
Abstract
The Fourier transform infrared spectra and Fourier transform Raman spectra of Linezolid have been recorded in the regions 4,000–400 and 4,000–100 cm−1, respectively. Utilizing the observed Fourier transform infrared spectra and Fourier transform Raman spectra data, a complete vibrational assignment and analysis of the fundamental modes of the compound have been carried out. The optimum molecular geometry, harmonic vibrational frequencies, infrared intensities and Raman scattering activities, have been calculated by density functional theory with 6-31G(d,p), 6-311G(d,p) and M06-2X/6-31G(d,p) levels. The difference between the observed and scaled wavenumber values of most of the fundamentals is very small. A detailed interpretation of the infrared and Raman spectra of Linezolid is reported. Mulliken’s net charges have also been calculated. Ultraviolet–visible spectrum of the title molecule has also been calculated using time-dependent density functional method. Besides, molecular electrostatic potential, highest occupied molecular orbital and lowest unoccupied molecular orbital analysis and several thermodynamic properties have been performed by the density functional theoretical method.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In the last decade, quantum mechanical ab initio and density functional theory (DFT) computational methods have gained importance in vibrational spectroscopy and also in the investigations of molecular structure. Vibrational spectroscopy and normal mode calculations for molecules and similar chemical species have become standard features of many quantum chemical program packages. Linezolid [(S)-N-({3-[3-fluoro-4-(morpholin-4-yl)phenyl]-2-oxo-1,3-oxazolidin-5-yl}methyl)acetamide] is a synthetic antibiotic with the molecular formula C16H20FN3O4. Linezolid is a member of the oxazolidinone class of drugs, that are resistant to several other antibiotics [1–3]. In the scientific literature, Linezolid has been called a “reserve antibiotic”—that it remains effective as a drug of last resort against potentially intractable infections [4, 5]. To our knowledge, the vibrational spectra and the theoretical calculations of Linezolid have not been reported except in our work. Recently, the DFT methods have evolved to a powerful quantum chemical tool for the determination of the electronic structure of molecules. IR, Raman spectroscopic studies along with highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO) analysis has been used to elucidate information regarding charge transfer within the molecule.
2 Experimental details
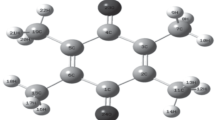
The pure sample was obtained from M/s. Sigma Aldrich Co., and was used as such without further purification. The Fourier transform infrared spectrum of the title compound was recorded using (Perkin Elmer) spectrometer in KBr dispersion in range 4,000–400 cm−1. The FT-Raman spectrum of Linezolid was recorded in 1,054 line of a Nd:YAG laser as excitation wavelength in region 4,000–100 cm−1 on a (Bruker model IFS 66V) spectrophotometer equipped with FRA 106 FT-Raman module accessory. The optical properties of the Linezolid were examined using UV–visible spectrophotometer at room temperature. UV–visible spectrum was recorded in the range of 190–800 nm in a (Perkin Elmer-Lambda 950-UV–visible) spectrometer. To measure the UV visible absorption, the Linezolid particles were dispersed in deionized water and measured. The optimized molecular structure of Linezolid has been given in Fig. 1. For visual comparison, the experimental and calculated FTIR of Linezolid at DFT–B3LYP method using 6-31G(d,p) and 6-311G(d,p) levels are shown in Fig. 2(a)–2(c) and the experimental and calculated FT Raman spectra of Linezolid at DFT–B3LYP method using 6-31G(d,p) and 6-311G(d,p) levels are shown in Fig. 3(a)–3(c).

Geometry of Linezolid optimized at DFT/B3LYP/6-31G(d,p) level

Comparison of FT-IR spectra of Linezolid (a) experimental, (b) calculated with B3LYP/6-31G(d,p) and (c) calculated with B3LYP/6-311G(d,p) levels

Comparison of FT-Raman spectra of Linezolid (a) experimental, (b) calculated with B3LYP/6-31G(d,p) and (c) calculated with B3LYP/6-311G(d,p) levels
3 Computational details
In the frame work of DFT approach, different exchange and correlation functions were routinely used. Among these, the B3LYP combination [6, 7] was the most used since it proved its ability in reproducing various molecular properties, including vibrational spectra. In order to provide information with regard to the structural characteristics and the normal vibrational modes of Linezolid, the DFT–B3LYP correlation functional calculations were carried out. The experimental molecular geometric data of Linezolid [8] was used for optimization and the entire calculations were performed at ab initio Hartree–Fock (HF) and DFT method using B3LYP/6-31G(d,p), B3LYP/6-311G(d,p) and M06-2X/6-31G(d,p) levels [9–11] on a Pentium V/1.6 GHz personal computer using GAUSSIAN 09W software package [12]. The Raman activities (S i ) calculated with the GAUSSIAN 09W program were subsequently converted to relative Raman intensities (I i ) using the following relationship derived from the basic theory of Raman scattering [13, 14],
where ν 0 denoting exciting frequency in cm−1, ν i denoting vibrational wave number of ith normal mode in cm−1, h, c, k and T being Planck and Boltzmann constants, speed of light and temperature in Kelvin and f takes a constant equal to 10−12 and chosen as common normalization factor for all peak intensities.
4 Results and discussion
The optimized geometrical parameters have been calculated at B3LYP with 6-31G(d,p) and B3LYP with 6-311G(d,p) levels. The calculated geometric parameters (bond lengths and bond angles) for Linezolid are listed in Table 1. The calculated values have been compared with X-ray diffraction results [15]. From the structural data, it is found that most of the optimized bond lengths are larger than the experimental values. This is due to the fact that the theoretical calculations result from isolated molecules in gaseous phase, while the experimental results are from molecule in solid state. Comparing bond lengths and bond angles of B3LYP/6-31G(d,p) and 6-311G(d,p) levels for Linezolid, it is also observed that the geometrical parameters are found to be almost same at B3LYP/6-31G(d,p) and B3LYP/6-311G(d,p) levels. However, the B3LYP/6-31G(d,p) level of theory, in general slightly over estimates bond lengths but it yields bond angles in excellent agreement with the B3LYP/6-311G(d,p) level.
As can be seen in Table 1, it is observed that the C–C bond distances in the molecule varies in the narrow range 1.377–1.534 Å calculated by B3LYP/6-311G(d,p) method. The C–C bond length in the ring R1 ranges from 1.377 to 1.405 Å. The C5–C6 bond length is 1.521 Å. This is smaller than the C–C bond distances of R3 ring. This clearly shows that there is a delocalization of the p-electrons of N–C towards the ring of R3. The inter nuclear distance determined by B3LYP/6-311G(d,p) method between O11 and H34, N9 and H36 in the optimized geometries are 2.207 and 2.674 Å, respectively. This longer distance is not in favor for the formation of intra molecular hydrogen bond of the type N–H and O–H. The calculated bond angle of ring R1 by B3LYP/6-311G(d,p) method lie in the range 118.34°–124.14°. The bond angle of C10–N9–C3, N9–C10–O11 and C14–C15–C17 are the highest, where the nitrogen Oxygen atom and Flourine atom are attached and are equal to 125.741, 128.961 and 124.145, respectively. This variation in bond angle depends on the electro negativity of the atom. If the electro negativity of the atom increases, bond angle increases.
4.1 Vibrational assignments
The molecule Linezolid has 44 atoms with 126 normal modes of vibration. According to group theory the point group symmetry for the molecule is C1 and all 126 fundamental modes of vibrations are active both in the Infrared absorption and Raman scattering. The detailed vibrational assignments of fundamental modes of Linezolid along with calculated IR and Raman frequencies and normal mode are reported in Table 2.
The main focus of the present investigation is the proper assignment of the experimental frequencies to the various vibrational modes of the title molecule at DFT/B3LYP/6-31G(d,p) level and DFT/B3LYP/6-311G(d,p) level. Comparison of the frequencies calculated by DFT–B3LYP method with the experimental values reveals the overestimation of the calculated vibrational modes due to neglect of anharmonicity in real system. Normally, the over estimation of unscaled frequencies when, compared to observed frequencies, were prominent only in the higher frequency region [16]. The results indicate that the B3LYP/6-311G(d, p) calculations approximate the observed fundamental frequencies much better than the B3LYP/6-31G(d, p) results. Therefore, it is customary to scale down the calculated harmonic wavenumber in order to improve the agreement with the experimental values. The harmonic vibrational frequencies have been scaled by 0.9712 and 0.9738 for B3LYP/6-31G(d,p) B3LYP/6-311G(d,p) and 0.9701 for M06-2X/6-31G(d,p) levels [17].
4.1.1 N–H vibrations
In mono fluorinated amines the stretch frequency of NH may vary and it not so intense. When the hydroxyl bonds on the same region are less intense the amines have low intense peaks due to halogens presence. According to Socrates [18], the stretching of amino group appears around 3,500–3,000 cm−1. The position of absorption in this region depends upon the degree of the hydrogen bonding, and hence upon the physical state of the sample or the polarity of the solvent. In the present study, the N–H stretching frequency has been observed at 3,542 and 3,543 cm−1 from DFT/B3LYP/6-31G(d, p) level and M06-2X level. The corresponding band in FT-IR spectrum is observed at 3,543 cm−1. Computational data for N–H in plane bending is assigned at 1,404 cm−1 by B3LYP/6-31G(d,p) level and experimentally it is assigned at 1,444 cm−1 in FT-IR and 1,444 cm−1 in FT Raman respectively. Theoretical data for N–H out of plane bending are presented in Table 2 and are found to agree well within the characteristic region [19].
4.1.2 Carbon–hydrogen vibrations
The heteroaromatic organic compounds and its derivatives are structurally very close to benzene and commonly exhibit multiple weak bands in the region 3,100–3,000 cm−1 due to C–H stretching vibrations [20]. In our study, the vibrational frequencies found at 3,173, 3,140, 3,128 and 3,060 cm−1 are assigned to C–H stretching vibrations of the molecule by B3LYP/6-31G(d,p) and B3LYP/6-311G(d,p) levels. The vibrational frequencies found at 3,146, 3,114, 3,100 and 3,067 cm−1 are assigned to C–H stretching vibrations of the molecule by M06-2X/6-31G(d,p) level. In FT-IR, frequency vibrations occurring at 3,180, 3,155 and 2,852 cm−1 are assigned to C–H vibrations and it shows good agreement with the literature data. The C–H in-plane and out-of-plane bending vibrations generally lie in the range 1,300–1,000 and 1,000–750 cm−1 [21, 22], respectively. The FTIR bands at 1,273, 1,220, 1,143, 1,078 and 1,022 cm−1 and FT-Raman bands at 1,207, 1,118, 1,077, 1,049 and 1,024 cm−1 are assigned to C–H in-plane bending vibrations of the molecule. The theoretically computed values by B3LYP/6-31G(d,p) and B3LYP/6-311G(d,p) levels are 1,245, 1,239, 1,216, 1,201, 1,046, 1,025 and 1,007 cm−1, which is in good agreement with literature values. The peaks at 935, 906, 756 in FTIR and 937,871 and 754 cm−1 in FT-Raman confirm the C–H out of plane bending vibrations, which agrees with the above said literature values. In general, the C–H vibrations (stretching, in plane and out-of-plane bending vibrations) calculated theoretically are in good agreement with experimental values.
4.1.3 Carbonyl group vibration
The characteristic infrared absorption frequencies of carbonyl group has been investigated earlier and the C=O stretching vibration are expected in the region 1,750–1,680 cm−1 [23, 24]. In Linezolid, the C=O stretching vibration is calculated at 1,690 cm−1 in B3LYP/6-31G(d,p) level and 1,775 cm−1 at M06-2X/6-31G(d,p) level. A strong band in IR spectrum at 1,747 cm−1 and a band in FT-Raman spectrum at 1,729 cm−1 is assigned to C=O stretching vibration. The carbon–oxygen bond is formed by pπ–pπ* between carbon and oxygen and the lone pair of electron on oxygen also determines the nature of carbonyl group. The higher degree of conjugation is due to the maximum overlap of p-orbital, occurring because of the planarity of the group (–CH=C=(C=O)–C=CH–). The conjugation increases the single bond character of C=O bond [25] and consequently lowers the wave number of the carbonyl absorption. The calculated values always overestimate the observed values due to anharmonicity in the real system.
4.1.4 Carbon–nitrogen vibrations
The identification of C–N vibrations is a very difficult task, since mixing of several bands are possible in this region. Silverstein et al. [26] assigned C–N stretching absorption in the region 1,382–1,266 cm−1 for aromatic amines. In this study, the bands observed at 1,323 and 1,204 cm−1 in B3LYP/6-31G(d,p) are assigned for C–N stretching. A band observed at 1,236 cm−1 IR and at 1,235 cm−1 at FT-Raman has been assigned to C–N stretching vibration of Linezolid. The slight shift in wavenumber is due to the fact that force constants of the C–N bond increases due to resonance with the ring. Pyrimidine absorb strongly in the region 1,600–1,500 cm−1 due to the C=C and C=N ring stretching vibrations [27]. The C–N in-plane and out-of-plane bending vibrations have also been identified and are listed in Table 2.
4.1.5 Ring vibrations
The ring stretching vibrations ν (Ring) are complicated combinations of stretching of C–N, C=C, C–O and C–C bonds. Many ring modes are affected by the substitution in the aromatic ring. In the present study, the rings are named as R1 (six_membered ring), R2 (Morpholin ring) R3 (oxazolidinone ring). The bands absorbed at 1,627, 1,567, 1,506, 1,339, 923 cm−1 in R1 and 1,465, 1,456, 1,488, 1,392, 1,376, 1,362, 1,332, 1,296, 1,104, 923 cm−1 for R2 and 1,386, 1,201, 961 cm−1 in R3 have been designated to ring stretching and bending modes respectively. The most important ring stretching vibration is the ring breathing vibration, where all bonds of the rings appear to stretch and contract in-phase with each other [30]. In the experimental infrared spectrum of Linezolid, this mode appears at 850 cm−1 both in FTIR and FT-Raman spectrum. The ring breathing vibrations are observed at 1,339 cm−1 in R1, 838 cm−1 in R2 and 866 cm−1 (mode 78) in R3 respectively [28].
4.1.6 C–X (F) vibrations
Vibrations belong to C–X (F, Br) bonds, which are formed between the ring and the halogen atoms are interesting since mixing of vibrations are possible due to the lowering of the molecules symmetry and the presence of heavy atoms. The assignments of C–F vibrations have been made by comparison with halogen substituted benzene derivatives. C–F stretching vibrations appear in the lower range of frequencies i.e. 1,000–1,400 and 760–505 cm−1 and C–F deformation occur in the range of 700–500 and 300–140 cm−1 respectively. The bands of observed spectra identified the stretching and bending vibrations arising at 1,220, 1,143, 493 cm−1 in FTIR and 1,207 cm−1 in FT-Raman. The calculated values for stretching and bending vibrations by B3LYP/6-31G(d,p) are found at 1,201, 1,135 cm−1 and 560, 491 cm−1 respectively for Linezolid [29].
4.1.7 CH2 group vibrations
The title molecule consists of rings R1, R2 and R3. For the assignments of CH2 group frequencies, basically six fundamentals can be associated to each CH2 group, namely, CH2 symmetrical stretching, CH2 asymmetrical stretching, CH2 scissoring stretching, CH2 rocking vibrations, which belong to in-plane vibrations. In addition to that CH2 wagging modes and CH2 twisting modes of CH2 group would be expected to be depolarized for out-of-plane bending vibrations. The asymmetric CH2 stretching vibration of CH2 are observed at 3,059, 3,047, 3,044, 3,028 and 3,021 cm−1 in all the rings and the symmetric CH2 stretching vibration of CH2 are observed at 2,975, 2,969, 2,949, 2,914, 2,910, 2,894, 2,876 cm−1 in all rings. The observed band at 2,978 cm−1 in IR spectrum is assigned to asymmetric CH2 stretching vibration. The observed band at 2,852, 2,831 cm−1 in IR spectrum and the bands observed at 2,980, 2,934, 2,897, 2,853 cm−1 in FT-Raman are assigned to symmetric CH2 stretching vibration. The CH2 wagging is observed at 1,339 cm−1 in DFT/B3LYP/6-31G level and it is observed a band at 1,376 cm−1 in FT-IR spectrum and in Raman spectrum, it is observed a band at 1,367 cm−1 that is assigned to CH2 wagging. The band assigned for CH2 scissoring are observed at 1,444 cm−1 in both FT-IR and Raman spectrum and the calculated band for CH2 scissoring is 1,404 cm−1. The CH2 rocking vibrations are observed at 1,204 cm−1 from DFT/B3LYP/6-31G level and 1,236 cm−1 in FT-IR spectrum and 1,235 cm−1 in Raman spectrum. The twisting mode of CH2 vibrations is observed at 1,245 cm−1 from DFT/B3LYP/6-31G level and from FT-IR spectrum it is found to be 1,273 cm−1. In Raman spectrum the twisting mode of CH2 vibrations is observed at 1,270 cm−1 [30].
5 Other molecular properties
5.1 Electrostatic potential, total electron density and molecular electrostatic potential
In the present study, the electrostatic potential (ESP), electron density (ED) and the molecular electrostatic potential (MEP) map figures for Linezolid calculated at DFT/B3LYP/6-311G(d,p) level are shown in Fig. 4(a)–4(c) respectively. It can be seen from the ESP figures, that while the negative ESP is localized more over the oxygen atoms and is reflected as a yellowish blob, the positive ESP is localized on the rest of the molecules. Molecular electrostatic potential (MEP) is related to the electronic density and is very useful descriptor in understanding sites for electrophilic attack and nucleophilic reactions as well as hydrogen bonding interactions [31, 32]. The negative (red)1 regions of MEP are related to electrophilic reactivity and the positive (blue) ones to nucleophilic reactivity, as shown in Fig. 4. As seen from the figure, the negative region is mainly localized on oxygen atoms (O3, O11, O12 and O23) whereas nucleophilic reactivity of the molecule is mainly localized on proton of C=O–NH group.

(a) Electrostatic potential (ESP), (b) electron density (ED) and (c) the molecular electrostatic potential (MEP) map for Linezolid calculated at B3LYP/6-311G(d,p) level. (Color figure online)
The MEP map in molecule suggests that potential swings widely between oxygen atoms of nitro group (dark yellow) and proton of C–N–OH group (dark blue) in the five membered ring (blue). The oxygen atoms reflect the most electronegative region and have excess negative charge, and the hydrogen atoms attached to the C–N–OH group bear the brunt of positive charge (blue region). The different values of the electrostatic potential at the surface are represented by different colors. Potential increases in the order red < orange < yellow < green < blue. The color code of these maps is in the range between −0.1 a.u. (deepest red) to +0.1 (deepest blue) in all compounds, where blue indicates strongest attraction and red indicates the strongest repulsion.
5.2 Atomic net charges
The total atomic charges of Linezolid obtained by Mulliken [29] using B3LYP/6-31G(d,p) and B3LYP/6-311G(d,p) levels have been listed in Table 3. The charge distribution on the molecule has an important influence on the vibrational spectra. The corresponding Mulliken’s plot is shown in Fig. 5 and it gives us information about the charge shifts relative to title molecule.

Mulliken’s plot of Linezolid
More charge density has been found at C10 than that of other ring carbon atoms. The high positive charge at C10 in Mulliken atomic charges are due to the effect of oxygen atom attached with C10. The nitrogen (N9) present in the five membered ring group has more negative charge than the nitro group nitrogen (N4, N20), which leads to the redistribution of electron density. The electron withdrawing character of the oxygen atom in the title molecule is demonstrated by a decrease of electron density on C5 and C6 atoms. In this study molecule, the Mulliken atomic charges show more negative values for the carbon atoms C21, C22 and C25 in B3LYP/6-311G(d,p) level. The above result shows that in B3LYP/6-311G(d,p) level the atomic charge changes are more sensitive to the changes in the molecular structure than B3LYP/6-31G(d,p) level.
6 UV–vis spectral studies and frontier molecular orbitals (FMOs)
In the UV–vis region with high extinction coefficients, all molecules allow strong π–π* and σ–σ* transition [33]. The experimental UV spectrum of Linezolid is plotted in Fig. 6. In an attempt to understand the nature of electronic transitions in terms of their energies and oscillator strengths, time-dependent DFT (TDDFT) calculations involving configuration interaction between the singly excited electronic states have been conducted. The calculated results involving the vertical excitation energies their molecular orbital contribution, oscillator strength (f) and wavelength are reported in Table 4. Due to the Frank–Condon principle, the maximum absorption peak (λmax) in an UV–vis spectrum corresponds to vertical excitation. TD-DFT calculations predict three transitions in the near ultraviolet region for Linezolid molecule. The strong transitions at 4.4282 eV (279.99 nm) with an oscillator strength f = 0.06312 and the other one 4.7495 eV (261.05 nm) with an oscillator strength f = 0.5014 have been observed. The maximum absorption wavelength corresponds to the electronic transition from the HOMO to (LUMO with 70 % contribution.

Experimental UV spectrum of Linezolid
The electronic absorption corresponds to the transition from the ground to the first excited state and is mainly described by one electron excitation from the HOMO to the LUMO. Commonly, the atom occupied by more densities of HOMO should have stronger ability for detaching electrons, whereas the atom with more occupation of LUMO should be easier to gain electrons. In order to evaluate the energetic behavior of the title molecule, the energies of HOMO, LUMO, LUMO +1, HOM −1 and their orbital energy gaps using B3LYP/6-31G(d,p) and 6-311G(d,p) basis sets are calculated and presented in Table 5. The pictorial illustrations of frontier molecular orbitals namely HOMO, HOMO −1, LUMO and LUMO +1, HOMO −2, HOMO −3, HOMO −4 for Linezolid are shown in Fig. 7. The HOMO lying at −5.09 eV is delocalized over π orbital and includes mixing of lone pairs of electrons on all the oxygen as well as nitrogen atoms. The H-1, 1.37 eV below the HOMO is localized over the oxygen and nitrogen atoms on the rings. The H-2 is 1.45 eV below the homo is localized mainly on the carbon atoms of the six membered rings. The H-3 is 1.56 eV below the HOMO and has mainly of π bonding character along with some sigma bond character, whereas the LUMO lying at −0.5483 eV, is a π* orbital, delocalized mainly on the carbon atoms of the six membered ring with some anti-bonding characters. From Table 5, it is evident that the absorption band centered around 220–260 nm arises mainly due to the four electronic transitions given by H → L + 1, H → L + 2, H → L + 3, H → L + 4 are assigned as π–π* type, whereas the more intense band around 260–280 nm, having maximum oscillator strength (0.6312), corresponds to H → L transition and is mainly characterized as n–π* type. The higher intensity of band corresponding to the n–π* type transition is attributed to the presence of large number of free lone pairs of electrons available on four oxygen and three nitrogen atoms. It can be seen from the plots that the HOMO is spread more over the nitrogen and the carbon atoms in the six and five membered rings that shows appreciable amount of π bonding character. The LUMO is mainly found to be uniformly distributed over the carbon atoms in the six membered ring and reflects a lot of anti-bonding π character. The energy gap of HOMO–LUMO explains the eventual charge transfer interaction within the molecule and the frontier orbital energy gap in case of Linezolid is found to be −4.9356 and −5.0956 eV obtained at B3LYP/6-31G(d,p) and B3LYP/6-311G(d,p) levels, respectively.

The atomic orbital compositions of the frontier molecular orbital for Linezolid
7 Thermodynamic properties
Using the DFT/B3LYP with large (6-311G(d,p)) and small (6-31G(d,p)) basis set calculations, several thermodynamic properties like heat capacity, zero point energy, entropy of Linezolid have been calculated and are presented in Table 6. The difference in the values calculated by both the methods is only marginal. Scale factors have been recommended for an accurate prediction in determining the zero point vibration energy (ZPVE) and the entropy (S). The variation in the ZPVE seems to be insignificant. The total energies are found to decrease with the increase of the basis set dimension. Dipole moment reflects the molecular charge distribution and is given as a vector in three dimensions. Therefore, it can be used as descriptor to depict the charge movement across the molecule. Direction of the dipole moment vector in a molecule depends on the centers of positive and negative charges. The total dipole moment of Linezolid determined by B3LYP method using 6-31G(d,p) and 6-311G(d,p) levels are 3.6321 and 2.3520 Debye, respectively. All the thermodynamic data provide helpful information for the further study on the Linezolid. They can be used to compute the other thermodynamic energies according to relationships of thermodynamic functions and estimate directions of chemical reactions according to the second law of thermodynamics in thermo chemical field [33].
8 Conclusions
The present investigation thoroughly analyzes the vibrational spectra, both infrared and Raman of Linezolid. The optimized geometries, harmonic vibrational wavenumbers and intensities of vibrational bands of Linezolid have been carried out using the DFT/B3LYP method using the standard 6-31G(d,p),6-311G(d,p) and M06-2X/6-31G(d,p) levels. The theoretical results have been compared with the experimental vibrations and it can be seen that the values obtained from M06-2X/6-31G(d,p) levels matches well with the experimental values. This DFT based quantum mechanical approach provides the most reliable theoretical information on the vibrational properties of Linezolid. The HOMO–LUMO analyses, MESP surface drawn and the electronic transitions identified for UV–Vis spectra may lead to understanding of properties and activity of Linezolid and the results are of assistance in the quest of the experimental and theoretical evidence for the title molecule in reaction intermediates and pharmaceuticals.
References
S J Brickner Curr. Pharm. Des. 2 175 (1996)
Wroe David (2002-02-28). "An antibiotic to fight immune bugs". The Age Retrieved 2009-05-16.
A P Wilson, J A Cepeda, S Hayman, T Whitehouse, M Singer and G Bellingan J. Antimicrob. Chemother. 58 470 (2006)
J P Abraham, I H Joe, V George, O F Nielson and V S Jayakumar Spectrochim. Acta Part A 59 193 (2003)
J Binoy, J P Abraham, I H Joe, V S Jayakumar, J Aubard and O F Nielson J. Raman Spectrosc. 36 63 (2005)
A D Becke J. Chem. Phys. 98 5648 (1993)
C Lee, W Yang and R G Parr Phys. Rev. B 37 785 (1988)
G Brancatelli, F Nicoló, S D Grazia, A M Monforte and A Chimirr ActaCryst. E 67 o1083 (2011)
A K Jissy, S Konar and A Datta Chem. Phys. Chem. 14 1219 (2013)
A K Jissy and A Datta J. Phys. Chem. B 117 8340 (2013)
A K Jissy and A Datta J. Phys. Chem. Lett. 4 1018 (2013)
M J Frisch et al. Gaussian 09 (Wallingford: Gaussian Inc.) (2009)
P L Polavarapu J. Phys. Chem. 94 8106 (1990)
K Rajalakshmi, S Gunasekaran and S Kumaresan Indian J. Phys. 88 733 (2014)
T A Olszak, O M Peteres, N M Blaton and C J De Ranter Acta Crystallogr. C 51 1304 (1995)
V Krishnakumar and R J Xavier Spectrochim. Acta Part A 61 253 (2005)
D Sajan, H J Ravindra, N Misra and I H Joe Vib. Spectrosc. 54 72 (2010)
G Socrates Infrared and Raman Characteristic Group Frequencies—Tables and Charts 3rd edn. (Chichester: Wiley) (2001)
S Sebastian and N Sundaraganesan Spectrochim. Acta Part A 75 941 (2010)
M M El-Nahass, M A Kamel, A F El-deeb, A A Atta and S Y Huthaily Spectrochim. Acta Part A 79 443 (2011)
N Sundaraganesan, C Meganathan and M Kurt J. Mol. Struct. 891 284 (2008)
V Arjunan, S Mohan, P Ravindran and C V Mythili Spectrochim. Acta Part A 72 783 (2009)
M S Weiss, M Brandi, J Suhnel, D Pal and R Hilgenfeld Trends Biochem. Sci. 26 521 (2001)
M Szafran, A Komasa and E B Adamska J. Mol. Struct. Theochem. 827 101 (2007)
S Gunasekaran, K Rajalakshmi, S Kumaresan Spectrochim. Acta Part A 112 351 (2013)
R M Silverstein, G C Bassler and T C Morrill Spectrometric Identification of Organic Compounds 4th edn. (New York: Wiley) p 245 (1981)
V M Geskin, C Lambert and J L Bredas J. Am. Chem. Soc. 125 15651 (2003)
V Arjunan, I Saravanan, P Ravindran and S Mohan Spectrochim. Acta Part A 74 642 (2009)
V Krishnakumar, R Sangeetha, R Mathammal and D Barathi Spectrochim. Acta Part A 104 77 (2013)
S Gunasekaran, K. Rajalakshmi and S Kumaresan Indian J. Phys. 87 723 (2013)
M Arivazhagan, S Jeyavijayan and J Geethapriya Spectrochim. Acta Part A 104 14 (2013)
R S Mulliken J. Chem. Phys. 23 1833 (1955)
R Zhang, B Dub, G Sun and Y Sun Spectrochim. Acta Part A 75 1115 (2010)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rajalakshmi, K., Gunasekaran, S. & Kumaresan, S. Density functional theory, comparative vibrational spectroscopic studies, highest occupied molecular orbital and lowest unoccupied molecular orbital analysis of Linezolid. Indian J Phys 89, 525–538 (2015). https://doi.org/10.1007/s12648-014-0618-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12648-014-0618-z