
Abstract
Materials with versatile physical properties are essential for contemporary physical society, especially in electronics, renewable energy, transportation, medicine, and more. This intact capability holds the potential for a revolutionary shift towards environmentally friendly renewable energy sources. Consequently, the exploration of materials that encompass multiple functions becomes highly imperative. This study is concentrated on comprehending the physical characteristics of elastic and optoelectronic materials to propose novel, highly efficient materials suitable for photovoltaic device applications. Within this paper, the fundamental study of fluoroperovskite properties in the context of density functional theory is undertaken, employing the full potential linearized augmented plane wave approach. Specifically, fluoroperovskite ZnXF3 (X = Sr, Ba) is scrutinized concerning its structural, electronic, optical, and elastic attributes. The optimized crystal structural parameters for both compounds are determined as 4.41 Å for ZnSrF3 and 4.52 Å for ZnBaF3, employing the Birch-Murnaghan fitting approach for the unit cell energy versus unit cell volume. All fundamental physical properties are subsequently calculated using these optimized lattice constants. To address strongly correlated electron systems, the recently developed Modified Beck-Johnson potential is employed in this research. The tolerance factor “τ” is computed for both materials, yielding values of 0.98 for ZnSrF3 and 0.86 for ZnBaF3, affirming the structural stability of these perovskite crystal structures. The analysis of electronic properties reveals that both compounds exhibit a metallic behavior, for ZnXF3 (X = Sr, Ba) fluoroperovskites. Furthermore, the research explores the potential of these selected compounds by computing their optical properties within the energy range of 0–14 eV for incident photons, with a focus on potential optoelectronic applications. Additionally, mechanical properties for both materials are assessed using the IRelast package, with results indicating that ZnXF3 (X = Sr, Ba) fluoroPerovskites are mechanically stable, resistant to abrasion, ductile, and anisotropic. The precision and accuracy of the reported findings provide strong support for the potential applications of zinc-based ZnXF3 (X = Sr, Ba) fluoroperovskites in photovoltaic and modern semiconductor industries.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Computational science and its resources have evolved steadily over the last few decades. As a result, simulation tools can now perform thorough and precise computations on a growing and complex array of materials. For instance, understanding the effect of interacting atoms that may be triggered by an external field. Computations can be performed through different methods, from classical approaches to quantum physics [1,2,3]. For example, the classical mechanical technique is based on a semi-empirical framework and requires extensive input parameters to produce nearer experimental results. On the other hand, the first principle method is based on quantum mechanical theories, which can treat smaller unit cells, and no experimental data is required to carry out the computations. Therefore, the DFT, which is based on the quantum mechanical simulation of periodic systems, has been largely employed for the investigation of various characteristics of different materials over the past few decades [4,5,6].
Perovskites are among the compounds that have been widely investigated through the DFT [7,8,9]. Calcium Titanate (CaTiO3) was the first Perovskite compound that was found in 1839 by Gustav Rose in the Ural Mountains of Russia and then named after Russian mineralogist Lev Perovski. Perovskites (ABX3) are a broad class of substances, where A is called an organic cation, B is considered a divalent metallic cation, and X is a halide anion. They exhibit electrical properties ranging from insulators to superconductor, which depends on the elements A, B, and X. Perovskites are commonly used in 3D printers, solar cells, light-emitting nanoantennas, and X-ray detectors. Also, they have wider optoelectronic applications due to their high radiative performance, lengthy carrier diffusion lengths, higher optical absorption, tolerance for defects, and ability to tune the band gap. Moreover, they are used in LEDs, lasers, photodetectors, and other active devices [10,11,12]. A wide variety of substances are found in the Perovskites family, ranging from nitride to halides. Many studies have been conducted on cubic Perovskites with different compositions and structures [13,14,15]. One of the most substantial Perovskites is known as the fluoroperovskites (ABF3), where elements A represents alkali metals, and B represent alkaline earth metals. Fluoroperovskites compounds have attracted a lot of attention due to their use in a variety of technological fields, including lithography, optoelectronic, semiconducting devices, piezoelectricity, and thermoelectricity [16]. The fluoroperovskites have a large band gap, making them particularly desirable materials with great transparency in the deep and vacuum ultraviolet spectrum and infrared regions [17,18,19,20]. Rigorous investigations have been conducted to anticipate the physical, optoelectronic, and mechanical properties of fluoroperovskites for applications in high-energy scenarios [21,22,23,24,25,26,27,28]. Rahman et al. [29] employed the FP-LAPW method to calculate the Mg-based fluoroperovskite XMgF3 (X = Ga, Al, and In) compounds. The WIEN2k method was used to investigate the various physical properties of these substances. The investigation discovered that, within reasonable limits, the structural features of these compounds matched prior literature results. GaMgF3 and InMgF3 compounds were revealed to be anisotropic, ductile, and mechanically stable, with GaMgF3 being stiffer and more compressible than InMgF3. The mechanical instability of the third chemical under investigation, AlMgF3, was also established. AlMgF3 and InMgF3 showed semiconductor electronic band structures with an indirect (M—X) band gap of 2.49 eV and 2.98 eV, respectively, however, GaMgF3 was discovered to be an insulator. The findings also indicated that the atoms' bonding force was largely ionic, with barely a hint of covalent nature. Finally, the research found that these chemicals can be employed to build electrical devices.
Therefore, this study aims to research the structural, electronics, optical, and elastic properties of Zn-based fluoroperovskite using DFT and the full potential linear augmented plane wave (FP-LAPW) approach.
1.1 Computational approach and modeling
The cubic fluoroperovskite structural compounds, known as ABF3, are characterized by the Pm-3 m (#221) space group. Our focus lies in the examination of Zn-based fluoroperovskites, specifically ZnXF3 (X = Sr, Ba), within the larger group of fluoroperovskite compounds. These materials adopt a cubic structure, with atoms located at specific coordinates: (0, 0, 0) for X, (1/2, 1/2, 1/2) for Zn, and (0, 1/2, 1/2) for F. For our investigations, we employ an ab initio FP-LAPW method, a density functional theory-based approach, implemented through the WIEN2K [30] software. Our primary objectives include studying optimized lattice constants, ground state volume, and energy. To achieve this, we fit the energy-volume relationship using the Murnaghan equation of state. The improved lattice constants resulting from this process are used to create a new structure, with self-consistent iterations taking into account the exchange–correlation effects [31] via a generalized gradient approximation. The TB-mBJ (Tran–Blaha modified Becke–Johnson) exchange potential is utilized to explore the electronic and optical properties of Zinc-based ZnXF3 (A = Sr, Ba) fluoroperovskite compounds. This lays the groundwork for further computations, such as elastic constants, band gap energy, density of states, and optical properties. To ensure our findings align with reported experimental results, we carefully select parameters, including the muffin-tin sphere radius (RMT) and the plane wave cutoff (Kmax). In the context of structural optimization, integrals across the Brillouin zone are computed, with up to 2000 k-points calculated throughout the entire Brillouin zone.
2 Results and discussion
2.1 Structural attributes
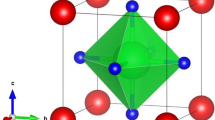
The cubic crystal structure of Zn-based compounds, specifically ZnXF3 (where X = Sr, Ba), is depicted in Fig. 1. This structure belongs to the space group Pm-3 m (#221). In this arrangement, X is positioned at the Wyckoff coordinates (0, 0, 0), and Zn is located at (0.5, 0.5, 0.5). Additionally, the F atoms occupy positions at (0.5, 0, 0.5), (0.5, 0.5, 0), and (0, 0.5, 0.5). The lattice constants for these materials, namely ZnSrF3 and ZnBaF3, have been determined using the TB-mBJ method and are found to be approximately 4.41 A0 and 4.52 A0 for both compounds.

Optimized unit cells (Pm-3 m (#221)) of ZnXF3 (X = Sr, Ba)
The phase of a perovskite material can be characterized using a parameter known as the tolerance factor. When the tolerance factor falls within the range of 0.9–1.0, the perovskite material is classified as cubic [32]. In the current scenario, the calculated tolerance factor is determined to be 0.98 for ZnSrF3 and 0.862 for ZnBaF3. Consequently, the crystal structure of all these perovskite materials is indeed cubic. The cubic crystal structure of ZnXF3 (X = Sr, Ba) is illustrated in Fig. 1.
Examining crystal structural properties involves minimizing the total energy of cubic ZznXF3 compounds (X = Sr, Ba) as volume changes. The Murnaghan equation of state (EOS) [33, 34] is employed to fit the energy-volume data, resulting in equilibrium lattice parameters, bulk modulus, pressure derivative, ground state energy (E0), and the corresponding ground state volume. The analysis of the optimization curve reveals a consistent pattern across all investigated substances. Figure 2 illustrates an initial energy decrease with increasing volume, culminating in the achievement of ground-state energy, representing the lowest attainable energy level. Examination of the structural characteristics of the chosen materials affirms that both possess a stable cubic crystalline structure, with detailed structural attributes outlined in Table 1.

Optimization curve of fluoroperovskites compounds of ZnXF3 (X = Sr, Ba)
2.2 Electronic properties
2.2.1 Band structure
In Fig. 3, we observe the computed band structures (BS) of ZnXF3 (X = Sr, Ba) within the Brillouin zone, covering higher symmetry directions. In these plots, there is an overlap between the valence band (VB) and conduction band (CB) states. However, the shapes of the energy bands differ due to the presence of Sr and Ba cations, among others, within the lattice. These cations possess distinctive attributes that play a role in distinguishing and characterizing the electronic structure of the substances under research. Every cation has a distinct electronic configuration because of variations in valence electrons, leading to differing behaviors of the materials’ cations. Through these theoretical investigations of ZnXF3 (X = Sr, Ba), it becomes apparent that these compounds exhibit metallic characteristics. This is attributed to the overlap of the valence and conduction bands at the Fermi level, and notably, there is no discernible forbidden energy gap.

Calculated band structure of ZnXF3 (X = Sr and Ba) compounds
2.2.2 Density of states (DOS)
In this section, the primary focus is on the exploration of potential electron states within the system. The Density of States (DOS) serves as a comprehensive tool for delineating the material's intricate structure, shedding light on the roles played by states within both the valence and conduction bands. The Fermi level, represented by EF and indicated by a dashed line in Fig. 4, acts as the demarcation point. The region to the left of the Fermi level constitutes the valence band, while the area to the right signifies the conduction band. In Fig. 4(a), we are presented with a graphical representation of the partial density of states (PDOS) and total density of states (TDOS) for ZnSrF3. This depiction offers a holistic view of the overall density of states for the entire compound and dissects the contributions of individual atoms such as Zn, Sr, and F to the overall DOS. The energy range is effectively divided into two classes: from − 4 to 0 eV (constituting the valence band) and from 0 to 7 eV (representing the conduction band), based on the PDOS distribution. It becomes evident that the major contribution to the DOS in the conduction band (within the 0–7 eV range) is attributed to Zn–s, Zn-d, and Zn-f states. Turning our attention to Fig. 4(b), we observe the TDOS and PDOS for ZnBaF3. Remarkably, on the right side of the Fermi level, the most prominent peaks in the TDOS emerge at 4 eV, with Zinc (Zn) being the primary contributing element. This signifies that the TDOS of ZnBaF3 is primarily influenced by states originating from the Zn atom. It is evident from Fig. 4 that no significant peaks are observed on the left side of the Fermi level, and (Zn) alone is responsible for the entirety of the contribution. The most significant DOS peaks in the conduction band (above the Fermi level) are notably located at 4 eV, further emphasizing that the TDOS of ZnBaF3 is predominantly shaped by the states associated with the Zn atom.

Total density of states of ZnSrF3 and ZnBaF3 using TB-mBj approximation
DOS represents the number of electronic states per unit of energy at each energy level. TDOS is the sum of the DOS contributions from all atoms in the material. Both the compounds are metallic systems with CB-VB overlap, a continuous distribution of states is observed across a broad energy range. The DOS and TDOS plots show a non-zero density of states in the region where CB and VB overlap, indicating the availability of states for electrons to move freely. This demonstrates that electrons have a wide range of energy levels available for movement, contributing to the metallic conductivity of the material. Since Zn, Sr, Ba, and F are the constituent elements, their atomic orbitals contribute to understanding the electronic structure near the VB and CB. The dominance in the valence band arises from the 2p and 2 s states of fluorine (F) atoms, whereas zinc (Zn) atoms contribute significantly through their 3d states. The uppermost valence band primarily consists of fluorine (F) 2p states, mainly concentrated in the energy range of − 2–0 eV. Additionally, Zn 3d states form bands extending from − 2–− 0.5 eV, as illustrated in the inset of Fig. 5. Conversely, the conduction band is characterized by the predominant influence of the 5 s and 4d states of strontium (Sr) and barium (Ba) atoms, consistent with previous studies [35].

Real and imaginary components of the complex dielectric function of ZnXF3 (X = Sr and Ba)
2.3 Optical properties
Analyzing the optical properties of composites, including their dielectric function, refractive index, extinction coefficient, conductivity, absorption, reflectivity, and energy loss function is a method for achieving a more profound understanding of the interaction between light and matter at the interface. Employing the following equations, one can determine various optical characteristics such as reflectivity, refractive index, extinction coefficient, and absorption coefficient [36, 37].
2.3.1 Dielectric function
The complex dielectric function, denoted as ε(ω) = ε1(ω) + iε2(ω), is a crucial tool used to explain the optical behavior of substances that absorb light [38]. In this context, the spectra of ε1(ω) and ε2(ω) for ZnSrF3 and ZnBaF3 are visually represented in Fig. 4(a) and (b). For the static dielectric constant ε1(ω), the values are 4.2 and 3.1 for ZnBaF3 and ZnSrF3, respectively. These ε1 (ω) values initially increase with higher photon energy, reaching peaks at 1.87 eV for ZnSrF3 and 1.73 eV for ZnBaF3, before gradually declining. The peak values of the real components are 3.57 and 3.45, while the peak values of the imaginary components occur at 3.7 (at 2.14 eV) and 3.2 (at 2.22 eV) for ZnSrF3 and ZnBaF3, respectively.
2.3.2 The refractive index and extinction coefficient
The refractive index, denoted as η(ω), is a crucial parameter for quantifying light refraction and plays a significant role in various photoelectric applications. It is characterized by both real and imaginary elements. The real component, representing the refractive index, is denoted as η(ω), while the imaginary part is designated as K(ω) and signifies the extinction coefficient, as shown in Fig. 6(a). ZnBaF3 exhibits a constant refractive index η(0) of 1.7, while ZnSrF3 registers around 2. The refractive index is larger than one because photons experience delays as they traverse a material as a result of electron interactions. In essence, the higher the refractive index of a material, the greater the delay experienced by photons during their passage through it. In general, any technique that raises the electron density of a material also elevates the η(ω) (refractive index), which is closely associated with bonding. Ionic compounds tend to have a lower refractive index compared to covalent compounds. In covalent bonding, electrons are predominantly shared among ions, leading to greater electron dispersion throughout the structure, and these electrons have interactions with the incident photons, causing delays. As for the imaginary part, K(ω), which represents the extinction coefficient, as depicted in Fig. 6(b), the figures reveal that the highest absorption in the investigated substances occurs at 2.1 eV for ZnBaF3 and 2.2 eV for ZnSrF3.These absorption peaks are associated with specific electronic transitions or energy levels within the materials. The energy values (2.1 eV and 2.2 eV) represent the photon energies at which the materials exhibit the highest absorption, indicating the onset of significant electronic transitions or absorption bands in these energy ranges.

The refractive index η(ω) and extinction coefficient k(ω) for Pd-based ZnXF3 (X = Sr, Ba) compounds
The extinction coefficient has a non-zero value in the energy range from 0 to 0.5 eV, which indicates that the material has absorption or scattering properties within that energy range. This means that the material is capable of absorbing or interacting with photons of energy within this range. A non-zero extinction coefficient suggests that the material is not transparent to light in the given energy range, and some fraction of incident light is absorbed or scattered.
2.3.3 Optical conductivity and absorption coefficient
The optical conductivity, denoted as σ(ω), characterizes the flow of electrons induced by using an electromagnetic field. In Fig. 7, we can observe the analysis of σ(ω) for our ZnBaF3 (X = Sr, Ba) compounds. Within the optical conductivity range spanning from 0 to 14 eV, there is a notable surge in electron conduction at the critical threshold of 10 eV for both substances, reaching a peak of 2400 for ZnBaF3 and 2200 for ZnSrF3. The absorption coefficient, denoted as I(ω), is influenced by both components of the dielectric function. It quantifies the amount of light absorbed by a material per unit length and is indicative of interactions between electrons and photons, including inter-band and intra-band transitions. Figure 7 makes it evident that these research substances exhibit substantial absorption characteristics. The threshold point signifies the onset of immediate electromagnetic radiation absorption by a compound. For ZnBaF3, the active absorption is notably recorded at 80 at 10.25 eV, while for ZnSrF3, it registers at 70 at 10.6 eV.

Optical conductivity and Absorption Coefficient of ZnXF3 (X = Sr, Ba)
2.3.4 Reflectivity R(ω)
Reflectivity is a crucial property of substances, allowing us to evaluate how effectively a material reflects incident energy. It helps us understand the material's ability to bounce back incoming radiation. We can determine the reflectivity constant by examining the graph presented in Fig. 8. This graph, derived from the dielectric function and depicting R(ω), covers the entire energy spectrum from 0 to 14 eV, as visually represented in Fig. 8. The zero-frequency reflectivity, represented as R(0), is measured at 0.12 for ZnBaF3 and 0.08 for ZnSrF3. As we increase the photon energy, the reflectivity also increases, reaching its peak at approximately 0.26 at 2.2 eV for PdBeF3 and around 0.25 at 2.2 eV for ZnSrF3.

Computed optical reflectivity R(ω) of ZnXF3 (X = Sr, Ba) compounds
2.3.5 Energy loss function
The interaction between electrons and material, resulting in inelastic processes, is intricately linked to several key factors. Among these factors, the energy-loss function plays a pivotal role in defining the potential for such inelastic scattering events. When we consider this in conjunction with energy-loss distribution and scattering angular distribution, these elements collectively provide valuable insights into the inelastic interactions of electrons within a material. Essentially, the energy-loss function furnishes information about how a solid reacts to an external electromagnetic disturbance. Remarkably, in both of these materials, the primary response to such an external disturbance occurs when the incident photon energy falls within the lower energy range. This trend is depicted in Fig. 9, which outlines the energy loss function spanning from 0 to 14 eV. In summary, Zn-based ZnXF3 (X = Sr, Ba) materials exhibit significantly elevated levels of optical conductivity, refractive index values, and energy loss functions, particularly at lower incident photon energies. These distinctive characteristics render these materials exceptionally well-suited for a wide array of contemporary optoelectronic industries, catering to a diverse range of device applications.

Energy loss Function of Pd-based fluoroperovskite compound ZnXF3 (X = Sr, Ba)
2.4 Elastic properties
The mechanical properties of Zinc-based fluoroperovskites, specifically ZnXF3 (X = Sr, Ba), are investigated by calculating elastic parameters. These elastic parameters play a crucial role in recognizing the types of bonding forces and the mechanical stability of the materials. To determine the elastic parameters C11, C12, and C44 by analyzing the relationship between strain and volume, as described in reference [39,40,41]. For cubic compounds, the Born stability principles are defined as C11 − C12 > 0, C11 > 0, C44 > 0, and C11 + 2C12 > 0 [42, 43].
The calculated elastic parameters, as displayed in Table 1, satisfy the criteria for Max Born stability. C11 characterizes resistance in the x-direction under linear compression, as detailed in references [44, 45]. Similarly, C44 provides information about the material's ability to resist shear distortion when subjected to shear stress. On the whole, C44 is associated with the material's hardness, as mentioned in reference [46]. With the knowledge of elastic constants, various elasticity-related parameters, including elastic anisotropy (A) and elasticity moduli such as bulk modulus (B), Young’s modulus (E), and shear modulus (G), can be readily derived using the following equations as outlined in reference [47]. The shear modulus G refers to a material property that describes its response to shear stress. It quantifies a material's resistance to deformation under shear forces. The shear modulus is denoted by the symbol G and is an important parameter in understanding a material's mechanical behavior. The symbol υ represents Poisson's ratio, a dimensionless quantity that characterizes the deformation behavior of a material. It is defined as the ratio of lateral contraction to longitudinal extension when a material is stretched or compressed. Poisson's ratio denoted by ν is a fundamental parameter in elasticity.
Bulk modulus (B) [47] plays a role in assessing a material's resistance to volume changes under pressure. This calculation can be easily conducted using the elastic constants found in Equations, Young's modulus (E) predicts a material's stiffness, while shear modulus (G) evaluates its ability to withstand permanent deformation under shear tension. G is a more subtle measure of hardness than B. All the computed elastic moduli are documented in Table 2. Notably, ZnSrF3 displays higher values for both shear modulus and Young's modulus compared to ZnBaF3, in line with its elevated C44 value, making ZnBaF3 the harder material. The evaluation of Pugh's ratio (B/G) [48, 49] is employed to determine the ductility or brittleness of a material, with values above 1.75 indicating ductility and values below 1.75 indicating brittleness. In the context of our study, both compounds are identified as ductile materials. Another critical parameter is the anisotropic ratio (A), which quantifies the level of a material's elastic anisotropy. In isotropic materials, A equals 1, while A not equal to 1 signifies anisotropy. The findings indicate A ≠ 1, confirming that the studied materials are indeed anisotropic. To assess the dynamic stability of the examined materials, we also calculated the shear modulus (G). The data presented in Table 2 reveal that the shear modulus for both compounds is greater than zero, indicating the dynamic stability of these materials. Additionally, we derived the Cauchy pressure to probe the bonding structure of the compounds. Positive and negative values of Cauchy pressure correspond to ionic and covalent bonding, respectively. The positive Cauchy pressure values for ZnSrF3 and ZnBaF3 confirm the presence of ionic bonding in these materials. To sum it up, the evaluation shows that the investigated compounds display mechanical ductility, anisotropy, toughness, and a significant resistance to fractures. These conclusions, drawn from their elastic properties, readily suggest their potential utility in diverse modern electronic technologies.
Similar to Pugh’s ratio, Poisson’s ratio (ν) categorizes crystalline materials as either brittle or ductile. A material is designated as brittle or ductile based on whether its Poisson's ratio falls below or exceeds the threshold of ν = 0.26 (ν < 0.26 = Brittle; ν > 0.26 = Ductile). For both ZnSrF3 and ZnBaF3, their Poisson's ratio values surpass the specified threshold, with ZnSrF3 having a value of 0.40 and ZnBaF3 having a value of 0.39. This confirms the ductile nature of both compounds. Furthermore, Poisson’s ratio provides insight into a crystal's resistance to shearing forces. A lower Poisson ratio suggests increased stability against shearing. Additionally, by examining Poisson’s ratio, one can anticipate whether the stability of a material is influenced by non-central or central forces. Typically, a material with a Poisson’s ratio falling within the range of 0.25–0.50 is classified as a central force crystal, indicating its stabilization by central forces. On the contrary, a material with a Poisson’s ratio exceeding the specified range is labeled as stabilized by non-central forces, termed a non-central force crystal [50,51,52]. The observed results indicate that both ZnSrF3 and ZnBaF3 fall into the category of central crystals, with Poisson’s ratios of 0.40 and 0.39, respectively.
We employed the Nose–Hoover heat bath scheme [53,54,55] in the ab initio molecular dynamics (MD) simulation, with a time step of 1 fs. To ascertain the thermodynamic stability of the discussed ZnXF3 (X = Sr, Ba), ab-initio finite temperature molecular dynamics (MD) simulations were conducted at 500 K for 8000 steps, using a time step of 1 fs and the Nose–Hoover heat bath scheme [56,57,58]. A supercell of 5 × 5 × 2 was employed to minimize periodic constraints. The total energy fluctuations of the compounds over simulation steps were monitored, revealing nearly constant average values. The atomic structures remained well-sustained throughout the simulation, as illustrated in Fig. 10.

The changes in the total energy as a function of simulation steps at 500 K for ZnXF3 (X = Sr, Ba). The structures are shown in the inset 8000 steps
Figure 10 demonstrates minimal energy fluctuations throughout the simulation, with both perovskite structures maintaining their original geometries without any discernible structural distortions.
The calculation of formation energy is a crucial step in understanding the stability and properties of chemical compounds [59,60,61,62]. The provided expression is employed to calculate the formation energies of the cubic-phase ternary compounds ZnSrF3 and ZnBaF3 halide perovskites. This calculation serves to confirm the structural stability of these complex compounds.
In the mentioned Equation-13, Hf represents the energy of formation, Etotal (ZnXF3) corresponds to the optimal total energy of ZnXF3 (X = Sr and Ba), EX denotes the energy of the ground state of the “Sr and Ba” atoms, EZn signifies the lowest energy state of the “Zn” atoms, and EF represents the lowest energy state of the fluorine atom. The formation energy for ZnSrF3 is -6.21 eV, while for ZnBaF3, it is -5.59 eV. This confirms the structural stability of both materials.
3 Conclusions
DFT calculations were carried out using the TB-mBJ approximations in the WIEN2K software to investigate the structural, optical, elastic, and electronic characteristics of fluoroperovskite compounds ZnXF3 (X = Sr, Ba). The outcomes regarding cohesive energy, tolerance factor, and structural optimization suggest these materials possess robust structural stability, indicating their potential for synthesis under laboratory equilibrium conditions. Our optimization process led to well-defined results, with the optimal volume corresponding to the energy minimum point. Key parameters at the ground state, including optimized lattice constants, bulk moduli, optimal volume, and energy, were determined. The optimized lattice parameters were found to be 4.41 Å for ZnSrF3 and 4.52 Å for ZnBaF3. The metallic nature of these substances was determined by the near convergence of the valence and conduction bands around the Fermi level. These perovskite materials were expected to exhibit uniform behavior at low energies, and their maximal conductivity was confirmed to occur in proximity to the Fermi level based on TDOS and PDOS graphs. Elastic property calculations using the IRelast package revealed that ZnSrF3 and ZnBaF3 are characterized as brittle, incompressible, anisotropic, and mechanically stable. Optical calculations demonstrated that the highest conductivity/absorptivity occurred in the low-energy range. Based on these findings, the potential applications of these materials in fields such as scintillation and various modern electronic devices are evident.
References
M Wang, C Jiang, S Zhang, X Song, Y Tang and H Cheng Nat. Chem. 10 667 (2018)
S Mu, Q Liu, P Kidkhunthod, X Zhou, W Wang and Y Tang Nat. Sci. Rev. 8 nwaa178 (2021)
Z Huang, P Luo, S Jia, H Zheng and Z Lyu J. Phys. Chem. Solids 167 110746 (2022)
Z Huang, P Luo, Q Wu and H Zheng J. Phys. Chem. Solids 161 110479 (2022)
J Wang, P Wang, W Chen, F Wan, Y Lu, Z Tang and Z Zhang Sens. Actuators B: Chem. 380 133350 (2023)
M Li, T Wang, F Chu, Q Han, Z Qin and M J Zuo IEEE Transact, Ind. Electr. 68 8777 (2021)
G C Papavassiliou and J Solid State Chem. 4 330 (1981)
G C Papavassiliou J. Mol. Struct. 79 395 (1982)
X Zhang, Y Tang, F Zhang and C Lee Adv. Energy Mater. 6 1502588 (2016)
A I L Ekimov, A L Efros and A A Onushchenkoet Solid State Commun. 56 921 (1985)
B Bakri, Z Driss, S Berri and R Khenta J. Phys. 91 1513 (2017)
H W Jang, S H Baek, D Ortiz, C M Folkman, R R Das, Y H Chu, P Shafer and J X Zhang Rev. Lett. 101 107602 (2008)
T L Phan, S G Min, S C Yu and S K Oh J. Magn. Mater. 304 e778 (2006)
A Moure, T Hungría, A Castro, J Galy and O Peña Chem. Mater. 22 2908 (2010)
J Chen, Z Zhang and H Lu Surf. Interfaces 33 102289 (2022)
J M García-Lastra, J Y Buzare, M T Barriuso, J A Aramburu and M Moreno Rev. B Condens Matter Mater. Phys. 75 155101 (2007)
C N Avram, M G Brik, I Tanaka and N M Avram Phys. B: Condens. Matter. 355 164 (2005)
S Korbel, M A L Marques and S Botti J. Mater. Chem. C. Mater. 4 3157 (2016)
M Husain, N Rahman, M Amami, T Zaman, M Sohail, R Khan, A A Khan, S A Shah Khan and A H Reshak Opt. Quant. Electr. 55 536 (2023)
Z H Fu, B J Yang, M L Shan, T Li, Z Y Zhu, C P Ma and W Gao Corrosion Sci. 164 108337 (2020)
A Habib et al. Materials 15 2669 (2022)
G Ayub et al. ACS Omega 8 17779 (2023)
M Husain, N Rahman, R Khan, M Sohail, A A Khan, H O Elansary, T K Z El-Abedin, E A Mahmoud, S A M Abdelmohsen and A Khan Semicond. Sci. Technol. 73 075004 (2021)
F T Tahir, M Husain, N Sfina, A A Rached, M Khan and N Rahman RSC Advances. 13 18788 (2023)
M Manzoor et al. Comput. Theor. Chem. 1217 113928 (2022)
J Y Al-Humaidi et al. Journal of Inorganic and Organomettalic Polymers and Materials. (2023)
A Algahtani, N U Khan, J Iqbal, V Tirth, S Abdullaev, M S Refat, A M Alsuhaibani, A M Henaish, A Zaman and H Fetooh Inorg. Chem. Commun. 1 111542 (2023)
N Chouit, S A Korba, M Slimani, H Meradji, S Ghemid and R Khenata Phys. Scripta 88 035702 (2013)
N Rahman et al. Fluoride 53 542 (2020)
P. Blaha et al. WIEN2k: An Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties. (2001)
E Fetil, Ş Özkan, T Ilknur, Y Erdem, B Lebe and A T Güneş Int. J. Dermatol. 41 892 (2002)
M Usman, M Usman, J Rehman, M B Tahir, A Hussain, H Alrobei, M Alzaid and A Dahshan Sci. Semicond. Process. 160 107399 (2023)
F D Murnaghan Proc. Nat. Acad. Sci. 30 9 (1944)
Q Zhu, J Chen, G Gou, H Chen and P Li J. Mater. Process. Technol. 246 267 (2017)
S M Rasul, D R Saber and S B Aziz Results Phys. 38 105688 (2022)
Y Lu, M Stegmaier, P Nukala, M A Giambra, S Ferrari, A Busacca and R Agarwal Nano Lett. 17 150 (2017)
S Bouhmaidi, A Marjaoui, A Talbi, M Zanouni, K Nouneh and L Setti Comput. Condense Matter. 31 e00663 (2022)
X Zhao, T Tang, Q Xie, L Gao, L Lu and Y Tang New J. Chem. 45 15857 (2021)
S A Dar, V Srivastava, U K Sakalle, V Parey and G Pagare Mater. Res. Exp. 4 106104 (2017)
C Lu, R Ren, Z Zhu, G Pan, G Wang, C Xu and K Sun Chem. Eng. J. 472 144878 (2023)
Z Huang, Y Zhang, H Wang and J Li Appl. Phys. Lett. 123 103501 (2023)
A L Selgin Int. J. Hydrogen Energy 44 3 (2019)
Born, Max, and R D Misra Cambridge University Press. 36 4 (1940)
S K Mitro, K M Hossain, R Majumder and M Z Hasan J. Alloys Compd. 15 157088 (2021)
C Jiang, Z Deng, B Liu, J Li, Z Han, Y Ma and Y Ma ACS Photon. 9 3089 (2022)
H H Raza, G Murtaza, N Muhammad and S M Ramay Int. J. Quant. Chem. 120 e26419 (2020)
S F X C I I Pugh The London, Edinburgh, Dublin Philosoph. Magazine J. Sci. 45 823 (1954)
R M Khalil, S Hayat, M I Hussain, A M Rana and F Hussain (2021) AIP Advances. 11 2
Y Zhang, X Liu, M Song and Z Qin Materials 16 2247 (2023)
S Al-Qaisi et al. Electronics 55 1015 (2023)
S Al-Qaisi et al. J. Comput. Chem. 44 2442 (2023)
M Khuili et al. J. Solid State Chem. 322 123955 (2023)
S Nose J. Chem. Phys. 81 511 (1984)
X Li, S Aftab, A Abbas, S Hussain, M Aslam, F Kabir and M Z Ansari Nano Energy 118 108979 (2023)
B Zhang, M Hao, Y Yao, J Xiong, X Li, A B Murphy and H B Ambalampitiya J. Phys. D: Appl. Phys. 56 134001 (2023)
H S Waheed et al. Phys. Status Solidi 260 2200267 (2023)
D Behera, S Al-Qaisi, M Manzoor, R Sharma, V Srivastava, M M Al-Anazy, E El Al-Shiekh and S K Mukherjee Mater. Sci. Eng. B. 297 116765 (2023)
B Zheng, D Lin, S Qi, Y Hu, Y Jin, Q Chen and R Yan Phys. Fluids 35 125129 (2023)
M A Ali, M M Saad H-E, A M Tighezza, S Khattak, S Al-Qaisi and M Faizan, Journal of Inorganic and Organometallic Polymers and Materials. (2023)
Q Mahmood et al. J. Phys. Chem. Solids 182 111584 (2023)
X Song, S Yang, G Wang, J Lin, L Wang, T Meier and W Yang Opt. Exp. 31 18862 (2023)
L Kong, Y Liu, L Dong, L Zhang, L Qiao, W Wang and H You Dalton Transact. 49 1947 (2020)
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University Abha 61421, Asir, Kingdom of Saudi Arabia for funding this work through the Large Groups Project under the grant number RGP.2/545/44.
Funding
Deanship of Scientific Research at King Khalid University Abha 61421, Asir, Kingdom of Saudi Arabia through the Large Groups Project under the grant number RGP.2/545/44.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ullah, W., Nasir, R., Husain, M. et al. Revealing the remarkable structural, electronic, elastic, and optical properties of Zn-based fluoropervskite ZnXF3 (x = Sr, Ba) employing DFT. Indian J Phys 98, 3559–3570 (2024). https://doi.org/10.1007/s12648-024-03146-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12648-024-03146-y