of the survey), rainfall was observed 8:30–10:30 and 13:30–14:30. With the rain, the concentration of SPM in river water increased until the evening of that day with the increase in flow amount of the river (Fig. 1f). The rainfall may have caused an inflow of surrounding surface soil to the river and/or the resuspension of river sediment. With the inflow of surrounding relatively low-level contaminated soil particles, the SPM-Hg (mg kg−1) decreased but the absolute amount of SPM increased, which caused an increase in P-Hg (μg L−1). Thus, the fluctuation in P-Hg concentration reflects mercury not only discharged from the work sites but also from other sources, such as inflow and resuspension of particles caused by rainfall.

The temporal variations in mercury concentrations in the rainy season (Mar. 2015): a Hg0; b Hg2+; c FT-Hg; d P-Hg; e Hg in SPM; and f SPM content
In August, the concentrations of FT-Hg and P-Hg showed wide fluctuations (Fig. 2), while the river water flow exhibited no significant variations with time. Since no rain was observed during our August sampling and the variations of FT-Hg and P-Hg concentration were synchronized with variations in SPM, the variations in mercury concentration and SPM may reflect the results of mining activity. The concentrations of FT-Hg and of P-Hg were 161 ± 167 μg L−1 and 142 ± 93 μg L−1, respectively, and were significantly higher than the values observed in the rainy season (March).

Temporal variations in mercury concentrations during the dry season (Aug. 2015): a FT-Hg; b P-Hg; c Hg in SPM; and (d) SPM content
The correlations between different mercury species
The correlation between different mercury species is shown in Fig. 3 for the March sampling period. Significant linear relations were observed among dissolved mercury species (Fig. 3a–c).

Correlations between a FT-Hg and Hg2+; b FT-Hg and Hg0; c Hg2+ and Hg0; d SPM and P-Hg; e SPM and FT-H; f FT-Hg and P-Hg in the rainy season (Mar. 2015)
Insolation is speculated to play an important role in the formation/consumption of dissolved gaseous mercury in both river water and seawater (Katarina Gårdfeldt et al. 2001; Zhang and Lindberg 2002; Jardim et al. 2010). The content of organic matter, water temperature, and wind can also be important factors. For example, dissolved gaseous mercury levels in nearshore surface waters of Whitefish Bay exhibited diurnal trends probably caused by wind-induced mixing and decreased insolation associated with the frontal passage (Zhang and Lindberg 2002). Mercury evasion measured for both river and seawater surfaces exhibits a consistently diurnal pattern with maximum evasion during the daytime and minimum during the night. At the freshwater site, mercury evasion is strongly correlated with the intensity of net insolation (Katarina Gårdfeldt et al. 2001).
Although the diurnal pattern was also observed in our study (Fig. 1), the significant relationship observed between FT-Hg and Hg0 suggests that workplace waste is the source of Hg0 and the variations in its concentration was caused by the extraction of gold from ores. The Hg0 concentration observed in this study was several orders of magnitude higher than reported values for creek water and nearshore surface water (Zhang and Lindberg 2002). This observation suggests that river water is not the source of Hg0.
The discharged Hg0 can be considered to be released from the river waters into the atmosphere during the downstream flow. For this case, the correlation between Hg0 and other dissolved Hg species would vanish. A significant correlation was also observed between SPM and P-Hg (Fig. 3d). The increase in SPM caused an increase in P-Hg concentrations. On the other hand, significant correlations for SPM vs. FT-Hg and for Ft-Hg vs. P-Hg were not observed in our plots. Since the waste from the work site includes mining waste, a linear relation might be observed. However, due to a supply of SPM from other than the work site, the correlations will be lost. As described above, on the afternoon of 8 March, an increase in SPM was caused by rainfall. When the data from the afternoon of March 8 were excluded from the SPM vs. FT-Hg and P-Hg vs. Ft-Hg plots, these plots showed significant linear relationships (p < 0.01) with R2 = 0.914 and R2 = 0.857, respectively. It can be said again that the variation in dissolved Hg reflects the discharge of mercury in real time and that the P-Hg concentration reflects not only the real-time discharge, but also the movement of mercury deposited in the surroundings. This is supported by the fact that significant correlations were observed for SPM vs. FT-Hg plots and for P-Hg vs. Ft-Hg plots in August, when rainfall was not observed (Fig. 4). On the other hand, a significant correlation was not observed between FT-Hg and Hg0 in August. The apparently higher Hg0 concentrations were observed in August. At such high concentration levels, Hg0 is unstable in water and may easily escape into the atmosphere. The datasets from March and August were used to generate the FT-Hg vs. Hg0 plots, Hg0, indicating that concentrations tended to be independent of FT-Hg concentrations in the higher concentration ranges (Fig. S4).

The correlations between a FT-Hg and Hg0; b FT-Hg and P-Hg; c SPM and P-Hg; and d SPM and FT-Hg in the dry season (Aug. 2015)
The variation of carbon and nitrogen concentrations and the relationship with mercury concentrations
The time fluctuations of TC, IC, TOC and TN are shown in Fig. S5 and the correlations between them are shown in Fig. S6. Significant linear relationships were observed between TC and TN, and between IC and TN. However, TOC showed no significant relationship with other species. Approximately 70% of the TC was inorganic carbon, thus a significant relationship could be observed between TC and IC. These observations suggest that the source of TOC and of IC is different. Their correlations with mercury species are shown in Fig. 5. The most interesting phenomenon is the high correlation of mercury species with IC and no significant correlation between mercury species and TOC. A correlation between mercury and organic carbon is typically observed (Brigham et al. 2009; Guentzel 2009), probably because mercury bonds with organic matter during river transport. Since the sampling location was in the ASGM-related village, it was thought that the river water directly reflected the properties of mining waste. In this village, workers add cyanide to slag that was used for gold extraction once to extract gold again. After the recovery of eluted gold by using activated charcoal, the waste water is discharged into the river. As a result, the high correlations would be observed between IC and mercury species. This can be supported by the significantly high linear relationship between IC and TN. On the other hand, TOC showed a significant linear relationship with P-Hg. The high P-Hg concentrations were observed during rainfall (8th March). At that time, high TOC values were recorded. As described above, the rainfall may have caused an inflow of surrounding surface soil to the river, which caused the increase in the TOC and P-Hg. We previously reported a relationship between Hg species and TOC in soils in this area (Tomiyasu et al. 2017). Throughout the investigation, it was observed that the soil around the village did not contain inorganic carbon. It follows that the inflow of surrounding surface soil does not contribute to the increase in IC concentrations in river water.

The dependencies of mercury species concentration on IC, TN, TOC and TC concentrations
Variations in concentrations of other ions
The variation of major anion concentrations showed no clear trend (Fig. S7). Concentrations of mercury species were independent of the ion concentrations. The sum of NO3ˉ and NO2ˉ content (as nitrogen), which was 16 ± 6% of TN, also showed no relationship with mercury concentration. This fact supports the notion that the dependency of mercury concentration on TN depends on cyanide. Some studies have reported that toxic elements such as lead and arsenic were present in the mining waste and caused pollution around the gold mining area (Edinger et al. 2008; Thorslund et al. 2012). In this study, a high concentration of these elements in river water was not observed. The elements showing significant correlations are tabulated in Table S1. Cu concentrations in filtered water (F-Cu) showed a significant linear relationship with FT-Hg. Copper contained in the ore could be extracted by mercury as an amalgam and be discharged with mercury. The average Cu concentration was 0.024 mg L−1, which is considerably lower than the standard for drinking water. Another significant linear relationship was observed between FT-Hg and Fe. In the grinding-extraction step, iron drums and bars were used and thus abraded iron might be carried into the waste water with mercury. Fe concentrations in unfiltered water (UF-Fe); F-Al; UF-Al; and UF-Ca showed a significant linear relationship with P-Hg. Since Fe, Al, and Ca showed a dependency on SPM, they could be components of the particulate matter. Generally, it can be stated that, in this area, water pollution has not occurred, except by mercury.
Amount of mercury transported by the river system
FT-Hg; P-Hg transferred and their sum in March was 0.62 ± 0.68 kg h−1; 1.5 ± 1.6 kg h−1; and 2.2 ± 1.8 kg h−1, respectively, and in August were 1.2 ± 1.2 kg h−1; 1.1 ± 0.7 kg h−1; and 2.4 ± 1.9 kg h−1, respectively. The temporal variation in these values is shown in Fig. 6. While the concentration of mercury species was several times higher during the dry season than during the rainy season, the quantity of transported mercury showed no significant difference between rainy and dry seasons. The amount of mercury discharged by ASGM activities can be considered to have been almost identical in March and August and that the decrease of river flow in the dry season caused an increase in the mercury concentration. The flow of transferred mercury, 2 kg h−1, observed in this study was of the same order of magnitude, as 39 kg day−1 and 55 kg day−1 transported by the Negro and Solimões Rivers, respectively (Maurice-Bourgoin et al. 2003).

The time variations in mercury transported by river: a rainy season (Mar. 2015) and b dry season (Aug. 2015)
No significant variation was observed in March and August for SPM concentrations (g L−1). If nearly all the SPM in river water is derived from the ASGM activities, the concentration should be greater in the dry season. In the rainy season, the river flow increases but the inflow of solid particles from surrounding surfaces and resuspension of river sediment occurs, which may cause the increase in SPM concentrations observed in river water. The average concentrations of SPM-Hg are 272 ± 45 mg kg−1 in March and 466 ± 136 mg kg−1 in August. For low river water levels, the high concentration of mercury causes large accumulations of mercury in the SPM. The Hg in SPM increased with an increase in FT-Hg concentration and Hg in SPM was virtually independent of FT-Hg at the higher concentration ranges of FT-Hg (Fig. S8). A significant high linear relationship was observed for FT-Hg vs. FT-Hg/Hg-SPM plot (Langmuir model). These observations suggest an adsorption equilibrium of mercury between water and SPM.
Conclusions
Mercury concentration in river water varied with weather and working processes and was significantly higher in the dry season than in the rainy season. On the other hand, since the water level is higher in the rainy season, there was no significant difference in the net flow of transported mercury, i.e., 2 kg h−1, between rainy and dry seasons. Approximately half of the transported mercury was particulate mercury, the concentration of which became > 600 mg kg−1 in the dry season. The deposition of such highly contaminated particles from irrigation water on rice fields can cause serious contamination of the rice. Moreover, significant linear correlations observed between inorganic carbon, total nitrogen content, and dissolved mercury concentrations suggest the stabilization effect by cyanide for dissolved mercury. Cyanide can extract deposited mercury into the water phase again, making it available for biological activity. A continuous survey, including the impact on ecosystems, is required.
References
Appleton JD, Williams TM, Breward N, Apostol A, Miguel J, Miranda C (1999) Mercury contamination associated with artisanal gold mining on the island of Mindanao, the Philippines. Sci Total Environ 228:95–109
Ashe K (2012) Elevated mercury concentrations in Humans of Madre de Dios, Peru. PLoS One 7:e33305
Bloom SN, Crecelius AE (1983) Determination of mercury in seawater at sub-nanogram per liter levels. Mar Chem 14:49–59
Brigham ME, Wentz DA, Aiken GR, Krabbenhoft DP (2009) Mercury cycling in stream ecosystems. 1. Water column chemistry and transport. Environ Sci Technol 43:2720–2725
Committee for the Mercury Analysis Manual (2004) Mercury analysis manual. Ministry of the Environment, Japan
Diringer SE, Feingold BJ, Ortiz EJ, Gallis JA, Araújo Flores JM, Berky A, Pan WKY, Hsu-Kim H (2015) River transport of mercury from artisanal and small-scale gold mining and risks for dietary mercury exposure in Madre de Dios, Peru. Environ Sci Process Impacts 17:478–487
Edinger EN, Azmy K, Diegor W, Siregar PR (2008) Heavy metal contamination from gold mining recorded in Porites lobata skeletons, Buyat-Ratototok district, North Sulawesi, Indonesia. Mar Pollut Bull 56:1553–1569
Eisler R (2004) Mercury hazards from gold mining to humans, plants, and animals. Rev Environ Contam Toxicol 181:139–198
Gårdfeldt K, Feng X, Sommar J, Lindqvist O (2001) Total gaseous mercury exchange between air and water at river and sea surfaces in Swedish coastal regions. Atmos Environ 35:3027–3038
Grandjean P, White RF, Nielsen A, Cleary D, Santos ECO (1999) Methylmercury neurotoxicity in Amazonian children downstream from gold mining. Environ Health Perspect 107:587–591
Guentzel JL (2009) Wetland influences on mercury transport and bioaccumulation in South Carolina. Sci Total Environ 407:1344–1353
Hidayati N, Juhaeti T, Syarif F (2009) Mercury and cyanide contaminations in gold mine environment and possible solution of cleaning up by using phytoextraction. J Biosci 16:88–94
Jardim FW, Bisinoti MC, Fadini SP, da Silva SG (2010) Mercury redox chemistry in the Negro River Basin, Amazon: the role of organic matter and solar light. Aquat Geochem 16:267–278
Logar M, Horvat M, Akagi H, Ando T, Tomiyasu T, Fajon V (2001) Determination of total mercury and monomethylmercury compounds in water samples from Minamata Bay, Japan: an interlaboratory comparative study of different analytical techniques. Appl Organometal Chem 15:515–526
Marrugo-Negrete J, Norberto Benitez L, Olivero-Verbel J (2008) Distribution of mercury in several environmental compartments in an aquatic ecosystem impacted by gold mining in Northern Colombia. Arch Environ Contam Toxicol 55:305–316
Maurice-Bourgoin L, Quemerais B, Moreira-Turcq P, Seyler P (2003) Transport, distribution and speciation of mercury in the Amazon River at the confluence of black and white waters of the Negro and Solimoes Rivers. Hydrol Process 17:1405–1417
Mol JH, Ramlal JS, Lietar C, Verloo M (2001) Mercury contamination in freshwater, estuarine, and marine fishes in relation to small-scale gold mining in Suriname, South America. Environ Res Sect A 86:183–197
Moreno-Brush M, Rydberg J, Gamboa N, Storch I, Biester H (2016) Is mercury from small-scale gold mining prevalent in the southeastern Peruvian Amazon? Environ Pollut 218:150–159
Niane B, Moritz R, Guédron S, Ngom PM, Pfeifer HR, Mall I, Poté J (2014) Effect of recent artisanal small-scale gold mining on the contamination of surface river sediment: case of Gambia River, Kedougou region, southeastern Senegal. J Geochem Explor 144:517–527
Olivero J, Solano B (1998) Mercury in environmental samples from a waterbody contaminated by gold mining in Colombia, South America. Sci Total Environ 217:83–89
Pfeiffer WC, Lacerda LD, Malm O, Souza CMM, Silveira EG, Bastos WR (1989) Mercury concentrations in inland waters of gold-mining areas in Rondonia, Brazil. Sci Total Environ 87(88):233–240
Pinedo-Hernández J, Marrugo-Negrete J, Díez S (2015) Speciation and bioavailability of mercury in sediments impacted by gold mining in Colombia. Chemosphere 119:1289–1295
Reichelt-Brushett AJ, Stone J, Howe P, Thomas B, Clark M, Male Y, Nanlohy A, Butcher P (2017) Geochemistry and mercury contamination in receiving environments of artisanal mining wastes and identified concerns for food safety. Environ Res 152:407–418
Roth DA, Taylor HE, Domagalski J, Dileanis P, Peart DB, Antweiler RC, Alpers CN (2001) Distribution of inorganic mercury in Sacramento river water and suspended colloidal sediment material. Arch Environ Contam Toxicol 40:161–172
Slowey AJ, Rytuba JJ, Brown JRGE (2005) Speciation of mercury and mode of transport from placer gold mine tailings. Environ Sci Technol 39:1547–1554
Thorslund J, Jarsjö J, Chalov SR, Belozerova EV (2012) Gold mining impact on riverine heavy metal transport in a sparsely monitored region: the upper Lake Baikal Basin case. J Environ Monit 14:2780–2792
Tomiyasu T, Kono Y, Kodamatani H, Hidayati N, Rahajoe JS (2013) The distribution of mercury around the small-scale gold mining area along the Cikaniki river, Bogor, Indonesia. Environ Res 125:12–19
Tomiyasu T, Kodamatani H, Hamada YK, Matsuyama A, Imura R, Taniguchi Y, Hidayati N, Rahajoe JS (2017) Distribution of total mercury and methylmercury around the small-scale gold mining area along the Cikaniki River, Bogor, Indonesia. Environ Sci Pollut Res 24:2643–2652
Wade L (2013) Gold’s dark side. Science 341:1448–1449
Yustiawati M, Syawal MS, Terushima M, Tanaka S (2006) Speciation analysis of mercury in river water in West Java. Indonesia. Tropics 15(4):425–428
Zhang H, Lindberg ES (2002) Trends in dissolved gaseous mercury in the Tahquamenon River watershed and nearshore waters of Whitefish Bay in the Michigan Upper Peninsula. Water Air Soil Pollut 133:381–391
Acknowledgements
This work was supported by Grants-in-Aid (Nos. 22404002 and 25257302) for Scientific Research from the Japan Society for the Promotion of Science (JSPS).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12665_2019_8727_MOESM3_ESM.pdf
Fig. S3 Temporal variations in (a) pH, (b) temperature, (c) redox potential, and (d) electrical conductivity (PDF 44 kb)
12665_2019_8727_MOESM8_ESM.pdf
Fig. S8 The relationship between FT-Hg and Hg in SPM: (a) FT-Hg vs. Hg in SPM plot; (b) FT-Hg vs. FT-Hg/Hg-SPM plots (Langmuir model); and (c) ln(FT-Hg /g L-1) vs. ln(Hg-SPM /mg kg-1) plots (Freundlich model) (PDF 42 kb)
Rights and permissions
About this article

Cite this article
Tomiyasu, T., Hamada, Y.K., Baransano, C. et al. Time variation in transfer amounts of mercury by a river system near an artisanal and small-scale gold mining area in West Java, Indonesia. Environ Earth Sci 78, 686 (2019). https://doi.org/10.1007/s12665-019-8727-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12665-019-8727-9