
Abstract
This research paper presents a thorough investigation of the structural, elastic, electronic, and optical properties of AuXF3 (X = Ca, Sr) compounds using density functional theory (DFT). The study utilizes the WEIN2K software, enabling the determination of various material characteristics. The analysis reveals that both AuCaF3 and AuSrF3 exhibit stable and cubic structures, as evidenced by Birch Murnaghan curve optimization, with computed lattice constants of 4.4270 Å and 4.7191 Å, respectively. The evaluation of elastic properties, including elastic constants, Young's moduli, anisotropy factors, Poisson's ratios, bulk moduli, and Pugh's ratios, indicate that both compounds possess ductile behavior, show anisotropy, and display mechanical stability. Furthermore, band structure calculations demonstrate wide indirect energy band gaps of 3.11 eV (M-Γ) for AuCaF3 and 3.35 eV (M-Γ) for AuSrF3, suggesting their semiconducting nature. An in-depth analysis of the partial and total density of states provides insights into the contributions of different elemental states to the band structure. The investigation of optical characteristics within the energy range of 0 eV to 15 eV encompasses refractive indices, absorption coefficients, and reflectivity. The compounds exhibit notable optical properties at higher energies, indicating potential applications such as energy storage and high-energy applications. Overall, this research offers a comprehensive computational analysis of AuXF3 (X = Ca, Sr) compounds, shedding light on their structural, elastic, electronic, and optical properties, and presenting opportunities for energy storage and high-energy applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Perovskite materials have garnered significant attention in recent years due to their remarkable properties and their potential applications in various fields, including optoelectronics, photovoltaics, and catalysis. The perovskite structure, characterized by the general formula ABX3, consists of an A-site cation, a B-site transition metal or rare earth cation, and an X-site anion (Zhao et al. 2020; Hazen 1988; Zhu et al. 2021). The unique arrangement of these constituents results in intriguing physical and chemical properties that make perovskites highly desirable for next-generation materials (Kojima et al. 2009; Li et al. 2022a; Huang, et al. 2023). The quest for innovative materials with remarkable properties has greatly expanded the area of materials research in recent years. Transition metal compounds have drawn a lot of interest among these materials because of their exceptional physical and chemical characteristics, which may be tuned for a variety of purposes (Stranks and Snaith 2015; Husain et al. 2023a; Liu et al. 2023).
Fluoroperovskites, a subclass of perovskite materials, have emerged as particularly fascinating compounds. By replacing the oxygen anion (O) with fluorine (F), these materials exhibit distinctive properties and behaviors. This substitution introduces several notable changes that distinguish fluoroperovskites from their oxygen-based counterparts (Rehman et al. 2022a; Muhammad et al. 2022; Wang et al. 2020). The introduction of fluorine can modify the lattice parameters and alter the structural stability of the perovskite material (Husain et al. 2022a). The smaller size of the fluorine atom compared to oxygen leads to a reduction in the lattice constant, resulting in a tighter and more compact crystal structure. This structural modification can have profound implications for the material's mechanical properties, such as hardness, elasticity, and thermal stability (Husain et al. 2022b; Wang et al. 2018; Yang et al. 2023). Fluoroperovskites also exhibit unique optical characteristics. The replacement of oxygen with fluorine affects the band structure and electronic transitions within the material. The strong electronegativity of fluorine enhances the polarization of the electronic states, leading to changes in the energy band gaps and optical absorption properties. These alterations make fluoroperovskites attractive for optoelectronic applications, including solar cells, light-emitting diodes (LEDs), and photodetectors (Husain et al. 2023b; Guo et al. 2023; Wan, et al. 2023). In addition to their optical properties, fluoroperovskites possess intriguing electronic properties. The presence of fluorine influences the charge transfer and mobility of electrons within the crystal lattice, affecting the material's conductivity and electronic transport properties. This can result in enhanced electrical performance and improved device efficiency (Rahman et al. 2022; Yang et al. 2022). Scientists and engineers are actively investigating these perovskite materials to uncover their potential in various fields, including energy harvesting, optoelectronics, catalysis, and sensing (Li et al. 2022b; Jameel et al. 2022; Usman et al. 2023; Usman et al. 2022a; Usman et al. 2022b; Rehman et al. 2023; Rehman et al. 2021; Rehman et al. 2022b; Rehman et al. 2022; Rafique et al. 2023; Kabli and J. ur Rehman et al. 2021; Usman and J. ur Rehman et al. 2022; Noreen, et al. 2022) (Zhong et al. 2023). Among fluoroperovskites, AuXF3 (X = Ca, Sr) compounds will attract significant attention due to the inclusion of gold (Au), which imparts exceptional properties. By employing DFT calculations, this research paper aims to provide a thorough understanding of the structural, electronic, mechanical, and optical properties of AuXF3 (X = Ca, Sr) compounds, which will provide valuable insights into their potential applications in diverse technological fields. Density Functional Theory (DFT) (Arif et al. 2022; Burke 2012) has revolutionized the field of materials science by enabling detailed investigations into the properties and behavior of various compounds, including AuXF3 (X = Ca, Sr) compounds. One of the key advantages of DFT is its ability to accurately model and predict the properties of materials by considering the behavior of electrons within the material. DFT employs a set of mathematical equations to describe the electron density distribution, which in turn provides insights into various properties (Kresse and Furthmüller 1996; Linh et al. 2019). In the case of AuXF3 (X = Ca, Sr) compounds, DFT calculations can shed light on their structural stability. By optimizing the geometries of the compounds using DFT, researchers can determine the most energetically favorable arrangement of atoms, indicating the compounds' stability (Wang et al. 2012). Furthermore, DFT calculations enable the analysis of electronic band structures, which are critical for understanding the compounds' electronic properties. The band structure reveals the energy levels and allowed electronic states within the material, providing insights into its electrical conductivity and optical behavior (Kohn et al. 1996; Herman 1958). Mechanical properties, such as the response to external forces, can also be investigated using DFT. Elastic constants, Young's modulus, and anisotropy factors can be computed, providing information about the compounds' mechanical stability, ductility, and response to strain (Hearmon 1946). Lastly, DFT allows for a comprehensive examination of the optical characteristics of AuXF3 (X = Ca, Sr) compounds. By calculating the absorption coefficient, reflectivity, refractive index, and other optical properties, researchers can evaluate the compounds' interaction with light. This information is crucial for potential applications in optoelectronic devices and optical coatings (Kumar and Radhakrishnan 2018; Mahmood et al. 2021; Saliba et al. 2018; Howard et al. 2019).
2 Computational details
The WIEN2K package's (Blaha et al. 2001, 2020) all-electron full-potential linearized augmented plane wave (FLAPW) approach (Blaha et al. 1990) was used to investigate the structural, electrical, elastic, and optical properties of AuXF3 (X = Ca, Sr) compounds. To take into consideration the exchange–correlation effects, the generalized gradient approximation (GGA) in the Perdew-Burke-Ernzerhof (PBE) (Perdew et al. 1996) form was used to calculate the structural properties and TB-mBJ potential (Tran and Blaha 2009) was used to calculate the electronic properties. The crystal structure of the compounds is obtained by selecting the optimized lattice constants, atomic positions, and space group. The structure was optimized by using structure optimization features in WIEN2K. Self-consistent computations were done to assess the electronic properties. The Brillouin zone's high-symmetry sites were used to determine the electronic band structures, which revealed details on the energy levels and band gaps of the AuXF3 (X = Ca, Sr) compounds (Weinert et al. 1982). Finite variations in the strain tensor were used to the optimized crystal structures to determine the elastic characteristics. The IRelast package (Jamal et al. 2018) for elastic properties interface in WIEN2K was used to analyze the resulting modifications in the total energy and stress tensors in order to derive the elastic constants, bulk modulus, shear modulus, and Poisson's ratio (Norris 2006). For the analysis of such compounds the RMT value is selected to be 5, the value of k-mesh is considered as 2000. The cut-off energy RMT × Kmax is a critical parameter in WIEN2K because it affects the accuracy and convergence of your calculations. A higher cut-off energy leads to a larger basis set, allowing for a more accurate description of the electronic wave functions. The energy cut-off for plane waves for both compounds is chosen to be 300 eV. The optical properties were examined by investigating the complex dielectric function as a function of frequency. By analyzing the real and imaginary components of the dielectric function, the absorption spectra, reflectance, and refractive index were determined (Ahamed et al. 2022). The calculations were carried out using high-performance computing resources that possess ample CPU cores and memory to accommodate the computational requirements of WIEN2K calculations.
3 Results and discussions
In this section, we comprehensively discuss and elaborate on the conclusions drawn from the investigation of AuXF3 (X = Ca, Sr) using the aforementioned research methods. We conduct a thorough analysis and offer comprehensive insights into each specific physical characteristic individually.
3.1 Structural properties
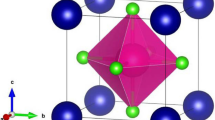
Using the widely utilized computational tool WIEN2K, we conducted an investigation into the structural characteristics of the AuXF3 (X = Ca, Sr) fluoroperovskites compounds. These compounds display a perfect cubic crystal structure, with lattice constants of 4.4270 and 4.7191 for AuXF3 (X = Ca, Sr) respectively. They belong to the Pm-3 m (#221) space group. In these compounds, the Ca and Sr atoms are positioned at coordinates (0.5, 0.5, 0.5), while the Au atoms are located at the origin (0, 0, 0). The “F” atoms are situated at coordinates (0, 0.5, 0.5), (0.5, 0, 0.5), and (0.5, 0.5, 0). The obtained crystal structure is shown in the Fig. 1 below.

Model crystal structure of AuXF3 (X = Ca, Sr), in which green color depicts the X atom, blue shows F, and red represents the Au atom
To determine the structural stability the Goldsmith tolerance factor is extensively used which is given by
where rA, rB and rX are the ionic radii of each ion. Putting the values of the respective parameters we find the tolerance factor of AuCaF3 and AuSrF3 equal to 0.89 and 0.93. Therefore, the tolerance coefficients of the compounds are within the defined range of cubic symmetry (cubic: 0.9 to 1), so both compounds exhibit stable cubic structures (Bartel, et al. 2019; Li et al. 2016; Travis et al. 2016).
To investigate the structural properties of the less-studied elements, we consider various attributes such as the ground state energy (E0), ground state volume (V0), bulk modulus (B), optimal lattice constants, and the pressure derivative of bulk modulus (B′). These parameters are determined using the volume optimization method, which aims to achieve stability for the entire system. The volume optimization procedure involves minimizing the total energy of the unit cell with respect to its volume. This process is incorporated into the Birch-Monaghan equation of states, thereby ensuring the overall stability of the system (Tyuterev and Vast 2006). In order to optimize the performance, the generalized gradient approximation (GGA) is employed along with the exchange–correlation potential term, which effectively considers the influence of both exchange and correlation effects. This approach plays a crucial role in characterizing various properties of specific materials. By utilizing this method in conjunction with the Birch-Murnaghan equation of state, the optimization curve, also known as the parabolic curve of volume and energy, is generated. This curve is obtained by fitting the energy values of different states to their corresponding volumes.
Figure 2 indicate a consistent trend of decreasing unit cell starting energy across the compounds studied. Analysis of these plots reveals that this pattern is observed in all the compounds. As the system's energy decreases, it eventually reaches the energy E0 at the ground state. The ground state corresponds to the optimized volume, which is the volume associated with the lowest energy. Upon reaching the ground state, the system loses its relaxed condition as the energy and volume of the unit cells increase.

Variation of energy against volume for a AuSrF3 b AuCaF3
The results of the structural parameters in their optimized states are presented in Table 1. The bulk modulus "B" serves as a measure of the compound's strength, while its pressure derivative B' helps identify its thermoplastic properties. Analyzing both parameters is valuable. Table 1 demonstrates a decreasing trend in the calculated bulk modulus when transitioning from AuCaF3 to AuSrF3. AuSrF3 exhibits the lowest bulk modulus value, whereas AuCaF3 possesses the highest value. This decrease in bulk modulus can be attributed to the increase in volume (Husain et al. 2023c).
3.1.1 Formation energy
Formation energy is a measure of the energy change associated with the formation of a compound from its constituent elements. It is a concept commonly used in the field of materials science and solid-state chemistry. The formation energy is defined as the energy difference between the compound in question and the isolated atoms of its constituent elements in their standard states. The standard state typically refers to the most stable form of the element at a given temperature and pressure, such as the most common allotrope or crystal structure. Mathematically, the formation energy (ΔEf) can be expressed as (Al-Qaisi et al. 2022a; Al-Qaisi et al. 2023):
where Etotal represents the total energy of the compound, \(\mathop \sum \limits_{x} E_{{{\text{total}}}} \left( x \right)\) represents the summation of the energies of the isolated atoms of the constituent elements. The formation energy provides insight into the stability and feasibility of a compound's formation. A negative formation energy indicates that the compound is thermodynamically stable and energetically favorable to form from its constituent elements. A positive formation energy suggests that the compound is energetically unfavorable or unstable, and it may tend to decompose or react with other species. Our calculated values of formation energy for both the materials possess a negative values reported in Table 1.
3.2 Elastic properties
The elastic properties of AuXF3 (X = Ca, Sr) are investigated to assess their mechanical stability, deformability, and resilience against external pressures. This examination will encompass evaluating various factors such as elastic constants, bulk modulus, young modulus, shear modulus, and Poisson's ratio, Pough ratio. These parameters yield valuable insights into how the materials respond to mechanical stresses and provide a deeper understanding of their mechanical behavior (Wang et al. 2019). The work being presented utilizes the IRelast package, which is integrated into the code of WIEN2k. This package is employed to assess the mechanical stability of the system. In a cubic system, the criteria \(C_{44} > 0\), \(C_{11} {-} C_{12} > 0\), \(C_{12} < B < C_{11}\), and B > 0 for mechanical stability impose restrictions on the elastic constants (Born 1940). Our investigation reveals that the investigated materials confirm to all the specified mechanical stability criteria, establishing their mechanical stability. Table 2 presents the significant elastic constants, including Poisson's ratio (v), shear modulus (G), Young's modulus (E), and anisotropy factor (A). These parameters are calculated by using the equations given below (Saddique et al. 2022):
It is noteworthy from the Table 2 that AuCaF3 exhibits a higher C11 value compared to the other materials, indicating a higher degree of elasticity. The C44 value signifies the stiffness of a material. Additionally, the elastic anisotropy factor (A) of the crystals is closely linked to their propensity to develop micro-cracks, which holds important implications for engineering studies. In the case of isotropic materials, an anisotropy factor (A) value of 1 is expected. Any value deviating from 1 indicates the presence of anisotropy. In our investigation, both compounds were found to be anisotropic, as evidenced by the deviation of A from 1. Table 2 presents the calculated values of A for AuCaF3 (0.1545) and AuSrF3 (0.092). Several characteristics are employed to determine the ductility or brittleness of a material. The key parameters like Pugh's ratio (B/G), Poisson's ratio (ʋ) play a significant role in assessing the material's behavior. According to Pugh's criteria, a material is considered brittle if its B/G ratio is less than 1.75, while it is regarded as ductile if the ratio exceeds 1.75. Frantsevich's rule utilizes Poisson's ratio to differentiate between ductile and brittle materials, with a critical value of 0.26. In essence, if a material exhibits a value lower than the specified threshold, it is categorized as brittle, while a value exceeding the threshold designates the material as ductile. With Poisson ratio values of 0.36 for AuSrF3 and 0.35 for AuCaF3, it is evident that both materials exhibit ductile nature. When the Poisson ratio registers at 0.10, it signifies a material primarily bound through covalent interactions. Conversely, a Poisson ratio value of 0.33 corresponds to materials belonging to the metallic bonding class. Furthermore, the Poisson ratio is also a criterion for categorizing solid materials into two groups: central force solids and non-central force solids. Central force solids typically have Poisson ratios either below 0.25 or above 0.50, whereas non-central force solids typically fall within the range of 0.25 to 0.50 (Al-Qaisi 2023; Al-Qaisi et al. 2021). The distinct Poisson ratio values of approximately 0.33 for both materials indicate that the atoms within the compounds being studied exhibit a metallic bonding nature. Our computed Poisson ratio values categorize both compounds as non-central force solids, as the Poisson ratios for AuSrF3 and AuCaF3 are 0.36 and 0.35, respectively.
In our study, both compounds, AuCaF3 and AuSrF3, exhibit positive Cauchy's pressure values, with AuCaF3 at 110.27 GPa and AuSrF3 at 101.05 GPa. AuCaF3 has Pugh ratio of 3.25, while AuSrF3 has a value of 3.16, indicating that AuCaF3 is slightly more ductile compared to AuSrF3. Furthermore, both compounds exhibit Poisson's ratios higher than 0.26, with AuCaF3 at 0.3606 and AuSrF3 at 0.354, further supporting their ductile characteristics. In conclusion, our investigation reveals that these compounds possess good crack resistance, display anisotropy, exhibit toughness, and demonstrate mechanical ductility. Considering their elastic properties, we envision significant implications for various electronic technologies.
3.3 Electronic properties
The electronic properties of AuXF3 (X = Ca, Sr) were investigated through band structure and density of states (DOS) calculations, utilizing the GGA approximation. These properties provide valuable insights into the electronic and optical behavior of the materials.
3.3.1 Band structure
In the realm of solid-state physics, electronic band structures play a crucial role in understanding the properties of various materials. They elucidate the distribution of energy levels at symmetrical points and greatly influence a material's magnetic, electrical, and thermal characteristics. The Fermi energy level, denoted as EF and represented by dotted lines in the band structure, holds significant importance. The conduction band lies above the Fermi level, the valence band lies below it, and the core region corresponds to the lower portion of the valence band. The position of the Fermi level determines whether a material is a conductor, insulator, or semiconductor. If the Fermi level falls between the conduction and valence bands, the material is a semiconductor or an insulator. On the other hand, if it lies within either band, the compound is considered metallic (Hamioud et al. 2016).
Based on these principles, we employed the TB-mBJ method to compute the electronic band structure, using a mesh of 2000 k-points to account for the effects of exchange and correlation. Figure 3 a and b illustrate the calculated electronic band structure and high symmetry directions within the first Brillouin zone at the equilibrium volume for AuXF3 (X = Ca, Sr). It is observed that AuCaF3 possesses an indirect band gap (M-Γ) of 3.11 eV, with the conduction band minima occurring at Γ-symmetry and the valence band maxima at M-symmetry. AuSrF3, on the other hand, exhibits a wider band gap of 3.35 eV (M-Γ), indicating behavior characteristic of wide band gap semiconductors. These materials hold potential for applications in energy storage devices and the semiconductor industry, depending on the desired electronic band gap. It is important to note that, to the best of our knowledge, no theoretical or experimental data regarding the band gap of these materials have been reported for comparison with our findings. Therefore, our research can serve as a foundation for future studies on these compounds.

a Estimated band structure and DOS of AuSrF3 b Estimated Band structure and DOS of AuCaF3
The density of states (DOS) analysis provides valuable insights into the electronic structure of the materials, revealing the distribution of possible states for the system's electrons. It allows us to examine the composition and structure of both the valence band (VB) and conduction bands (CB), shedding light on the bonding characteristics and properties of the compound. By studying the DOS, we can gain a comprehensive understanding of the band structure and other relevant electronic properties (Nazir et al. 2018). The characteristics of AuXF3 (X = Ca, Sr) compounds were analyzed through the calculation of total density of states (TDOS) and partial density of states (PDOS), as depicted in Fig. 3 a and b. The energy range considered for the DOS figures spans from − 8 to 8 eV. The valence band, labeled as region 1 (− 4–0 eV), and the conduction band, labeled as region 3 (0–6 eV), are distinguished. The Fermi level (EF) is represented by the dotted line at 0 eV. In the valence band of AuCaF3, the dominant contributions to the DOS arise from F-p, F-d, and F-f states, with minor contributions from Au-d, Au-f, and Ca-p states. Conversely, in the conduction band, the DOS is mainly influenced by the Au–s state. For AuSrF3, the valence band is primarily composed of F-p, F-d, and F-f states, with minor contributions from Au-d, Au-f, and Sr-p states. The conduction band, on the other hand, exhibits significant DOS contributions from the Au–s state. These findings provide insights into the electronic states and bonding characteristics of the AuXF3 (X = Ca, Sr) compounds, highlighting the significant contributions from specific orbitals within the valence and conduction bands.
3.4 Optical properties
When we talk about investigating the optical properties of a material, we are referring to the study of how that material interacts with light and the behavior it exhibits in response to it. Light can be absorbed, reflected, transmitted, or refracted by a material, and studying these interactions provides valuable information about the material's internal structure and electronic properties (Houari et al. 2020; Tahir et al. 2023). The optical properties of AuXF3 (X = Ca, Sr) compounds will be investigated, including their absorption, reflectance, and refractive index. These properties, essential for optoelectronic and photonic applications, will be determined through calculations of the dielectric function and optical spectra, shedding light on the compounds' potential in light manipulation and energy harvesting.
The real component ε1 (ω) and the imaginary component ε2 (ω) of ε(ω) play a crucial role in understanding a substance's electronic band structure and its absorptive effects. The ε1 (ω) describes how light is scattered or dispersed from the surface of an object, while ε2 (ω) indicates the substance's ability to absorb light. This complex function provides insights into both intra-band and inter-band transitions, which are important for understanding the substance's optical properties. The imaginary component ε2 (ω) specifically represents the absorption of light and reveals optical shifts occurring within the energy bands. By analyzing ε2 (ω), we can gain an understanding of how light is absorbed and transformed within the material (Makhdoom et al. 2019; Rahman 2023). The energy range considered for the analysis of optical properties spans from 0 to 15 eV, representing the range of incoming photon energies. It can be seen from the figure that at zero frequency, the static dielectric constant is determined, and for ε1 it is approximately 1.6, while ε2(ω) is measured to be around 1.47, as observed in the zero-frequency limit. In the spectra of AuCaF3 and AuSrF3, there are peaks ε1max at 2.5 occurring at 11.1 eV and 2.3 occurring at 9.7 eV, respectively. The negative real part of the dielectric function (ε(ω) < 0), typically refers to the fact that the material exhibits optical gain at certain frequencies. In other words, the material can amplify an incident electromagnetic wave at those frequencies rather than attenuate it. A negative real part of the dielectric function implies that the material can support optical modes (e.g., laser modes) that are sustained and amplified, which is crucial for laser devices and other applications where optical gain is required. It's important to note that not all materials have a negative real part of the dielectric function, and whether or not this occurs depends on the electronic and optical properties of the material and the specific frequency range of interest ((Al-Qaisi et al. 2022b; Al-Qaisi et al. 2022c)). The absorptive effect, characterized by ε2, is intimately associated with the electronic band structure of materials. According to the spectra of Ɛ(ω) presented in the Fig. 4 a, both AuCaF3 and AuSrF3 exhibit maximum values of 2.57 at 12.4 eV and 1.9 at 12.9 eV, respectively. The absorption spectra of these compounds demonstrate a broad range of absorption, indicating their potential suitability for diverse optical applications.

a Spectra of dielectric function for \({\text{AuXF}}_{3} \left( {{\text{X}} = {\text{Ca}}, {\text{Sr}}} \right)\). b Spectra of refractive index of AuXF3 (X = Ca, Sr). c Spectra of absorption coefficient of \({\text{AuXF}}_{3} \left( {{\text{X}} = {\text{Ca}}, {\text{Sr}}} \right)\). d Spectra of reflectivity of \({{AuXF}}_{3} \left( {{{X}} = {{Ca}},\user2{ Sr}} \right)\)
The refractive index, represented by n(ω), plays a vital role in utilizing optical materials for photonic and optical devices. It is an optical characteristic that describes the interaction of light with a substance, and its value depends on the frequency of the incident light beam. The refractive index indicates how light propagates through the material, specifically how it changes direction and speed when entering and exiting the substance. By understanding the refractive index, one can determine important properties such as the material's ability to bend or refract light, its transparency, and its reflective properties. This knowledge is crucial for designing and optimizing optical devices, such as lenses, fibers, and waveguides, as it enables precise control and manipulation of light within the material. The relationship between the refractive index n(ω) and energy for AuXF3 (X = Ca, Sr) compounds is depicted in Fig. 4b. The spectra of the refractive index demonstrate that the values of n at zero frequency, n (0), for AuCaF3 and AuSrF3 are 1.261 and 1.217, respectively. Furthermore, AuCaF3 exhibits a maximum refractive index of 1.6806 at 11.182 eV, while AuSrF3 has a maximum refractive index of 1.545 at 9.702 eV.
The absorption coefficient of a substance quantifies the amount of light that is absorbed by an optical system as it traverses through a material over a given distance. It arises due to various electronic transitions occurring between different energy bands within the material, as well as interactions between photons (light particles) and electrons within the material. The absorption coefficient determines the extent to which light is absorbed by the material and does not pass through it. Materials with low absorption coefficients are considered poor absorbers and are transparent to light, allowing most of the incident light to pass through unaffected. On the other hand, materials with high absorption coefficients have a significant capacity to absorb light, resulting in a reduced amount of transmitted light. It is a key factor in determining the optical properties of a substance, including its transparency or opacity to different wavelengths of light (Rahman et al. 2021; Rahman et al. 2023a). Figure 4c illustrates the absorption coefficient for the investigated compounds, AuCaF3 and AuSrF3. In the graph, the threshold value indicates the energy level at which the material begins to rapidly absorb electromagnetic radiation. For AuCaF3, the active absorption is observed to be 157.959 at 13.186 eV, indicating that the material strongly absorbs light at this energy. Similarly, for AuSrF3, the active absorption is measured to be 144.305 at 13.428 eV, indicating significant absorption at this energy level. These values demonstrate the ability of the compounds to effectively absorb electromagnetic radiation in the given energy range.
The reflectivity or reflection coefficient, denoted as R(ω), is an important optical characteristic that describes the ratio of reflected power to incident power on a material's surface. It quantifies how much of the incident light is reflected back from the surface of the material. Understanding the reflectivity helps in optimizing the efficiency and performance of optical systems by controlling the amount of light that is reflected or transmitted through a material's surface. Figure 4d illustrates the reflectivity (R(ω)) of the two perovskite compounds under investigation, AuCaF3 and AuSrF3. The reflectivity values for both compounds at zero frequency (static reflectivity) are 0.01, indicating a low reflectivity in the absence of radiation frequency. The reflectivity spectra show that AuCaF3 exhibits a maximum reflectivity value of 0.5, while AuSrF3 has a maximum reflectivity of 0.4. These reflectivity values in specific frequency ranges suggest that these materials possess low energy regions, particularly in the visible and infrared spectrums. This characteristic makes them suitable for applications where minimizing reflection is desirable, such as in the development of anti-reflective coatings. By employing these compounds as anti-reflective coatings, they can improve the efficiency and performance of various optical devices, including lenses, windows, and displays, by minimizing the loss of incident light due to reflection.
3.4.1 Extinction coefficient
The extinction coefficient is a measure of the absorption properties of a material in the optical or UV regions of the electromagnetic spectrum. It quantifies the fraction of incident light that is absorbed per unit distance and is related to the imaginary part of the complex refractive index of the material (Rahman et al. 2023b; Husain et al. 2021). The extinction coefficient, denoted as k (ω), has been determined by performing calculations along the xx-direction, as illustrated in Fig. 5a. At 0 eV, the local maxima of the extinction coefficient, k (ω), in the material AuCaF3 and AuSrF3 correspond to the zeros of static extinction coefficient. A zero value of the static extinction coefficient indicates that the material exhibits no absorption of electromagnetic radiation at a particular wavelength or energy. It suggests that the material is highly transparent or has minimal absorption properties at that particular energy level. The value of the extinction coefficient can vary with the wavelength or frequency of the incident light, indicating the material's absorption behavior across different parts of the electromagnetic spectrum.

a Investigated spectra of extinction coefficient of \(AuXF_{3} \left( {X = Ca, Sr} \right)\). b Computed spectra of optical conductivity for AuXF3 (X = Ca, Sr) ternary perovskites compounds versus incident photon energy range
Optical conductivity is a property that describes the ability of a material to conduct electric current in response to the presence of electromagnetic radiation, particularly in the optical frequency range. It quantifies the relationship between the applied electric field and the resulting current density induced by the radiation. The optical conductivity is typically represented by the symbol "σ" and is often described as a complex quantity. It is utilized in the design and characterization of materials for applications such as solar cells, photo-detectors, transparent conductive coatings, and optical devices. Figure 5b shows the investigated spectra of optical conductivity for the AuXF3 (X = Ca, Sr) ternary perovskites compounds versus incident photon energy range.
3.5 Phonon calculations
Highlighting the significance of comprehending phonon dispersion holds crucial importance in capturing the complex interplay between various dynamic properties, thermodynamics, and vibrational Raman spectroscopy in crystalline solids. To tackle this aspect, we employ the well-accepted and efficient method called density functional perturbation theory (DFPT), which is incorporated within the pseudo-potential framework of Quantum Espresso (Lemmens et al. 2021; Schuld et al. 2021). This approach empowers us to investigate the dynamic stability within the fundamental unit cells of AuXF3 (X = Sr, Ca) perovskites. As we delve into the dynamic characteristics of these molecular crystal structures, it becomes clear that a dynamically stable compound inherently displays three acoustic branches at the Γ-point with zero frequency, corresponding to k = 0 in reciprocal space. The remaining branches, characterized by non-zero frequencies, are recognized as optical phonons. Specifically, in the context of acoustics, we observe a distinctive longitudinal acoustic (LA) mode and two transverse acoustic (TA) modes. For a crystal composed of a unit cell with N atoms, the count of optical modes amounts to 3N–3. Shifting our attention to the current investigation, the configuration of 10 constituent atoms leads to the emergence of 30 distinct frequency branches. Among these, precisely 3 branches converge at the Γ point and are classified as acoustic, while the remaining 27 are labeled as optical, as visually illustrated in Fig. 6. Notably, the lower-frequency branches can be attributed to the pronounced vibrations of the heavier elements within the structure, whereas the optical branches mainly arise from the vibrational tendencies of the lighter atoms. By applying factor theory principles, we discern that the optical branches encompass a range of Raman, Infrared, and Silent modes based on their respective frequency bands. Of particular importance, the absence of negative or imaginary frequencies in the subsequent band dispersions highlights the intrinsic dynamic stability of the systems under study.

Phonon dispersion profiles within the irreducible Brillouin zone for the unit cell AuXF3 (X = Sr, Ca)
4 Conclusion
This study focuses on the computational analysis of Au-based fluoroperovskites compounds, specifically AuXF3 (X = Ca, Sr). Using the WEIN2K software and density functional theory (DFT), the structural, optical, elastic, and electronic properties of these materials are investigated. The structural properties of AuCaF3 and AuSrF3 are determined to be cubic and stable through Birch Murnaghan curve optimization. The optimized lattice constants are found to be 4.4270 Å and 4.7191 Å for AuCaF3 and AuSrF3, respectively, indicating the stability of the compounds at these lattice constants. Elastic characteristics are calculated using the IRelast package, which provides elastic constants and related properties such as Young's moduli, anisotropy factors, Poisson's ratios, bulk moduli, and Pugh's ratios. The results indicate that both compounds are ductile, anisotropic, and mechanically stable. The band structure calculations reveal that AuCaF3 and AuSrF3 possess wide indirect energy band gaps, indicating their semiconducting properties. The energy band gaps are determined to be 3.11 eV (M-Γ) for AuCaF3 and 3.35 eV (M-Γ) for AuSrF3. The partial density of states (PDOS) and total density of states (TDOS) analysis provide insights into the contributions of different elemental states to the band structure. The investigation of the optical characteristics of these compounds reveals high refractive indices, absorption coefficients, and reflectivity at high energies. The analysis covers the energy range from 0 to 15 eV. The transparency of these compounds to photons suggests their potential applications as anti-reflection coatings and lenses. Overall, this study presents a systematic and comprehensive computational analysis of AuXF3 (X = Ca, Sr) compounds, providing valuable insights into their structural, optical, elastic, and electronic properties. The findings contribute to a better understanding of the capabilities and potential applications of Au-based fluoroperovskites, representing a significant advancement in the field.
References
Ahamed, M.I., Boddula, R. and Altalhi, T.: Optical properties and applications of semiconductors. CRC Press, (2022)
Al-Qaisi, S., et al.: Structural, elastic, thermodynamic, electronic, optical and thermoelectric properties of MgLu2X4 (X= S, Se) spinel compounds from ab-initio calculations. Mater. Sci. Semicond. Process. 128, 105766 (2021)
Al-Qaisi, S., et al.: First-principles investigations of Ba2NaIO6 double Perovskite semiconductor: material for low-cost energy technologies. Mater. Chem. Phys. 275, 125237 (2022a)
Al-Qaisi, S., et al.: First-principles calculations to investigate electronic, structural, optical, and thermoelectric properties of semiconducting double perovskite Ba2YBiO6. Micro. Nanostruct. 170, 207397 (2022b)
Al-Qaisi, S., et al.: First-principles investigations of Na2CuMCl6 (M= Bi, Sb) double perovskite semiconductors: Materials for green technology. Mater. Sci. Semicond. Process. 150, 106947 (2022c)
Al-Qaisi, S., et al.: Tuning of band gap by variation of halide ions in K2CuSbX6 (X= Cl, Br, I) for solar cells and thermoelectric applications. J. Phys. Chem. Solids 174, 111184 (2023)
Al‐Qaisi, S.: et al. A theoretical investigation of the lead‐free double perovskites halides Rb2XCl6 (X= Se, Ti) for optoelectronic and thermoelectric applications, J. Comput. Chem. (2023)
Arif, M., et al.: Density functional theory based study of the physical properties of cesium based cubic halide perovskites CsHgX3 (X═ F and Cl). Int. J. Energy Res. 46(3), 2467–2476 (2022)
Bartel, C.J., et al.: New tolerance factor to predict the stability of perovskite oxides and halides. Sci. Adv. 5(2), eaav0693 (2019)
Benmhidi, H., Rached, H., Rached, D., Benkabou, M.: Ab initio study of electronic structure, elastic and transport properties of fluoroperovskite LiBeF 3. J. Electron. Mater. 46(4), 2205–2210 (2017)
Blaha, P., Schwarz, K., Sorantin, P., Trickey, S.B.: Full-potential, linearized augmented plane wave programs for crystalline systems. Comput. Phys. Commun. 59(2), 399–415 (1990)
Blaha, P., Schwarz, K., Tran, F., Laskowski, R., Madsen, G.K.H., Marks, L.D.: WIEN2k: An APW+ lo program for calculating the properties of solids. J. Chem. Phys. 152(7), 74101 (2020)
Blaha, P., Schwarz, K., Madsen, G.K.H., Kvasnicka, D and Luitz, J.: wien2k, An Augment. Pl. wave+ local orbitals Progr. Calc. Cryst. Prop., vol. 60, (2001)
Born, M.: On the stability of crystal lattices. I. Math. Proc. Cambridge Philos. Soc. 36(2), 160–172 (1940)
Burke, K.: Perspective on density functional theory. J. Chem. Phys. 136(15), 150901 (2012)
Guo, R., Zhang, S., Gao, H., Murugan, G.S., Liu, T., Cheng, Z.: Blazed subwavelength grating coupler. Photon. Res. 11(2), 189–195 (2023)
Hamioud, F., AlGhamdi, G.S., Al-Omari, S., Mubarak, A.A.: Ab initio investigation of the structural, electronic, magnetic and optical properties of the perovskite TlMnX3 (X= F, Cl) compounds. Int. J. Mod. Phys. B 30(7), 1650031 (2016)
Hazen, R.M.: Perovskites. Sci. Am. 258(6), 74–81 (1988)
Hearmon, R.F.S.: The elastic constants of anisotropic materials. Rev. Mod. Phys. 18(3), 409 (1946)
Herman, F.: Theoretical investigation of the electronic energy band structure of solids. Rev. Mod. Phys. 30(1), 102 (1958)
Houari, M., et al.: Semiconductor behavior of halide perovskites AGeX 3 (A= K, Rb and Cs; X= F, Cl and Br): first-principles calculations. Indian J. Phys. 94, 455–467 (2020)
Howard, I.A., et al.: Coated and printed perovskites for photovoltaic applications. Adv. Mater. 31(26), 1806702 (2019)
Huang, Q.: et al. Highly active and durable triple conducting composite air electrode for low-temperature protonic ceramic fuel cells, Nano Res., pp. 1–9, (2023)
Husain, M., et al.: Insight into the physical properties of the inter-metallic titanium-based binary compounds. Eur. Phys. J. plus 136(6), 1–10 (2021)
Husain, M., et al.: Structural, electronic, elastic, and magnetic properties of NaQF3 (Q= ag, Pb, Rh, and Ru) flouroperovskites: A first-principle outcomes. Int. J. Energy Res. 46(3), 2446–2453 (2022a)
Husain, M., et al.: Exploring the exemplary structural, electronic, optical, and elastic nature of inorganic ternary cubic XBaF3 (X= Al and Tl) employing the accurate TB-mBJ approach. Semicond. Sci. Technol. 37(7), 75004 (2022b)
Husain, M., et al.: Insight into the physical properties of beryllium-based ternary centro-symmetric fluoroperovskites BeXF3 (X= Bi, Y, Al, Sc, and In) compounds using the first-principle approach. Opt. Quantum Electron. 55(10), 908 (2023a)
Husain, M., et al.: DFT-based computational investigations of structural, mechanical, optoelectronics, and thermoelectric properties of InXF3 (X= Be and Sr) ternary fluoroperovskites compounds. Phys. Scr. 98(7), 75905 (2023b)
Husain, M., et al.: The comparative investigations of structural, optoelectronic, and mechanical properties of AgBeX3 (X= F and Cl) metal halide-perovskites for prospective energy applications utilizing DFT approach. Opt. Quantum Electron. 55(10), 920 (2023c)
Jamal, M., Bilal, M., Ahmad, I., Jalali-Asadabadi, S.: IRelast package. J. Alloys Compd. 735, 569–579 (2018)
Jameel, M.H., et al.: First principal calculations to investigate structural, electronic, optical, and magnetic properties of Fe3O4and Cd-doped Fe2O4. Comput. Condens. Matter 30, e00629 (2022)
Kabli, M.R., Ur Rehman, J., Tahir, M.B., Usman, M., Ali, A.M., Shahzad, K.: Structural, electronics and optical properties of sodium based fluoroperovskites NaXF3 (X= Ca, Mg, Sr and Zn): First principles calculations. Phys. Lett. a. 412, 127574 (2021)
Kohn, W., Becke, A.D., Parr, R.G.: Density functional theory of electronic structure. J. Phys. Chem. 100(31), 12974–12980 (1996)
Kojima, A., Teshima, K., Shirai, Y., Miyasaka, T.: Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 131(17), 6050–6051 (2009)
Kresse, G., Furthmüller, J.: Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54(16), 11169 (1996)
Kumar, N.R., Radhakrishnan, R.: Electronic, optical and mechanical properties of lead-free halide double perovskites using first-principles density functional theory. Mater. Lett. 227, 289–291 (2018)
Lemmens, L., De Vriendt, X., Tolstykh, D., Huysentruyt, T., Bultinck, P. and Acke, G.: GQCP: The Ghent quantum chemistry package, J. Chem. Phys., vol. 155, no. 8 (2021)
Li, Z., Yang, M., Park, J.-S., Wei, S.-H., Berry, J.J., Zhu, K.: Stabilizing perovskite structures by tuning tolerance factor: formation of formamidinium and cesium lead iodide solid-state alloys. Chem. Mater. 28(1), 284–292 (2016)
Li, Z., et al.: Directly printed embedded metal mesh for flexible transparent electrode via liquid substrate electric-field-driven jet. Adv. Sci. 9(14), 2105331 (2022a)
Li, J., et al.: First insights into mineralogy, geochemistry, and isotopic signatures of the Upper Triassic high-sulfur coals from the Thai Nguyen Coal field, NE Vietnam. Int. J. Coal Geol. 261, 104097 (2022b)
Linh, N.H., Tuan, N.H., Dung, D.D., Bao, P.Q., Cong, B.T.: Alkali metal-substituted bismuth-based perovskite compounds: a DFT study. J. Sci. Adv. Mater. Dev. 4(3), 492–498 (2019)
Liu, Y., Qin, J., Lu, L., Xu, J., Su, X.: Enhanced microwave absorption property of silver decorated biomass ordered porous carbon composite materials with frequency selective surface incorporation. Int. J. Miner. Metall. Mater. 30(3), 525–535 (2023)
Mahmood, Q., et al.: Study of optical and thermoelectric properties of ZYbI3 (Z= Rb, Cs) for solar cells and renewable energy; Modelling by density functional theory. J. Phys. Chem. Solids 155, 110117 (2021)
Mahmoud, N.T., Khalifeh, J.M., Mousa, A.A.: Effects of rare earth element Eu on structural, electronic, magnetic, and optical properties of fluoroperovskite compounds SrLiF3: First principles calculations. Phys. B Condens. Matter 564, 37–44 (2019)
Makhdoom, M., et al.: First-principles description of the different phases in the Li 2 NH compound: electronic structure and optical properties. J. Korean Phys. Soc. 74(12), 1140–1145 (2019)
Muhammad, I., Ali, A., Zhou, L., Zhang, W., Wong, P.K.J.: Vacancy-engineered half-metallicity and magnetic anisotropy in CrSI semiconductor monolayer. J. Alloys Compd. 909, 164797 (2022)
Nazir, G., et al.: Under pressure DFT investigations on optical and electronic properties of PbZrO3. Acta Phys. Pol. A 133(1), 105–113 (2018)
Noreen, S., et al.: Structural, mechanical, thermodynamic, electronic, magnetic and optical properties of ZnFe2O4 ferrite: A DFT study. Opt. Mater. Amst. 133, 112930 (2022)
Norris, A.N.: Poisson’s ratio in cubic materials. Proc. r. Soc. A Math. Phys. Eng. Sci. 462(2075), 3385–3405 (2006)
Perdew, J.P., Burke, K., Ernzerhof, M.: Generalized gradient approximation made simple. Phys. Rev. Lett. 77(18), 3865 (1996)
Rafique, A., Usman, M., Rehman, J.U., Nazeer, A., Ullah, H., Hussain, A.: Investigation of structural, electronic, mechanical, optical and hydrogen storage properties of cobalt-based hydride-perovskites XCoH3 (X= In, Mn, Sr, Sn, Cd) for hydrogen storage application. J. Phys. Chem. Solids 181, 111559 (2023)
Rahman, N., et al.: First principle study of structural, electronic, optical and mechanical properties of cubic fluoro-perovskites:(CdXF3, X= Y, Bi). Eur. Phys. J. plus 136(3), 1–11 (2021)
Rahman, N., et al.: First-principles calculations to investigate structural, elastic, optical, and thermoelectric properties of narrow band gap semiconducting cubic ternary fluoroperovskites barium based BaMF3 (M= Ag and Cu) compounds. J. Mater. Res. Technol. 21, 2168–2177 (2022)
Rahman, N., et al.: Appealing perspectives of the structural, electronic, elastic and optical properties of LiRCl 3 (R= Be and Mg) halide perovskites: a DFT study. RSC Adv. 13(27), 18934–18945 (2023a)
Rahman, N., et al.: Probing the physical properties of M 2 LiCeF 6 (M= Rb and Cs) double perovskite compounds for prospective high-energy applications employing the DFT framework. RSC Adv. 13(23), 15457–15466 (2023b)
Rahman, N., et al.: Exploring the structural, optoelectronic, elastic, and thermoelectric properties of cubic ternary Fluoro-perovskites sodium based NaMF3 (M= Si and Ge) compounds for heterojunction solar cells applications. Phys. Scr. 98(6), 065929 (2023)
Rehman, J.U., Usman, M., Tahir, M.B., Hussain, A., Rashid, M.: Investigation of structural, electronics, optical, mechanical and thermodynamic properties of YRu 2 P 2 compound for superconducting application. J. Supercond. Nov. Magn. 34, 3089–3097 (2021)
Rehman, J.U., et al.: First-principles calculations to investigate structural, electronics, optical and elastic properties of Sn-based inorganic Halide-perovskites CsSnX3 (X= I, Br, Cl) for solar cell applications. Comput. Theor. Chem. 1209, 113624 (2022a)
Rehman, J.U., et al.: Electronic and optical properties of nitrogen and sulfur doped strontium titanate as efficient photocatalyst for water splitting: A DFT study. Int. J. Hydrog. Energy 47(3), 1605–1612 (2022b)
Rehman, J.U., et al.: A DFT study to investigate structural, electronic, optical, mechanical and magnetic properties of NaGeBr 3 for photovoltaic and optoelectronic applications. Emerg. Mater. 6(2), 699–709 (2023)
Rehman, J.U., Usman, M., Tahir, M.B., Hussain, A., Rehman, M.A., Sagir, M., Alrobei, H., Ullah, S., Assiri, M.A.: First-principles calculations to investigate ultra-wide bandgap semiconductor behavior of NaMgF3 fluoro-perovskite with external static isotropic pressure and its impact on optical properties. Optik 252, 168532 (2022)
Saddique, J., et al.: Modeling structural, elastic, electronic and optical properties of ternary cubic barium based fluoroperovskites MBaF3 (M= Ga and In) compounds based on DFT. Mater. Sci. Semicond. Process. 139, 106345 (2022)
Saliba, M., Correa-Baena, J., Grätzel, M., Hagfeldt, A., Abate, A.: Perovskite solar cells: from the atomic level to film quality and device performance. Angew. Chemie Int. Ed. 57(10), 2554–2569 (2018)
Schuld, M., Sweke, R., Meyer, J.J.: Effect of data encoding on the expressive power of variational quantum-machine-learning models. Phys. Rev. A 103(3), 32430 (2021)
Stranks, S.D., Snaith, H.J.: Metal-halide perovskites for photovoltaic and light-emitting devices. Nat. Nanotechnol. 10(5), 391–402 (2015)
Tahir, F.T., Husain, M., Sfina, N., Rached, A.A., Khan, M., Rahman, N.: Probing the physical properties for prospective high energy applications of QMnF 3 (Q= Ga, In) halide perovskites compounds employing the framework of density functional theory. RSC Adv. 13(27), 18788–18798 (2023)
Tran, F., Blaha, P.: Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 102(22), 226401 (2009)
Travis, W., Glover, E.N.K., Bronstein, H., Scanlon, D.O., Palgrave, R.G.: On the application of the tolerance factor to inorganic and hybrid halide perovskites: a revised system. Chem. Sci. 7(7), 4548–4556 (2016)
Tyuterev, V.G., Vast, N.: Murnaghan’s equation of state for the electronic ground state energy. Comput. Mater. Sci. 38(2), 350–353 (2006)
Usman, M., et al.: First-principles calculations to investigate variation in the bandgap of NaSrF3 Fluoro-Perovskite with external static isotropic pressure and its Impact on optical properties. Comput. Theor. Chem. 1214, 113766 (2022a)
Usman, M., Rehman, J.U., Tahir, M.B., Hussain, A.: First-principles calculations to investigate the effect of Cs-doping in BaTiO3 for water-splitting application. Solid State Commun. 355, 114920 (2022b)
Usman, M., et al.: First-principles calculations to investigate structural, electronics, optical, and mechanical properties of Bi-based novel fluoroperovskites TBiF3 (T= Hg, Xe) for optoelectronic applications. Mater. Sci. Semicond. Process. 160, 107399 (2023)
Usman, M., ur Rehman, J., Tahir, M.B., Hussain, A.: Structural, electronics, magnetic, optical, mechanical and hydrogen storage properties of Ga-based hydride-perovskites X GaH3 (X= K, Li). Int. J. Energy Res. 46(11), 15617–15626 (2022)
Wan, D. et al. Hyperuniform disordered solids with morphology engineering, Laser Photon. Rev., 2300398, (2023)
Wang, Y., Lv, J., Zhu, L., Ma, Y.: CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun. 183(10), 2063–2070 (2012)
Wang, M., Jiang, C., Zhang, S., Song, X., Tang, Y., Cheng, H.-M.: Reversible calcium alloying enables a practical room-temperature rechargeable calcium-ion battery with a high discharge voltage. Nat. Chem. 10(6), 667–672 (2018)
Wang, W.Y., Li, J., Liu, W., Liu, Z.-K.: Integrated computational materials engineering for advanced materials: A brief review. Comput. Mater. Sci. 158, 42–48 (2019)
Wang, J., et al.: Evolution of crystallographic orientation, precipitation, phase transformation and mechanical properties realized by enhancing deposition current for dual-wire arc additive manufactured Ni-rich NiTi alloy. Addit. Manuf. 34, 101240 (2020)
Weinert, M., Wimmer, E., Freeman, A.J.: Total-energy all-electron density functional method for bulk solids and surfaces. Phys. Rev. B 26(8), 4571 (1982)
Yang, M., Liu, W., Liu, Z., Cai, C., Wang, Y., Yang, J.: Binocular vision-based method used for determining the static and dynamic parameters of the long-stroke shakers in low-frequency vibration calibration. IEEE Trans. Ind. Electron. 70(8), 8537–8545 (2022)
Yang, H., et al.: Sensing mechanism of an Au-TiO 2-Ag nanograting based on Fano resonance effects. Appl. Opt. 62(17), 4431–4438 (2023)
Zhao, C., Cheung, C.F., Xu, P.: High-efficiency sub-microscale uncertainty measurement method using pattern recognition. ISA Trans. 101, 503–514 (2020)
Zhong, J., Han, M., Li, C., Li, R., He, H.: Facile and scalable fabrication process of electroluminescent filament with high luminescent efficiency. Mater. Lett. 350, 134868 (2023)
Zhu, X., et al.: Templateless, plating-free fabrication of flexible transparent electrodes with embedded silver mesh by electric-field-driven microscale 3D printing and hybrid hot embossing. Adv. Mater. 33(21), 2007772 (2021)
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University Abha 61421, Asir, Kingdom of Saudi Arabia for funding this work through the Large Groups Project under grant number RGP.2/498/44.
Funding
Deanship of Scientific Research at King Khalid University Abha 61421, Asir, Kingdom of Saudi Arabia through the Large Groups Project under the grant number RGP.2/498/44.
Author information
Authors and Affiliations
Contributions
AJ, MH, SB, NR, VT wrote the main manuscript. AAR, Muhammad YK, MN, AK prepared figures, SNK and KI reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
No competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jehan, A., Husain, M., Bibi, S. et al. Insight into the structural, optoelectronic, and elastic properties of AuXF3 (X = Ca, Sr) fluoroperovskites: DFT study. Opt Quant Electron 55, 1242 (2023). https://doi.org/10.1007/s11082-023-05394-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-023-05394-4