
Abstract
Quaternary chalcogenide compositions have gained much attention because of their promising potential for various optoelectronic applications. The band structure, density of states, and optical properties of CuZn2InS4 and CuZn2GaS4 for kesterite and stannite structures are studied with the full-potential augmented plane wave method as implemented in the Wien2k code. The total energy in equilibrium is calculated for different possible crystal structures. The phase stability and transitions with p–d orbitals are also analyzed. Moreover, the absorption coefficient, dielectric function, and refractive index of these materials are explored within a broad range of energy. We compare the calculated band gap values with available experimental results.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In the studies of photovoltaic technologies, thermoelectrics, and optoelectronic devices, the quaternary semiconductors Cu2-II-IV-VI4 have attracted tremendous attention since their elements are earth-abundant, inexpensive, and environment-friendly in nature. In addition, their band gap offers a broad range of the optical spectrum. The designs of Cu2-II-III-VI4 quaternary compositions have resulted from cation cross-substitution of binary II-VI and ternary I-III-VI2. It offers an alternative route for exploring promising materials to be used as absorbers in photovoltaic applications. Originally, Goodman (1958) and Pamplin (1964) initiated the formation of quaternary chalcogenides from ternary I-III-VI2 systems through cation cross-substitution which obeys octet rule [1, 2]. There are two possibilities for these cation substitutions: One of them is forming an I2-II-IV-VI4 compound with one group-IV and one group-II atom by replacing two group-III atoms; another one is to replace one group-III atom and one group-I with two atoms of group-II, which formed I-II2-III-VI4 compound. These can also be defined by the II2x(I-III)1−xVI2 alloy at x = 0.5. Recently, the I2-II-IV-VI4 compound, especially Cu2-II-IV-VI4 quaternary semiconductors, has been studied both experimentally and theoretically due to its optimum value of band gap (≈1.5 eV) and high value of absorption coefficient (≈104–105 cm−1) [3,4,5,6,7]. However, in the literature, the studies of Cu-II2-III-VI4 quaternary semiconductors, for example, CuCd2InTe4, CuTa2InTe4, AgZn2InTe4, and CuTa2InTe4, are synthesized either in stannite or cubic structure and information on their various properties is comparatively limited [8,9,10,11,12,13,14,15,16,17]. Using many possible atomic variations, these emerging chalcogenide groups open a new era for finding materials with suitable characteristics. A cation cross-substitution of ternary and binary compounds has been studied by Chen et al. [8]. They evaluated the structural stability of various crystal phases and found that the stannite and kesterite structures have higher energy compared to the wurtzite structure after relaxation. Further, the crystalline quaternary chalcogenides CuZn2InTe4 and AgZn2InTe4 compounds have also been reported by Shi et al. [18]. Although semiconducting chalcogenides CuZn2AlSe4 are explored through cross-substitution, the total energy calculation gives the information of material properties, shifting of energy band and the conductivity of the material [19]. Moreover, a group of Cu2ZnAS4-x (x = 0.5 ± 0.3) and CuZn2AS4 (A = Al, Ga, In) nanocrystals are synthesized in wurtzite phase via hot injection method [9]. It has been found that the compounds have band gap values of range ~ 1.20–1.72 eV in the visible region and have high absorption values, which indicate that these compounds offer decent optical properties to be used as inexpensive and nontoxic active layers in photovoltaic applications. The structural stability, structure, composition, and band structure of CuZn2AlS4 have been reported earlier [10]. However, there is no systematic theoretical investigation to elucidate the structural, optoelectronic, and band structure properties of stannite and kesterite phases of CuZn2InS4 and CuZn2GaS4 compounds.
In the present report, we have studied the optoelectronic and structural properties of the kesterite (KS, space group I\(\overline{4}\)) and stannite (SS, space group I2 m)-type quaternary chalcopyrite compounds CuZn2MS4 (M = In and Ga) (Fig. 1(a) and (b)) via full-potential augmented plane wave method (FP-LAPW) as implemented in the Wien2k code. The band structure with partial and total density of states (DOS) is evaluated from optimized structural parameters at the lowest energies. The absorption spectra, dielectric function, and refractive index are reported and discussed. In addition, we have investigated the partial and total DOS of kesterite structure of CuZn2InxGa1-xS4 (where x = 0.5) to explore the transition from CuZn2InS4 to CuZn2GaS4. Our systematic studies could provide valuable knowledge of this quaternary chalcogenide group and enable to compare it with the related and vastly studied quaternary group of materials.

Crystal structures of (a) kesterite structure (KS) and (b) stannite (SS) structure of CuZn2GaS4, respectively
2 Computational methodology
To study the phase stability from total energy calculation and the electronic structure of CuZn2MS4 (M = In and Ga) compounds, the scalar-relativistic full-potential augmented plane wave method (FP-LAPW) is employed via WIEN2k code [20]. In this computational work, core and valence subsets split from the basis set located in the muffin–tin (MT) sphere. The core state contribution comes mainly from the spherical part of the potential where the charge density is spherically symmetric. On the other hand, the scalar-relativistic approach is applied for the valence states, although the full relativistic method with full relaxation is employed for the core states. Here, the valence states orbital configuration are Cu (3p6, 4s2, 3d9), Zn (3d10, 4s2), In (4d10, 5s2, 5p1), Ga (3d10, 4s2, 4p1), and S (3s2, 3p4). All these orbits are expanded within a potential in spherical harmonics. The Perdew–Burke–Ernzerhof (PBE) [21] potential in generalized-gradient approximation (GGA) is applied for crystal structure optimization, optimization of atomic positions, and defect formation energy evaluation. To avoid the charge leakage inside the core state, we have optimized the values of MT radii of all the atoms. Here, in connection with default values of the code and with respect to the computer time, RMT has been chosen. Generally, for doing faster calculations, radii can be taken as large as possible considering possible structural changes like force minimization or lattice parameters. So, we are able to perform all calculations of different properties with the same value of RMT. In our calculation, the MT radii for Zn, Cu, In, Ga, and S are defined to be 2.22, 2.20, 2.30, 2.28, and 1.90 a.u., respectively. The core and valence state separation is considered in the form of cutoff energy with a value of 6.0 Ry. The self-consistent convergence of the simulations set approximately in the range of 10−5 Ry. Further, the expansion of the basis is taken with RMT × KMAX equal to 7, where RMT represents the smallest atomic MT sphere radius and plane wave cutoff of k-vector defined as KMAX. The RMT set in a way that converges with the number of plane waves (PWs) remains the same for the smallest-sized atom as well as the other atoms of the compound. The magnitude of charge density (Gmax) in Fourier expansion is equal to 12 (Ry)1/2. In the calculation, the Brillouin zone integrations are performed with the tetrahedral method [22] with a 4 × 4 × 4 division k-point. To study the density of states (DOS) of CuZn2InxGa1-xS4 (where x = 0.5), we have structured a supercell from optimized kesterite CuZn2InS4 structure and optimized the lattice parameters of the system.
The absorption coefficient, α(ω), together with the real and imaginary part of the dielectric function [ε (ω) = ε1(ω) + iε2(ω)] is also analyzed using the following formulas [23]
where e and m represent the electronic charge and mass, respectively, i is the initial state, j is the final state, M represents the dipole matrix, Ei indicates ith state of the energy of electron, and fi is the Fermi distribution function for the ith state.
The real part contribution of dielectric function [ε1(ω)] is extracted from the Kramers–Kronig relation [24]
where ω is the frequency, ω’ represents the variable of integration, and p defines the principal value of the integral. The absorption coefficient [25] can be obtained by using the formula given as.
3 Results and discussion
3.1 Total energy
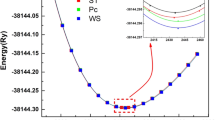
The kesterite and stannite phases of CZMS are found to be more stable compared to the wurtzite phase. The total equilibrium energy of wurtzite, kesterite, and stannite crystal phases are checked. The variation of total energy with the volume of the compound is shown in Fig. 2. In Fig. 2(a) and (b), both the vertical axes represent negative total energy values which indicate that the compounds are thermodynamically stable. From Fig. 2(a) and (b), we can see that kesterite phase has lower energy than that of stannite and wurtzite phases. This confirms that kesterites are more stable as compared to stannite and wurtzite counterparts. The equilibrium lattice constant is evaluated from the volume with the corresponding value of lowest energy. The Murnaghan’s equation of state, as given below, is used to calculate the lattice constants by fitting the calculated total energy.
where E0 represents the minimum energy at T = 0 K with volume V, B0 indicates the bulk modulus at the equilibrium volume, and V0 and B0' are the pressure derivative of bulk modulus at the equilibrium volume.

(a) represents volume vs. total energy for wurtzite, kesterite, and stannite structure of CuZn2InS4, and (b) represents volume vs. total energy for wurtzite, kesterite, and stannite structure of CuZn2GaS4
The optimized lattice parameters are evaluated as a = 5.529 Å, c = 11.058 Å for KS-CuZn2InS4; a = 5.533 Å, c = 11.066 Å for SS-CuZn2InS4; a = 5.417 Å, c = 10.833 Å for KS-CuZn2GaS4; and a = 5.421 Å, c = 10.842 Å for SS-CuZn2GaS4.
3.2 Density of states (DOS) and band structure
To analyze the electronic behavior of the quaternary compounds, the band structure and DOS of all the compounds in KS and SS structures are plotted in Figs. 3 and 4, respectively. Figure 3(a) represents the total and partial DOS of kesterite CuZn2InS4 (i.e., KS-CuZn2InS4), whereas the total and partial DOS of stannite CuZn2InS4 (i.e., SS-CuZn2InS4) are shown in Fig. 3(b), the total and partial DOS of kesterite CuZn2GaS4 (i.e., KS-CuZn2GaS4) are depicted in Fig. 3(c), and the total and partial DOS of stannite CuZn2GaS4 (i.e., SS-CuZn2GaS4) are displayed in Fig. 3(d). Here, we have shown the total DOS (represented by black color) together with the partial DOS of Cu-s and Cu-p orbitals (green and orange color, respectively), In-s and In-d orbitals (sky and pink color, respectively), Zn-s orbital (blue color plot), and S-s and S-p orbitals (green and red color, respectively). It is observed that the band gap decreases with increasing atomic number of group-III elements within the compound. As the conduction band minimum (CBM) is composed of group-III-s and anion p states, the band gap is reduced from Ga to In. From the DOS plots, it is observed that the valence band maximum (VBM) is coming from the Cu 3d and anion p states, as plotted in Fig. 3(a)–(d). Generally, the KS structure is more stable and has a lesser band gap as compared with SS structure of the material. Also, it shows that all the structures are direct-band semiconductors. Further, it is observed that going from Ga to In, the bonding and antibonding states of p−d orbitals overlap more. For all the compounds, it is clear from DOS plots that the VBM state leads by the antibonding p−d coupling between the group-I (i.e., Cu) and group-VI (i.e., S) atoms, while CBM states are dominated by the antibonding coupling between the group-III (i.e., Ga or In) s state and VI (i.e., S) s and p states. Therefore, a very little shifting of VBM occurs (as shown in total DOS plot at energy range 0 to −2.5 eV), and due to the remarkable downshifting of CBM from In to Ga, the band gap value decreases. In Fig. 4, the band structure is plotted for further understanding of the electronic properties. Here, Fig. 4(a) and (b) shows the band structure of CuZn2InS4 for kesterite (i.e., KS-CuZn2InS4) and stannite (i.e., SS-CuZn2InS4) structure, respectively, whereas Fig. 4(c) and d reflects the band structure of CuZn2GaS4 for kesterite (i.e., KS-CuZn2GaS4) and stannite (i.e., SS-CuZn2GaS4), respectively. It also shows the direct-band nature (i.e., Γ to Γ transition) of CZMS, as the conduction band minima and the valence band maxima are aligned in similar momentum positions and are located at high symmetry Γ-point. This is qualitatively similar to the reported results of CuZn2GaS4 [9] and CuZn2AlS4 compounds [10].

Partial density of states (DOS) and total DOS of (a) KS-CuZn2InS4, (b) SS- CuZn2InS4, (c) KS-CuZn2GaS4, and (d) SS-CuZn2GaS4. The ‘KS’ represents kesterite structure, and the ‘SS’ represents stannite structure (color figure online)

Band structure spectra for (a) KS-CuZn2InS4, (b) SS- CuZn2InS4, (c) KS-CuZn2GaS4, and (d) SS-CuZn2GaS4, respectively. The ‘KS’ represents kesterite structure, and the ‘SS’ represents stannite structure
Figure 5 depicts the overall total DOS together with the partial DOS of Cu-s, Cu-d, Zn-s, In-s, d, Ga-s, Ga-d, S-s, and S-p orbitals of CuZn2InxGa1-xS4 (where x = 0.5) in KS structure. The calculated band gap value is 1.52 eV. The relative band gap of CuZn2InxGa1-xS4 decreases in comparison with CZGS because of the downshifting of the conduction band side. This occurs due to the larger ionic radius of In compared to Ga. The trend of DOS, however, is similar for CZGS and CZIS structures. Around the Fermi energy (EF), the d orbital of Cu is hybridized with S-p in the edge of the valence band. Also, the admixture of Cu-d and the d-orbital of In and Ga hybridizes with S-p states in energy range of −6 to 0 eV. On the conduction band side, the major contribution takes place with In-s, Ga-s and S-p states.

Partial density of states (DOS) and total DOS of kesterite structure (KS) of CuZn2Ga0.5In0.5S4
3.3 Optical properties
The dielectric function of all the structures is depicted in Fig. 6 with energy from 0 to 10 eV. Here, Fig. 6(a) represents the real part of dielectric function, whereas imaginary contribution of dielectric function is reflected in Fig. 6(b). We have found that the dielectric function has quite a similar trend over a broader energy range. The real part of the dielectric function, ε1(ω), can be defined by the dispersion of the incident photons of the material, ε1(ω), whereas the quantity of the imaginary part of the dielectric function, ε2(ω), measured by the absorption of energy. In Fig. 6(a), for both the KS and SS structure of CuZn2InS4 and CuZn2GaS4, the peak is around 2 eV and occurs due to the transition of S-3p and Zn-4 s orbitals. In Fig. 6(b), the spectrum distribution follows the same sequence of the density of states.

Dielectric function spectra ε(ω) = ε1(ω) + iε2(ω) of CuZn2InS4 and CuZn2GaS4 in KS and SS structures. The caption (a) represents the real part of dielectric function, ε1(ω), and the caption (b) represents the imaginary part of dielectric function, ε2(ω). The ‘KS’ represents kesterite structure, and the ‘SS’ represents stannite structure
Another key parameter for defining the optical properties of the material is the refractive index. This is related to the polarizability of ions associated with the local field inside. The refractive indices are evaluated for both the kesterite and stannite crystal phases of CuZn2MS4 (M = In and Ga) compound from simulation (shown in Fig. 7(a)) and are obtained as 2.58, 2.60, 2.66, and 2.67, respectively. From the figure, we have found that the estimated refractive index for all the crystal phases in the visible region increases and attains the UV region with its maximum values. It further decreases with the increase in energy. Since with the increasing value of refractive index, band gap of the material decreases, this property helps the solar cell to respond more in the higher wavelength region of the solar spectrum.

(a) Refractive index, n(ω) and (b) absorption coefficient, α(ϵ) spectra of CuZn2InS4 and CuZn2GaS4 in KS and SS structures. The inset figure within (b) shows the Tauc plots of all structures, respectively. The ‘KS’ represents kesterite structure, and the ‘SS’ represents stannite structure
The full spectrum of the calculated absorption coefficient for both compounds is shown in Fig. 7(b). From Fig. 7(b), it can be realized that in the visible region, the value of the absorption coefficient is higher than 104 cm−1, which makes CuZn2InS4 and CuZn2GaS4 promising materials as highly efficient absorbers for photovoltaic applications. The band gap values (Eg) of CuZn2MS4 (M = In and Ga) compound are estimated from the Tauc’s plot. In this plot, we considered the absorption coefficient (α) as α2 ∝ (hν−Eg), where hν is the photon energy. (υ is the frequency, and h is the Planck’s constant.) Considering scissor correction, the calculated optical band gaps using PBE are 1.44 eV, 1.48 eV, 1.54 eV, and 1.57 eV for KS-CuZn2InS4, SS-CuZn2InS4, KS-CuZn2GaS4, and SS-CuZn2GaS4, respectively (inset figure of Fig. 7(b)). The absorption starts in the energy of the band gap region. From Fig. 7(b), we can also observe that the peak of the absorption is located in the energy region of 1.4–1.57 eV. This is due to the transition of the charge carriers from Cu-d and anion-p (i.e., S-p) hybridized states to the empty states in the CB.
4 Conclusions
In summary, a systematic investigation of the structural, optical, and electronic properties of CuZn2InS4 and CuZn2GaS4 in the KS and the SS phases is carried out using the density functional scalar-relativistic full-potential linear augmented plane wave method. The density of states, band diagram, and optoelectronic properties, such as absorption coefficient, dielectric function, and refractive index, are reported in detail. From the total energy calculations, we have found that the kesterite and the stannite crystal phases are more stable compared to the wurtzite crystal phase. In the case of heavier group-III atoms, the bonding and antibonding states of the compounds overlap more. Moreover, the optical spectra shifted from In to Ga toward the lower energy region. Our present study shows that these materials are very promising to be used as absorber materials in the context of optoelectronic including photovoltaic (PV) applications. The present systematic study will further aid for the experimental quest on the material structure of this novel class of quaternary chalcogenides.
Data and code availability
Not applicable.
References
C H L Goodman J. Phys. Chem. Solids 6 305 (1958)
B R Pamplin J. Phys. Chem. Solids 25 675 (1964)
S Palchoudhury, K Ramasamy and A Gupta Nanoscale Adv. 2 3069 (2020)
W Shi, A R Khabibullin and M Lilia Adv. Theory Simul. 2000041 1 (2020)
A S Najm, A Al-Ghamdi, M T Amin, A A Ghamdi, H Moria, A M Holi, A M Abed, A A AL-Zahrani, K Sopian, B Bais and A J Sultan Sci. Rep. 13 15418 (2023)
A Ghosh, R Thangavel and M Rajagopalan J. Mater. Sci. 50 1710 (2015)
A Ghosh, R Thangavel and M Rajagopalan J. Mater. Sci. 48 8259 (2013)
S Chen, X G Gong, A Walsh and S H Wei Phys. Rev. B 79 165211 (2009)
A Ghosh, S Palchoudhury, R Thangavel, Z Zhou, N Naghibolashrafi, K Ramasamy and A Gupta Chem. Commun. 52 264 (2016)
A Ghosh, R Thangavel and A Gupta New J. Chem. 40 1149 (2017)
O P Ojo, L Ma, W D C B Gunatilleke, A F May, L M Woods and G S Nolas Inorg. Chem. 62 16114 (2023)
A Ghosh, D K Chaudhary, A Mandal, S Prodhan, K K Chauhan, S Vihari, G Gupta, P K Datta and S Bhattacharyya J. Phys. Chem. Lett. 11 3 591 (2021)
O P Oja, W D C B Gunatilleke, H Poddig, H Wang, J Martin, D J Kirsch and G S Nolas Dalton Trans. 50 17611 (2021)
G Halder, A Ghosh, S Parvin and S Bhattacharyya Chem. Mater. 31 161 (2019)
D K Chaudhary, A Ghosh, R Thangavel and L Kumar Thin Solid Films 649 202 (2018)
L Ma, W Shi and L M Woods J. Alloy. Compd. 969 172399 (2023)
A Ghosh, R Thangavel and A Gupta J. Alloy. Compd. 694 394 (2016)
W Shi, A R Khabibullin, D Hobbis, G S Nolas and L M Woods J. Appl. Phys. 125 155101 (2019)
M V Jyothirmai and R Thapa ACS Omega 7 31098 (2022)
P Blaha, K Schwarz, G K H Madsen, D Kvasnicka, J Luitz, R Laskowski, F Tran, L D Marks, WIEN2k an Augmented Plane Wave + Local Orbitals Program for Calculating Crystal Properties, Karlheinz Schwarz, Techn. University at Wien, Austria, ISBN 3-9501031-1-2. (2023)
J P Perdew, K Burke and M Ernzerhof Phys. Rev. Lett. 77 3865 (1996)
H J Monkhorst and J D Pack Phys. Rev. B 13 5188 (1976)
C M I Okoye J. Phys. Condens. Matter 15 5945 (2003)
B Amin, M I Ahmad, S Maqbool, G Said and R Ahmad J. Appl. Phys. 109 023109 (2011)
S Ozaki and S Adachi J. Appl. Phys. 75 7470 (1994)
Acknowledgements
A. Ghosh acknowledges the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), Government of India (New Delhi). Also, A. Ghosh would like to thank SR University for providing the SEED grant to support the present work.
Author information
Authors and Affiliations
Contributions
AG and RT conceived and directed the project. AG did the simulation work and analyzed the data. AG and RT participated in the preparation of the manuscript and commented on its content.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ghosh, A., Thangavel, R. Structural stability, electronic band structure, and optoelectronic properties of quaternary chalcogenide CuZn2MS4 (M = In and Ga) compounds via first principles. Indian J Phys (2024). https://doi.org/10.1007/s12648-024-03162-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12648-024-03162-y