
Abstract
Fourier transform infrared (FT-IR) and Raman (FT-Ra) spectra of 1-(4-methoxyphenyl)-1H-imidazole were recorded and analyzed. The vibrational wavenumbers of the structure were computed by using the density functional theory (DFT)/Becke three Lee–Yang–Parr (B3LYP)/6–311 ++ g(d,p) basis set. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy values were determined to be − 1 eV and − 6.27 eV, respectively. The HOMO–LUMO energy gap value was found to be 5.27 eV. A large HOMO–LUMO gap means low reactivity in chemical reactions, indicating the high stability of the molecule. The HOMO–LUMO energy gap for the existing structure is relatively high, as a result the title molecule is hard. 1H, 13C NMR, and UV–Vis experimental and theoretical spectral analyses were given. Molecular electrostatic potential, Fukui functions, and charge analysis were performed to predict electrophilic and nucleophilic regions or atoms. The nonlinear optical properties of the structure were investigated, and it was found that the structure showed an important nonlinear optical property. Basic thermodynamic properties such as entropy (S), enthalpy changes (ΔH), heat capacity (Cp), Gibbs free energy (G), and zero-point vibration energy were calculated under constant pressure in the gas phase for different temperature values. It was determined that this compound has a significant antibacterial effect against some bacterial strains. Also, to support the antibacterial activity studies of the title molecule, molecular docking studies were carried out with protein structures of some microorganisms used in the antibacterial activity studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Imidazole is a 5-membered planar ring and is a highly polar compound. It is completely soluble in water and other polar solutions. Because the hydrogen atom can be found on any of the two nitrogen atoms, it occurs in two tautomeric forms (Verma et al. 2013). Many biologically active compounds have a five-membered nitrogen-containing heterocyclic ring in their structure (Shalini et al. 2010). Therefore, imidazole and its derivatives are one of the important heterocycles and are widely used in the field of pharmacology such as anti-inflammatory, analgesic, anti-convulsant, antitubercular, antimicrobial, anticancer, anti-Parkinson, and anti-HIV activities (Pandey et al. 2009; Boiani and González 2005; Spasov et al. 1999). Besides, imidazole and its derivatives play an important role in the charge transfer process in biological systems. The imidazole core is found naturally in many structures such as histidine, purine, histamine, and nucleic acid (Vijesh et al. 2011; Narasimhan et al. 2011). Imidazole is also found in the structure of many natural and synthetic drug molecules such as cimetidine and metronidazole. Cimetidine, a derivative of imidazole, is used in the treatment of stomach and intestinal ulcers. It is also used in the treatment of reflux disease caused by the backflow of stomach acid into the esophagus (Fınkelstein and Isselbacher 1978). Metronidazole is a drug that has been used for many years in the treatment of trichomoniasis, amebiasis, and giardiasis diseases. It is highly effective on anaerobic bacteria. Therefore, it is used in the treatment of anaerobic infections of the breast, head, gastrointestinal, and female genitourinary systems (Brogden et al. 1978).
Lu et al. (2012) synthesized a series of substituted 4-(2,6-dichlorobenzyloxy) phenyl thiazole, oxazole, and imidazole derivatives. In vitro antimicrobial studies of these derivative structures containing imidazole were performed on Staphylococcus aureus, Escherichia coli, Streptococcus Pneumoniae, and penicillin-resistant Streptococcus pneumoniae. Additionally, these derivatives were also investigated for in vitro antitubercular activity against Mycobacterium tuberculosis H37Rv in this study. The antifungal and antimycobacterial activities of 1-(3,5-Diaryl-4,5-dihydro-1H-pyrazol-4-yl)-1H-imidazole derivatives were investigated in vitro. These synthesized imidazole derivatives showed very important antifungal activity against the Candida albicans strain, which is a clinical strain. They showed antituberculosis activity against the M. tuberculosis H37Rv strain (Zampieri et al. 2008). In another study, amino-linked imidazole derivatives were synthesized from 4-aryloxazol-2-amine, 4-arylthiazol-2-amine, and 4-aryl-1H-imidazol-2-amine. The antibacterial effects of these synthesized derivatives on gram-positive bacteria Stophylococcus aureus and Bacillus subtilus with gram-negative bacteria Pseudomonas Aeruginosa and Klebsiella pneumonia were investigated. In this study, it was shown that compounds including thiazole and imidazole units exhibit significant activity (Padmavathi et al. 2011). 2-substituted-4,5-diphenyl-N-alkyl imidazole derivatives were synthesized (Jain et al. 2013). The antibacterial activities of these new structures were evaluated against S. aureus, Bacillus subtilus, and Escherichia coli. All synthesized compounds indicated moderate activity against these bacteria. Therefore, it was stated that the synthesized structures should be modified to increase the antimicrobial effect. Many new imidazole-(benz)azole and imidazole-piperazine derivatives were synthesized by Ozkay et al. (2010), and anticancer activity studies were conducted. As a result of the research, it was revealed that these synthesized structures are highly active compounds (Ozkay et al. 2010).
According to the literature, imidazole derivatives appear to have many biological activities in the field of pharmacology. Since 1-(4-methoxyphenyl)-1H-imidazole is an imidazole derivative, it is thought to be a very important biologically active structure. Therefore, it is very important to explain the chemical reactivity, electronic and optical properties as well as spectroscopic properties (FT-IR, FT-Ra, NMR, UV–visible) of this molecule. Density functional theory (DFT) in hybrid form (B3LYP) is one of the most accurate and efficient methods for calculating quantum chemical modeling of compounds.
Firstly, the optimized molecular structure of the 1-(4-methoxyphenyl)-1H-imidazole will be determined by the density functional theory method with the B3LYP hybrid functional using the 6–311 ++ G(d,p) basis set. In the vibrational spectral analysis section, vibrational frequencies will be calculated over the optimized structure, experimental FT-IR and FT-Ra spectra will be assigned, and theoretical and experimental vibration values will be compared with each other. In order to obtain information about the chemical reactivity of the title molecule, the energy values of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the optimized structure will be calculated and the energy gap between these two molecular orbitals will be determined. As well, the ionization potential, electron affinity, global hardness, chemical potential, electronegativity, global electrophilicity, the MEP map, Fukui functions, nonlinear optic properties, atomic charges, and thermodynamic properties are calculated at the same level of theory. The UV–Vis, and NMR spectra of the compound will be recorded and calculated theoretically. Finally, the antimicrobial activity of 1-(4-methoxyphenyl)-1H-imidazole will be determined and a molecular docking study performed.
Experimental
Instrumentation for recording spectra
1-(4methoxyphenyl)-1H-imidazole was supplied from Sigma-Aldrich Chemical Company. This structure is sold at 98% purity and the Cas and MDL numbers of the molecule are 10040–95-6 and MFCD00060492, respectively. Also, the melting temperature of the title molecule is given in the range of 59–67 ○C-lit in Sigma-Aldrich Company Safety Data Sheet 2006. FT-IR and FT-Raman spectra were recorded in the regions between 4000 and 400 and 4000 and 100 cm−1 without further purification. While Bruker FT-IR spectrometer with ATR equipment was used to obtain the FT-IR spectrum, Jasco FT-Raman spectrometer with NRS400 confocal microscope was used for the FT-Raman spectrum. Chemical shifts of the title molecule for 1H NMR and 13C NMR analysis were recorded in the range of 0–15 ppm and 100–200 ppm, respectively, in chloroform solvent at room temperature using the Bruker Ultrashield 300 MHz spectrometer. The UV–visible absorption spectrum was recorded in the range of 200–500 nm by using the Shimadzu UV-2101PC spectrophotometer.
Antibacterial and anti-quorum sensing activity by agar well diffusion method
The antibacterial activity of 1-(methoxyphenyl)-1H-imidazole was evaluated against thirteen different bacterial strains. These bacterial strains are the following: Staphylococcus aureus, Staphylococcus epidermidis, Bacillus cereus, Bacillus subtilis, Enterococcus feacalis, Listeria monocytogenes, Escherichia coli, Klebsiella pneumoniae, Shigella dynesteriae, Pseudomonas aeruginosa, Salmonella typhimurium, Vibrio anguillarum, and Enterobacter aerogenes. The test stock (60 mg) of the compound was dissolved in 1 ml of dimethyl sulfoxide (% 10 DMSO). The test compound was dropped into the well (6 mm in diameter) at a concentration of 60 mg/mL, and the plates were incubated for 24 h at 37° C for all bacteria. Antibacterial activity was measured based on the diameter of the inhibition zone in mm. Ampicillin was used as a standard antibacterial drug for the comparison of activities (CLSI 2007). Also, the anti-quorum activity of the compound was determined against Chromobacterium violeceum ATCC 12,472 in LB agar. C. violaceum (1 × 106) culture was spread on the LB agar surface by a swap. Then, wells were drilled in LB agar using a cork borer, and the tested compound (5 mg/mL) was applied to the wells after dissolving in 10% DMSO (McLean et al. 1997). The plates were incubated at 30 °C for 24 h to determine inhibition of pigment production around the well. The formation of a clear halo around the disc and the formation of bacterial growth inhibition was evaluated as positive. For statistical analysis, the title compound was subjected to a Duncan Test using the SPSS 21.0 statistical program to determine that it shows antibacterial activity on bacteria. As a result, this compound also gave statistically significant results (p < 0,01).
Antibacterial activity by minimum inhibitory concentration
All bacteria were grown in a nutrient broth (NB) medium. The stock solution of the compound was prepared in DMSO 1% at the final concentration and tested at concentrations 200, 100, 50, 25, 12.50, 6.25, 3.12, 1.56, and 0.78 mg/mL. Concentrations were determined in a sterile 96-well plate by using a broth micro-dilution technique The compound was prepared at a concentration of 200 mg/mL, its microtiter was determined, and it was incubated at 37 °C for 24 h. The lowest concentration at which no visible bacterial growth could be found was taken as the MIC value (Petrus et al. 2011).
Computational methods
Computational analyses were performed using the Gaussian 09 and GaussView 5.0.9 molecular visualization program package. The Density functional theory (DFT/B3LYP) at the 6–311 ++ G(d,p) basis set level was used to obtain the optimized structure of the title molecule (Çelik et al. 2020). Geometric parameters, vibrational wavenumbers, energy values of the HOMO–LUMO, Fukui functions, natural bond orbital (NBO) analysis, molecular electrostatic potential, UV spectrum analysis, and other properties were calculated over the optimized structure. 1H and 13C NMR analyses of the title molecule were performed by the DFT/B3LYP method using the 6–311+ G(2d,p) basis set with the GIAO method (Subashini and Periandy 2016).
Molecular docking investigations were performed using the Autodock Vina software (Trott and Olson 2009) and the AutoDockTools graphical interface (Morris et al. 2009). The 3D dimensional crystal structure of proteins was obtained from the Protein Data Bank (PDB ID:4URM, 4JCN, and 2RG7)(https://www.rcsb.org). The binding sites were centered on the selected protein and molecule, and a grid box was created with 40 × 40 × 40 points and 0.375 Å grid spacing. The outputs from Autodock Vina were visualized using the Discovery Studio Visualizer (https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/visualization/).
Results and discussion
Torsion angle
To obtain the most accurate results in quantum chemical calculations for the structure, firstly the most stable conformer structure was determined before the optimization process. Therefore, the torsion angles are calculated for the N–C bond between imidazole and phenyl ring and the C-O bond between methoxy group and phenyl ring. Firstly, the torsion angle (τ1) between the imidazole and phenyl ring was fixed at 180○ and changed in steps of 10○ from 0○ to 180○. In the second stage, the torsion angle (τ2) for the phenyl ring and the methoxy group was fixed at 180○ and changed in steps of 10○ between 0○ and 180○. The minimum energy was obtained to be -358.08992 × 103 kcalmol−1 for the dihedral angles τ1 and τ2, respectively, at 130○ and 180○ as shown in Fig. 1a, and 1b. As a result of this process, the molecular structure with the lowest energy was obtained and all other calculations were made after this structure was optimized (Ravindranath and Reddy 2020).

Absolute energy values as a function of rotation angle around C–N inter-bond in between phenyl and imidazole ring (I) and C–O inter-bond in between phenyl ring and methoxy group (II) of 1-(4-methoxyphenyl)-1H-imidazole were calculated at DFT/B3LYP/6–311 + + G(d,p) basis set
Molecular structure
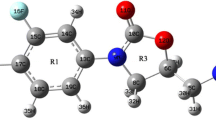
The title molecule was optimized at the DFT/B3LYP level of theory by using the 6–311 ++ g(d,p) basis set. The optimized molecular structure of 1-(4-methoxyphenyl)-1H-imidazole with atomic numbering is indicated in Fig. 2. Also, the geometrical values (bond lengths and bond angles) of the optimized molecule are listed in Table 1. These values were compared with the bond lengths and the bond angles of 2-chloro-1-(4-methoxyphenyl)-4,5-dimethyl-1H-imidazole (Thomas et al. 2018).

Optimized molecular structure and numbering of 1-(4-methoxyphenyl)-1H-imidazole at DFT/B3LYP/6–311 ++ G(d,p) basis set. The optimized structure gives the minimum energy level obtained for the title molecule
For 1-(4-methoxyphenyl)-1H-imidazole, the bond lengths of C4–C5, C4–C6, C5–C8, C6–C9, C7–C8, and C7–C9 bonds in the phenyl ring were determined to be 1.399, 1.392, 1.385, 1.396, 1.401 and 1.397 Å, respectively. While the bond lengths of C5–H14, C6–H15, and C8–H16 were found to be 1.083 Å, the bond length of C9–H17 was found to be 1.081 Å. Also, the imidazole ring has a single C–C bond with a bond length of 1.369 Å (C10–C12). The C-N bonds in the structure are as follows: N2–C4, N2–C10, N2–C11, N3–C11, and N3–C12. The bond lengths of these C–N bonds are 1.424, 1.385, 1.374, 1.312 and 1.376 Å, respectively. The C atoms in the imidazole ring make C10–H18, C11–H19, and C12–H20 bonds with H atoms. While the C10–H18 bond length was found 1.077 Å, the bond lengths of both C11–H19 and C12–H20 were found 1.079 Å. The bond length of the O1–C7 bond that connects the phenyl ring with the methoxy group is 1.362 Å. When the bond lengths of 1-(4-methoxyphenyl)-1H-imidazole are compared with that of 2-chloro-1-(4-methoxyphenyl)-4,5-dimethyl-1H-imidazole (CLMPDI), it is seen that the bond lengths are very close to each other (Thomas et al. 2018). Also, the bond angles of the title molecule and 2-chloro-1-(4-methoxyphenyl)-4,5-dimethyl-1H-imidazole (CLMPDI) are very close to each other. The reason for the small difference is that in Thomas et al. 2018, the chlorine (Cl) atom is attached to the C11 atom and CH3 (methyl)group is attached to the C10 and C12 atoms in the title molecule. The terms of root-mean-square deviations (RMSD) values were calculated for comparisons of bond lengths and angles. This value for bond lengths was obtained to be 0.0136. (Thomas et al. 2018). The RMSD value for the bond angles was found to be 0.527.
Assignment of vibrational spectra
The recorded FT-IR, FT-Raman wavenumbers, and theoretical scaled values are given in Table 2. Total energy distribution (TED) was calculated and fundamental vibrational modes were assigned by their TED (Erdoğdu 2010). Also, the experimental FT-IR and FT-Raman spectra are shown in Fig. 3. The theoretical wavenumber values are higher than the experimental values due to the use of a one-particle basis set and the electron correlation incomplete. Therefore, a scale factor is used to accommodate theoretical and experimental values. In this study, a scale factor of 0.983 was used for wavenumbers below 1800 cm−1 for theoretical values (Govindarajan et al. 2015), while a scale factor of 0.955 was used for wavenumbers above 1800 cm−1 (Çelik et al. 2020).

Experimental FT-IR (a) and FT-Raman (b) spectra of 1-(4-methoxyphenyl)-1H-imidazole
The C–H stretching vibrations for heteroaromatic molecules appear in the range of 3100–3000 cm−1. Besides, in-plane and out-of-plane C-H bending vibrations occur in the region of 1000–1300 cm−1 and 750–1000 cm−1, respectively (Erdogdu et al. 2010; Govindarajan et al. 2015; Saalem et al. 2011). The C–H stretching vibrations for phenyl and imidazole rings of the title molecule were observed at 3127 cm−1 (vw, IR)/3128 cm−1 (vw, Ra), 3106 cm−1 (m, IR)/3107 cm−1 (vw, Ra), 3060 cm−1 (vw, Ra), 3041 cm−1 (vw, Ra), and 2960 cm−1 (vw, IR) in the FT-IR and FT-Raman spectra. The C-H stretching vibrations were calculated at 3125 cm−1, 3099 cm−1, 3092 cm−1, 3063 cm−1, 3055 cm−1, 3042 cm−1, and 3040 cm−1. In a previous study, FT-IR, FT-Raman, and DFT simulations analysis of 4-(4-Fluoro-phenyl)-1H-imidazole were performed. In that study, the fluorophenyl C–H stretching vibrations were observed at 3056 cm−1, at 3004 cm−1 in the FTIR and at 3070 cm−1, at 3005 cm−1 in the FT-Raman, respectively (Erdogdu et al 2013). In the other study, the C–H stretching vibrations in the imidazole ring of 4-phenylimidazole were found at 3163 and 3135 cm−1 (Güllüoglu et al. 2011).
The C–H stretching vibrations for methyl and methylene groups define in between 2800 cm−1 and 3000 cm−1 (Sarıkaya and Dereli 2013). The symmetric stretching vibrations were observed at 2837 cm−1 (w, IR)/2838 cm−1 (vw, Ra) in experimental spectra and at 2871 cm−1 in theoretical calculation for the title molecule. In another study, the C–H stretching vibrations of the methyl group for 7-methoxy-4-methylcoumarin were determined at 2902 cm−1 and 2855 cm−1 in the FT-IR spectrum (Sarıkaya and Dereli 2013). The antisymmetric C-H stretching vibrations of methyl group of 1-(4-methoxyphenyl)-1H-imidazole were determined at 2996 cm−1 and 2928 cm−1, theoretically. This vibration was observed at 2960 cm−1 (vw, IR) in the FT-IR spectrum. While the CH3 antisymmetric stretching vibrations for 7-methoxy-4-methylcoumarin were observed at 3001 cm−1, 2954 cm−1, 2951 cm−1 in the FT-IR spectrum, these bands were observed at 3000 cm−1 and 2951 cm−1 in the FT-Ra spectrum (Sarıkaya and Dereli 2013).
The methyl group contains symmetric and asymmetric bending vibrations according to the in-plane and out-of-plane bending of the C-H bonds (Sarıkaya and Dereli 2013). These modes were calculated at 1450 cm−1 as symmetric, at 1478 cm−1 and 1467 cm−1 as asymmetric modes. The symmetric and asymmetric bending vibrations were observed at 1460 cm−1 (m, IR) and, 1450 cm−1 (vw, Ra), respectively. While the asymmetric bending vibration for 7-methoxy-4-methylcoumarin was observed at 1456 cm−1, 1441 cm−1 in the FT-IR spectrum, the symmetric bending vibration was observed at 1422 cm−1, 1384 cm−1, and 1376 cm−1 in the FT-IR and FT-Ra spectra (Sarıkaya and Dereli 2013). The in-plane rocking vibration that occurred in the methyl group molecule was determined at 1147 cm−1 from theoretical calculations. The out-of-plane rocking vibration for the methyl group was calculated at 1190 cm−1 and observed at 1181 cm−1 (w, IR)/1189 cm−1 (vw, Ra) in experimental spectra. The in-plane-rocking and out-of-plane-rocking vibrations of CH3 for 7-methoxy-4-methylcoumarin were observed at 1153 cm−1, 1038 cm−1, and 1134 cm−1, 979 cm−1 in the FT-IR spectrum (Sarıkaya and Dereli 2013).
The in-plane CCH bending modes of phenyl and imidazole rings were found at 1310 cm−1, 1254 cm−1, 1241cm−1, 1171 cm−1, 1112 cm−1, 1099 cm−1, 1053 cm−1, and 1008 cm−1 from the theoretical calculations for the title molecule. These modes are observed at 1257 cm−1 (vw, Ra), 1241 cm−1 (s, IR), 1109 cm−1 (m, IR)/1110 cm−1 (vw, Ra), and 1059 cm−1 (s, IR). The in-plane CCH bending modes of 4-phenylimidazole were observed in FT-IR as well as in the FT-Raman spectrum at 1309, 1183, 1157, 1062 cm−1 and 1310, 1181, 1154 cm−1 [25]. In the experimental FT-IR and FT-Ra spectra, the mixed out-of-plane bending modes for 1-(4-methoxyphenyl)-1H-imidazole were observed at 822 (vs, IR)/839 (vw, Ra), and 759 (s, IR). Also, these modes were found at 835 cm−1 and 723 cm−1 theoretically. Besides, the out-of-plane C–H bending vibration for 4-phenylimidazole was found at 895 cm−1 in the FT-IR spectrum and these vibrations were calculated to be 822 cm−1 and 799 cm−1 in theoretical calculations (Güllüoglu et al. 2011).
The C–C and C=C stretching vibrations in the aromatic molecule rings are found in the range of 1650–1200 cm−1 (Çelik et al. 2020). The C=C stretching vibration in the imidazole ring of the title molecule was calculated at 1508 cm−1. Also, these vibrations in the phenyl ring were found to be 1626 cm−1, 1591 cm−1, 1374 cm−1, and 1325 cm−1. In the experimental spectra, C–C and C=C stretching vibrations were observed at 1609 cm−1 (w, IR)/1612 cm−1 (m, Ra), 1590 cm−1 (w, IR)/1592 cm−1 (vw, Ra), 1363 cm−1 (not observed, IR)/1363 cm−1 (vs, Ra), and 1301 cm−1 (m, IR)/(not observed Ra). While fluorophenyl ring C–C stretching bands of 4-(4-Fluoro-phenyl)-1H-imidazole are observed at 1608 and 1563 cm−1 in FT-IR, the imidazole ring C–C stretching vibrations was observed in FT-Raman at 1513, 1293 cm−1 (Erdogdu et al. 2013).
The C-N stretching vibrations are generally found at 1342–1266 cm−1 (Srivastava et al. 2016). These vibrations of the title molecule were defined at 1374 cm−1, 1310 cm−1, 1303 cm−1, 1262 cm−1, 1125 cm−1, and 1053 cm−1. According to experimental spectra, the C-N stretching vibrations were indicated at 1363 cm-1(vs, Ra), 1321 cm−1 (w, IR)/ 1312 cm−1 (w/Ra), 1301 cm−1 (m, IR), and 1059 cm−1 (s, IR). According to Güllüoğlu et al. 2011, the C-N stretching vibration appeared at 1395 cm−1 for 4-phenylimidazole (Güllüoglu et al. 2011). These vibration for 4-(4-Fluoro-phenyl)-1H-imidazole was observed at 1459 cm−1, 1406 cm−1, 1313 cm−1, 1277 cm−1, and 1113 cm−1 in the FT-IR and at 1464 cm−1, 1403cm−1, 1312 cm−1, 1277 cm−1, and 1111 cm−1 in the FT-Raman (Erdogdu et al. 2013).
The C-O stretching vibration mode of 1-(4-methoxyphenyl)-1H-imidazole was calculated at 1039 cm−1 and observed at 1027 cm−1 as a strong band in the FT-IR spectrum.
1 H and 13C NMR analysis
1H NMR analysis provides information on the amounts of a number of protons as well as the nature of the immediate environment of these protons. 13C NMR analysis gives structural information about the different carbon atoms in the molecules. The experimental and computer calculation techniques for NMR analysis are widely used together to better understand and interpret the structure of molecules (Uzun et al. 2019). In the theoretical calculation for 1H and 13C NMR analysis of the title molecule, the density functional theory (DFT/B3LYP) method using the 6–311 + G(2d,p) basis set with the GIAO method has been selected as a computational method (Subashini and Periandy 2016). The experimental chemical shifts of hydrogen atoms of the 1-(4-methoxyphenyl)-1H-imidazole in chloroform were determined as follows: 1H NMR (300 MHz, CHCl3) δ/ppm: (14H-15H) 7.29, (16H-17H) 7.00, (18H) 7.32, (19H) 7.78, (20H) 7.00, (21H-22H-23-H) 3.85. Theoretical chemical shift values relative to TMS were found as follows: (14H) 7.74 ppm, (15H) 7.62 ppm, (16H) 7.31 ppm, (17H) 7.12 ppm, (18H) 7.36 ppm, (19H) 7.74 ppm, (20H) 7.31 ppm, (21H-22H) 3.80 ppm, and (23H) 4.10 ppm. In 13C NMR analysis, the chemical shift signals of carbons in the organic molecules are observed in the range of 100–160 ppm. The experimental chemical shifts of carbon atoms in chloroform of 1-(4-methoxyphenyl)-1H-imidazole were determined as follows: 13C NMR (300 MHz, CHCl3) δ/ppm: Also, theoretical chemical shift values for carbon atoms relative to TMS were determined as: (4C) 137.25, (5C) 129.59, (6C) 129.21, (7C) 166.17, (8C) 122.87, (9C) 113.3, (10C) 124. 65, (11C) 141.25, (12C) 135.83, (13C) 55.37. According to these results, experimental and theoretical chemical shift values of 1H and 13C NMR for the title molecule are in good agreement with each other. (See Fig. 4 a and b).

Experimental and theoretical a 1H and b 13C NMR spectra of 1-(4-methoxyphenyl)-1H-imidazole
HOMO–LUMO analysis
The electron donation and acceptance abilities of a molecule are determined according to HOMO and LUMO molecular orbital energy values. While HOMO is the highest energy molecular orbital occupied by electrons, LUMO is the lowest energy molecular orbital not occupied by electrons (Yazıcı et al. 2011; Ebrahimi et al. 2013). Chemical reactions take place by transferring electrons in the HOMO molecular orbital to the LUMO molecular orbital (Mumita et al. 2020). HOMO and LUMO molecular orbitals have an important role in electronic and optical properties (Demircioglu et al. 2015). Also, these molecular orbitals provide information on biological mechanisms in pharmaceutical studies (Kumar et al. 2018). Chemical properties of a molecule such as chemical hardness, chemical potential, reactivity, kinetic stability, chemical softness, electronegativity, electrophilicity, and optical polarizability can be explained by these molecular orbitals. According to HOMO and LUMO energy values of a molecule, the ionization potential, electron affinity, global hardness, chemical potential, electronegativity, and global electrophilicity can be calculated follows: \(I = - E_{{{\text{HOMO}}}}\), \(A = - E_{{{\text{LUMO}}}}\), \(\eta = {{\left( { - E_{{{\text{HOMO}}}} + E_{{{\text{LUMO}}}} } \right)} \mathord{\left/ {\vphantom {{\left( { - E_{{{\text{HOMO}}}} + E_{{{\text{LUMO}}}} } \right)} 2}} \right. \kern-\nulldelimiterspace} 2}\), \(\mu_{c} = {{\left( {E_{{{\text{HOMO}}}} + E_{{{\text{LUMO}}}} } \right)} \mathord{\left/ {\vphantom {{\left( {E_{{{\text{HOMO}}}} + E_{{{\text{LUMO}}}} } \right)} 2}} \right. \kern-\nulldelimiterspace} 2}\), \(\chi = - \mu_{c}\) and \(\omega = {{\mu_{c}^{2} } \mathord{\left/ {\vphantom {{\mu_{c}^{2} } 2}} \right. \kern-\nulldelimiterspace} 2}\eta\) (Kumar et al. 2019; Gece et al. 2012).
The HOMO and LUMO energy values of the title molecule in the gas phase and in water solvent were calculated at the B3LYP/6–311 ++ g (d,p) level and are given in Table 3 with the energy values of other chemical properties. While the HOMO and LUMO molecular orbital energy values in the gas phase were obtained as − 6.27 eV and − 1 eV, these energy values in water solvent were calculated as − 6.32 eV and − 0.90 eV, respectively. While the HOMO energy decreased with the increase of the dielectric constant, the LUMO energy increased. The HOMO–LUMO energy gap values in the gas phase and in water solvent were found to be 5.27 and 5.42 eV, respectively. According to a study in the literature, HOMO–LUMO energy gap values in different solvents were found to be higher than the energy value in the gas phase, and also support our study (Çelik et al. 2017). As a result, the energy gap value of the title molecule increased depending on the increase in the dielectric constant. Also, the ionization potential value of the title molecule in the water increased. This means that the transfer of electrons from HOMO to LUMO is easier in the gas phase. According to these energy gap values, we can say that the title molecule does not have high reactivity and that this molecule is a hard structure. Also, the values of ionization potential (I), electron affinity (A), global hardness (η), electronegativity (χ), chemical potential (µc), global electrophilicity (ω) were found as follows: 6.27, 1, 2.64, 3.64, − 3.64 and 2.51 eV for the gas phase, and 6.32, 0.9, 2.71, 3.61, − 3.61 eV, and 2.41 eV for the water solvent.
The HOMO and LUMO distributions of the molecule are given in Fig. 5. According to the HOMO distribution of the molecule, it is seen that the electron density is distributed over almost the entire molecule. On the other hand, LUMO is mostly distributed on the phenyl ring.

The calculated energy values in the gas phase of the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO) of the 1-(4-methoxyphenyl)-1H-imidazole
The simulated density state spectrum (DOS) is shown in Fig. 6. This spectrum gives states of molecular orbitals at different energy levels and helps to estimate optical transition probabilities. The molecular orbitals were calculated using Mulliken population analysis and the DOS spectrum was created. The blue lines in this spectrum allow us to see virtual orbitals better, while the green lines allow us to see occupied orbitals better (Çelik et al. 2020).

The simulated density of states spectrum (DOS) of 1-(4-methoxyphenyl)-1H-imidazole. The spectrum lines allow us to see occupied and virtual orbitals more, respectively
UV–Vis spectrum analysis
The ultraviolet spectrum analysis of the 1-(4methoxyphenyl)-1H-imidazole was researched by theoretical and experimental. The electronic absorption spectrum was measured in DMSO. The theoretical absorbance values for the title molecule were computed by using the DFT/B3LYP/6–311 ++ G(d,p) theory level. The experimental and theoretical UV spectra are given in Fig. 7, and the experimental and calculated wavelengths, excitation energies, oscillator strengths, and major contributions are listed in Table 4. The measured experimental spectrum shows an intense and broad-centered band at approximately 298 nm. According to the DFT calculation, two absorbed bands are found at 200.52 nm and 249.16 nm. The absorbed band is calculated at 200.52 nm with an oscillator strength f = 0.2655 which transition is due to the contributions of HOMO/LUMO + 6, HOMO-2/LUMO, HOMO-2/LUMO + 1. As can be seen from Table 4, the contribution to the charge transition in 249.16 nm with an oscillator strength f = 0.5511 is from the HOMO/LUMO and HOMO/LUMO + 1.

Experimental and calculated UV visible spectra of 1-(4-methoxyphenyl)-1H-imidazole
Molecular electrostatic potential
The molecular electrostatic potential is a powerful electronic density tool used to determine electrophilic and nucleophilic reaction regions and understand hydrogen bond interactions (Kumar et al. 2019; Saravanan et al. 2015). The MEP surface explains the neutral, negative and positive electrostatic potential with a color band that includes red, orange, yellow, green, and blue. While the red color is used to determine the negative electrostatic potential region on the MEP surface, the dark blue represents the positive electrostatic potential region. The yellow color represents the less negative potential region than the red color. The green color represents the neutral regions compared to the red and dark blue regions, while the light blue region represents the less positive potential region than the blue region (Mumita et al. 2020; Saravanan et al. 2015). Figure 8 shows the molecular electrostatic potential (MEP) map created for 1-(4-methoxyphenyl)-1H-imidazole. The electrophilic region (negative) is localized on the N3 atom in the imidazole ring. Especially, the C atom and the H atoms in the methoxy group and the other H atoms are located in the less positive potential region. The O atom in the methoxy group and the carbon atoms in the imidazole and phenyl ring are in the yellow region. In this case, we can say that these atoms are in the less electronegative region (Fig. 10).

Molecular electrostatic potential surface map for 1-(4-methoxyphenyl)-1H-imidazole. This map allows us to visualize variably charged regions of the title molecule
In addition, a two-dimensional contour map of molecular electrostatic potential surface values for 1-(4-methoxyphenyl)-1H-imidazole was designed (see Fig. 9). In this two-dimensional map, the all molecule is in the less negative potential region.

Two dimensions contour map of molecular electrostatic potential surface values (all in arbitrary unit) for 1-(4-methoxyphenyl)-1H-imidazole. The region around the oxygen and nitrogen atoms were found to be slightly electron rich

Thermodynamic properties (heat capacity, entropy enthalpy changes and Gibbs free energy) of 1-(4-methoxyphenyl)-1H-imidazole as function of temperature in the range 100–1000 Kelvin at constant pressure
Thermodynamic properties
Basic thermodynamic properties such as entropy (S), enthalpy changes (ΔH), heat capacity (Cp), Gibbs free energy (G), and zero-point vibration energy were calculated at the 6–311 ++ g(d, p) basis set under constant pressure in the gas phase for different temperature values. Thermodynamic properties were calculated in the temperature range of 100–1000 K. The zero-point vibration energy is constant at all temperatures and this property of the molecule is characteristic. The zero point vibration energy of 1-(4-methoxyphenyl)-1H-imidazole was found to be 481.59 kJmol−1. The values of entropy, heat capacity, and enthalpy changes for the title molecule showed a rise with increasing temperature. The change in entropy and enthalpy that occur with the increase in temperature shows that the structure changes its thermodynamic system. The G is defined by the formula given below (Çelik et al. 2020; Junchao et al. 2018):
where H, T, and S are enthalpy, temperature and, entropy, respectively. Enthalpy (H) can be expressed as
where E, P and V are internal energy, pressure, and volume, respectively. Therefore;
In solid and liquid phases, at atmospheric pressure the value of PV is far less than the Gibbs free energy value, so PV can be neglected.
The value of Gibbs free energy decreased because of the rise of molecular vibrations and entropy depends on increasing temperature. Thermodynamic properties at different temperatures are listed in Table 5 and are presented Fig. 10.
Fukui Functions
The Fukui function is one of the most basic and common reactivity indicators used to determine the reactive regions of the molecule. The Fukui function is defined as the change in the density function of the molecule as a result of adding or removing some amount of electrons to the molecule under the constraint of a constant external potential (Demircioglu et al. 2015; Ayers and Parr 2000). The Fukui functions are calculated using a finite-difference methodology for the neutral, cationic, and anionic states of the structure in the same molecular geometry. The Fukui functions of the molecule for three states are defined as follows:
In these equations, qk defines the atomic charge at the rth atomic site and N, N + 1, and N − 1 are the total number of electrons in the neutral, anion, and cation state of the molecule, respectively (Sethi et al. 2016; Pilepic and Urslic 2001). The dual descriptors Δf(r) provide an evident difference between the nucleophilic and electrophilic attacks at a particular site with their signs. The dual descriptor from Fukui functions is calculated by the following formula;
When Δf(r) > 0, the site is favored for a nucleophilic attack, whereas when Δf(r) < 0, the site is favored for an electrophilic attack (Sheeba et al. 2021).
The dual descriptor positive values are in the following order: C9 > N3 > C8 > C12 > O1 > C11 > C7 > N2 > C4 > H20 > H19 > C5 > C6. The negative dual descriptor values are in the following order: C10 > H23 > H21 > C13 > H22 > H17 > H15 > H14 > H13. The data for the Fukui functions are given in Table 6. Figure 11a shows electrophilic and nucleophilic regions calculated from the dual descriptors. Also, Fig. 11b presents a graphical view of the Fukui function for 1-(4-methoxyphenyl)-1H-imidazole.

According to dual descriptors calculated from Fukui functions, electrophilic and nucleophilic regions of the title compound (a), Graphical view of Fukui function for 1-(4-methoxyphenyl)-1H-imidazole (b)
When comparing the results obtained from Fukui functions with the molecular electrostatic potential surface map, it seems that the data are compatible with each other. We have stated that the N atoms in the imidazole ring and the O atom in the methoxy group are in the negative electrostatic potential region (electrophilic) on the MEP surface. Besides, it was determined that C atoms in the imidazole and phenyl rings had lower negative electrostatic potential than N and O atoms from the MEP surface. Therefore, we can say that the results obtained from Fukui functions are compatible with the MEP surface map.
Nonlinear optical properties
Recently, there has been an increasing interest in technical applications of nonlinear materials. Research of nonlinear optical properties was initially made with inorganic materials such as quartz, potassium, dihydrogen phosphate, cadmium sulfide, cadmium selenite, cadmium germanium arsenite lithium niobate. Nowadays, NLO optical properties of organic and organometallic molecules are being investigated (Senge et al. 2007). Molecules with NLO properties are used in many fields such as telecommunications, data storage, computers, and imaging technologies. Therefore, it is very important to investigate new molecules with NLO properties (Eryılmaz et al.2016). The optical property of a substance is expressed as the response of electrons in the structure to exposure to an electric field. The dipole moment is reshaped in a molecule that is exposed to external electricity. The dipole moment of a molecule interacting with an external electric field is as follows:
where μ0 is the permanent dipole moment of the molecule, α molecular polarity, β first-order hyperpolarizability, γ quadratic hyperpolarizability (Leszczynski et al. 2006). Hyper polarizability is defined as the quantity that effects the nonlinear optical properties of the material. The permanent dipole moment (μ), the mean polarizability (\(\mathop \alpha \limits^{ - }\)), the anisotropy of the polarizability (\(\Delta \alpha\)) and first-order hyperpolarizability (β0) values are calculated as follows (Yamijala et al. 2015; Gunduz et al. 2017):
The nonlinear optical properties for 1-(4-methoxyphenyl)-1H-imidazole are given in Table 7.
The urea compound is used as a brink value in NLO characteristic analysis and the other structures are compared with this brink value. The first hyperpolarizability value of urea is 0.3728 × 10–30 esu (Çelik et al. 2020). This value obtained for 1-(4-methoxyphenyl)-1H-imidazole is 1.75 × 10–30 esu and this value is approximately 5 times greater than that of urea. Therefore, we can say that the title molecule has nonlinear optical properties (Çelik et al. 2020; Eryılmaz et al. 2016).
Charge analysis
The NBO analysis uses for examining molecular orbitals that give information about the reactivity of molecules. It is an effective method to explain the electronic charges of atoms and the binding potential of a molecule. Atomic charges affect molecular moment, molecular polarity, electronic structure, bond types, bond structures, and many other properties in a molecular system. Therefore, charge density values are very important to in quantum chemical calculations (Singh et al. 2018; Yurdakul et al. 2019; Büyükmurat and Akyüz 2001). Atomic charge distribution analysis was performed using 6–311 + + g(d,p) basis set on the optimized structure of 1-(4-methoxyphenyl)-1H-imidazole. NBO, Hirshfeld, and APT charge values are listed in Table 8 and presented Fig. 12. The magnitudes of all H atoms in the structure were found to be positive. According to three charge analysis types, the O1 atom in the methoxy group, the N2, N3 atom in the imidazole ring, and the C5, C6, C8, and C9 atoms in the phenyl ring are electronegative. Carbon atoms that have positive charge values were found as C4, C7, and C11 atoms. All these data are in accord with the results obtained from the MEP surface and Fukui functions.

Natural Bond Orbital (NBO), Hirshfeld and Atomic Polar Tensor (APT) electron charge distribution for 1-(4-methoxyphenyl)-1H-imidazole
Antibacterial and anti-quorum sensing property analysis
In this study, the Inhibition zone in mm of diameter and minimum inhibitory concentrations (MIC) in mg/mL for the antibacterial property of the tested compound are summarized in Table 8. The compound showed very high antibacterial activity against all bacteria at 60 mg/ml concentration. Also, the activity is greater compared to the standard drug ampicillin. The bacterial inhibition zone diameters of the compound are 30 ± 1 mm for S. epidermidis, S. dysenteriae, are 25 ± 1 mm for S. aureus, B. subtilis, K. pneumoniae, P. aeruginosa, V. anguillarum, and are 20 ± 1 mm for E. feacalis, L monocytogenes, E. coli, S. typhimurium, E. aerogenes. The results of the MIC of compound synthesized on all bacteria are shown in Table 9. The MIC values of the compound were obtained to be approximately 3.12 ± 0.12 mg / mL for C. violaceum, E. coli, V. anguillarum, and S. epidermidis, 1.56 ± 0.06 mg/mL for B. cereus, E. feacalis, K. pneumoniae, E. aerogenes in 200 mg/mL concentration. Also, this value was found 0.78 ± 0.08 mg/mL for S. aureus, B. subtilis, S. dysenteriae, P. aeruginosa, S. typhimurium, and 0.39 ± 0.09 mg/mL for L. monocytogenes. The overall results indicate that most of the title compound showed very high activity against different tested bacteria. In the previous antibacterial study, 2-(4-methoxynaphthalen-1-yl)-1-(4-methoxyphenyl)-1H-phenanthro[9,10- d] imidazole showed significant inhibitory activity against S. aureus, S. Typhi, and E. coli, whereas imidazole showed low inhibitory activity against P. Aeruginosa (Ramanathan 2017).
Anti-quorum sensing activity test of compound 1-(4-methoxyphenyl)-1H-imidazole was performed with C. violaceum ATCC 12472 producing Gram-negative and violeceum pigment. As a result of bacteria resistance to antibiotics, infections cannot be controlled. Therefore, there is a need to synthesize new compounds. Quorum sensing (QS) signaling molecule is used in intercellular interaction during infection of pathogenic bacteria. Pathogenic, virulence, and biofilm formation in many bacteria are regulated by QS signaling systems. In the QS inhibition system, it may cause a decrease in virulence in pathogenic bacteria and a defense against bacterial infections (Asif and Imran 2020). In our study, the compound 1-(4-Methoxyphenyl)-1H-imidazole created a very good anti-QS activity (zone diameter = 30 ± 1 mm) (Table 9).
Molecular Docking
Molecular docking is one of the most common applications used in structural molecular biology and computer-assisted drug design. The ligand–protein interaction predicts the dominant binding modes of the ligand to a protein (Amul et al. 2019). The interactions of the title molecule with 4URM, 4JCN, and 2RG7 proteins obtained from the protein data bank (PDB) (https://www.rcsb.org/) were performed in molecular docking simulations using the AutoDockTools graphical user interface. The binding affinities of the complexes were generated using the AutoDock Vina program (Haruna et al. 2019). The docking results for the 1-(4-methoxyphenyl)-1H-imidazole molecule are given in Table 10, 11, and 12. Also, the docked conformation and hydrogen bond interactions are presented in Fig. 13. The title molecule was docked into the reactive regions of the 4URM, 4JCN, and 2RG7 proteins, and the docking minimum binding affinity values were found to be -5.30 kcal/mol, -5.10 kcal/mol, and -5.90 kcal/mol, respectively.

Molecular docking and Hydrogen bond interaction between 1-(4-methoxyphenyl)-1H-imidazole and 4URM (a), 4JCN (b) and 2RG7 (c) proteins
The general assumption is that a good docking should be 2 Å according to the RMSD values (Stigliani et al. 2012). The docking results are presented revealed a binding site of the mode 1 that contains a pose with the most favorable binding based on its lowest free energy and RMSD ~ 0.00 Å.
As a result of the ligand interaction of the 4URM protein, it was determined that the N3 atom of 1-(4-methoxyphenyl)-1H-imidazole is located at a distance of 2.53 Å from the H atom of THR173. In the ligand interaction of the 4JCN protein, the H atom of ASN274 was found at a distance of 1.95 Å from the N3 atom of the ligand. For the 2RG7 protein, the hydrogen bond acceptor interaction was defined at a distance of 2.35 Å between the N3 atom of the ligand and TRP87. According to these results, we can say that the title molecule may exhibit biological activity and contribute to the synthesis and development of new drugs in the future.
Conclusion
A complete vibrational analysis of 1-(4-methoxyphenyl)-1H-imidazole was performed using the density functional theory basis by Becke-3-Lee–Yang–Parr (B3LYP) functional using 6–311 ++ G(d,p) basis set. The computational wavenumbers suggested by the TED calculations are in agreement with the observed wavenumbers from the FT-IR and FT-Ra spectra. The molecular electrostatic potential (MEP), Fukui Functions, and charge analysis are powerful electronic density tools used to determine electrophilic and nucleophilic reaction regions and understand hydrogen bond interactions. In these three methods, the N3 atom in the imidazole ring and the O1 atom in the methoxy group were found in the electrophilic region. Also, the experimental and theoretical 1H and 13C NMR analyses were performed to better understand and interpret the structure of the title molecule. It is determined that the experimental and theoretical chemical shift values are in good agreement with each other. The ultraviolet spectrum analysis of the 1-(4-methoxyphenyl)-1H-imidazole was performed by theoretical and experimental methods, and wavelengths, excitation energies, oscillator strengths, and major contributions were determined. While the values of entropy, heat capacity, and enthalpy changes for the title molecule showed a rise with increasing temperature, Gibbs free energy decreased because of the rise of molecular vibrations and entropy. The first hyperpolarizability value for 1-(4-methoxyphenyl)-1H-imidazole is 1.75 × 10–30 esu and this value is found to be approximately 5 times greater than that of urea. Therefore, this molecule has a high nonlinear optic property. Also, antibacterial studies revealed that the reported 1-(4-Methoxyphenyl)-1H-imidazole against S. epidermidis, C. violaceum, and S. dysenteriae showed considerable inhibition activity. Additionally, molecular docking experiments were carried out with the protein structures of certain microorganisms utilized in antibacterial activity research to support the antibacterial activity of the investigated title molecule. As a result of antibacterial and molecular docking studies, we can say that 1-(4-methoxyphenyl)-1H-imidazole may be promising a strong new antibacterial effect.
References
Amul B, Muthu S, Raja M, Sevvanthi S (2019) Spectral, DFT and molecular docking investigations on Etodolac. J Mol Struc 1195:747–761. https://doi.org/10.1016/j.molstruc.2019.06.047
Asif M, Imran M (2020) Effect of Quorum sensing inhibitor agents against Pseudomonas aeruginosa. Russ J Bioorgan Chem 46:194–164. https://doi.org/10.1134/S1068162020020041
Ayers PW, Parr RG (2000) Variational principles for describing chemical reactions: the fukui function and chemical hardness revisited. J Am Chem Soc 122:2010–2018. https://doi.org/10.1021/ja9924039
Biovia (2021) Visualization. https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/visualization/. Accessed 01 October 2021.
Boiani M, González M (2005) Imidazole and benzimidazole derivatives as chemotherapeutic agents. Mini-Reviews Med Chem 5:409–424. https://doi.org/10.2174/1389557053544047
Brogden RN, Heel RC, Speight TM, Avery GS (1978) Metronidazole in anaerobic infections: a review of its activity. Pharmacokinet Ther Use Drugs 16(5):387–417. https://doi.org/10.2165/00003495-197816050-00002
Büyükmurat Y, Akyüz S (2001) Theoretical and experimental IR spectra and assignments of 3-aminopyridine. J Mol Struct 563:545–550. https://doi.org/10.1016/S0022-2860(00)00801-2
CLSI Document M100-S17 (2007) Performance standards for antimicrobial susceptibility testing 17th Informational Supplement, Clinical and Laboratory Standards Institute, Wayne, Pennsylvania: CLSI, Table 1, p. 26
Çelik S, Alp M, Yurdakul S (2020) A combined experimental and theoretical study on vibrational spectra of 3-pyridyl methyl ketone. Spectrosc Lett 53(4):234–248. https://doi.org/10.1080/00387010.2020.1734840
Çelik S, Badoğlu S, Yurdakul S (2017) Vibrational spectroscopic and density functional study on 1,2,4- triazolo-[1,5-a]pyrimidine. Vibrat Spect 92:20–26. https://doi.org/10.1016/j.vibspec.2017.08.011
Demircioglu Z, Kastas CA, Büyükgüngör O (2015) The spectroscopic (FT-IR, UV-Vis), Fukui Function, NLO, NBO, NPA and Tautomerism Effect Analysis of (E)-2-[(2-hydroxy-6- methoxybenzylidene)amino]benzonitrile. Spectrochim Acta A 139:539–548. https://doi.org/10.1016/j.saa.2014.11.078
Ebrahimi H, Hadi JS, Al-Ansari HS (2013) A new series of Schiff bases derived from sulfa drugs and indole-3-carboxaldehyde: synthesis, characterization, spectral and DFT computational studies. J Mol Struct 1039:37–45. https://doi.org/10.1016/j.molstruc.2013.01.063
Erdogdu Y, Manimaran D, Güllüoglu MT, Amalanathan M, Joeb IH, Yurdakul S (2013) FT-IR, FT-Raman, NMR Spectra and DFT Simulations of 4-(4-Fluoro-phenyl)-1H-imidazole. Opt Spect 114(4):525–536. https://doi.org/10.7868/S0030403413040077
Erdoğdu Y, Unsalan O, Sajan D, Gulluoglu MT (2010) Structural conformations and vibrational spectral study of chloroflavone with density functional theoretical simulations. Spectrochim Acta A 76:130–136. https://doi.org/10.1016/j.saa.2010.02.043
Eryılmaz S, Akdemir N, İnkaya E (2016) The examination of molecular structure properties of 4,4′-oxydiphthalonitrile compound: combined spectral and computational analysis approaches. Spectrosc Lett 52:28–42. https://doi.org/10.1080/00387010.2018.1544569
Fınkelstein W, Isselbacher KJ (1978) Medical Intelligence. New England J Med 2:992–996
Gece G, Bilgiç S (2012) Molecular-level understanding of the inhibition efficiency of some inhibitors of zinc corrosion by quantum chemical approach. Ind Eng Chem Res 51:14115–14120. https://doi.org/10.1021/ie302324b
Govindarajan M, Ganasan K, Periandy S, Karabacak M, Mohan S (2015) Vibrational spectroscopic analysis of 2-chlorotoluene and 2-bromotoluene: A combined experimental and theoretical study. Spectrochim Acta A Mol Biomol Spect. https://doi.org/10.1016/j.saa.2010.08.038
Gunduz SK, Bicak B, Celik S, Akyuz S, Ozel AE (2017) Structural and spectroscopic investigation on antioxidant dipeptide, l-Methionyl-l-Serine: A combined experimental and DFT study. J Mol Struct. https://doi.org/10.1016/j.molstruc.2017.02.075
Güllüoglu MT, Erdogdu Y, Karpagam J, Sundaraganesan N, Yurdakul S (2011) DFT, FT-Raman, FT-IR and FT-NMR studies of 4-phenylimidazole. J Mol Struct 990(1–3):14–20. https://doi.org/10.1016/j.molstruc.2011.01.001
Hakiria R, Ameurb I, Abidb S, Derbela N (2018) Synthesis, X-Ray Structural, Hirshfeld surface analysis, FTIR, MEP and NBO analysis using DFT study of a 4-chlorobenzylammonium nitrate (C7ClH9N)+(NO3)-. J Mol Struct 1164:486–492. https://doi.org/10.1016/j.molstruc.2018.03.068
Haruna K, Kumar VS, Maray YS, Popoola SA, Thomas R, Roxy MS, Al-Saadi AA (2019) Conformational profile, vibrational assignments, NLO properties and molecular docking of biologically active herbicide1,1-dimethyl-3-phenylurea. Heliyon 5:e01987. https://doi.org/10.1016/j.heliyon.2019.e01987
Jain AK, Ravichandran V, Sisodiya M, Agrawal RK (2010) Syntthesis and antibacterial evaluation of 2-substituted-4,5-diphenylN-alkyl imidazole derivatives. Asian Pac J Trop Med. https://doi.org/10.1016/S1995-7645(10)60113-7
Junchao L, Han H, Zhang X, Li S, Ge S, Zhang G, Gao T (2018) First-principles study the structural, electronic, vibrational and thermodynamic properties of Zr1-xHfxCoH3. Int J Hydrog Energy 43(41):19152–19163. https://doi.org/10.1016/j.ijhydene.2018.08.130
Kumar S, Radha A, Kour M, Kumar R, Chouaih A, Pandey SK (2019) DFT studies of disubstituted diphenyldithiophosphates of nickel(II): Structural and some spectral parameters. J Mol Struct 1185:212–218. https://doi.org/10.1016/j.molstruc.2019.02.105
Kumar S, Saini V, Maurya IK, Sindhu J, Kumari M, Kataria R, Kumar V (2018) Design, synthesis, DFT, docking studies and ADME prediction of some new coumarinyl linked pyrazolylthiazoles: Potential standalone or adjuvant antimicrobial agents. Ploes One 13(4):0196016. https://doi.org/10.1371/journal.pone.0196016
Leszczynski J, Papadopoulos MG, Sadlej AJ (2006) Non-linear optical properties of matter. Springer, Dordrecht, The Netherlands
Lu X, Liu X, Wan B, Franzblau SG, Chen L, Zhou C, You, (2012) Synthesis and evaluation of anti-tubercular and antibacterial activities of new 4-(2,6-dichlorobenzyloxy)phenyl thiazole, oxazole and imidazole derivatives, Part 2. Eur J Med Chem 49:164–171. https://doi.org/10.1016/j.ejmech.2012.01.007
McLean KH, Winson MK, Fish L, Taylor A, Chhabra SR, Camara M, Daykin M, Lamb JH, Swift S, Bycroft BW, Stewart GS, Williams P (1997) Quorum sensing and Chromobacterium violaceum: exploitation of violacein production and inhibition for the detection of N-acyl homoserine lactones. Microbiology 143(12):3703–3711. https://doi.org/10.1099/00221287-143-12-3703
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptorflex-ibility. J Comput Chem 30:2785–2791. https://doi.org/10.1002/jcc.21256
Mumita MA, Pala TK, Alam MA, Islama AAM, Paulb S, Sheikh MC (2020) DFT studies on vibrational and electronic spectra, HOMO–LUMO, MEP, HOMA, NBO and molecular docking analysis of benzyl-3N-(2,4,5-trimethoxyphenylmethylene) hydrazinecarbodithioate. J Mol Struct 1220:128715. https://doi.org/10.1016/j.molstruc.2020.128715
Narasimhan B, Sharma D, Kumar P (2011) Biological importance of imidazole nucleus in the new millennium. Med Chem Res 20:1119–1140. https://doi.org/10.1007/s00044-010-9472-5
Ozkay Y, Isıkdag I, Incesu Z, Akalın G (2010) Synthesis of Synthesis of 2-subsituted-N-[4-(1-methyl-4,5-diphenyl-1H-imidazole-2-yl)phenyl]acetamide derivatives and evalutaion of their anticancer activity. Eur J Med Chem 45:3320–3328. https://doi.org/10.1016/j.ejmech.2010.04.015
Padmavathi V, Kumari CP, Venkatesh BC, Padmaja A (2011) Synthesis and antimicrobial activity of amido linked pyrrolyl and pyrazolyl-oxazoles, thiazoles and imidazoles. Eur J Med Chem 46:5317–5326. https://doi.org/10.1016/j.ejmech.2011.08.032
Pandey J, Tiwari VK, Verma SS, Chaturvedi V, Bhatnagar S, Sinha S, Gaikwad AN, Tripathi RP (2009) Synthesis and antitubercular screening of imidazole derivatives. Eur J Med Chem 44:3350–3355. https://doi.org/10.1016/j.ejmech.2009.02.013
Petrus EM, Tinakumari S, Chai LC, Ubong A, Tunung R, Elexson N, Chai LF, Son R (2011) A study on the minimum inhibitory concentration and minimum bactericidal concentration of Nano Colloidal Silver on food-borne pathogens. Int Food Res 18:55–66
Pilepic V, Urslic S (2001) Nucleophilic reactivity of the nitroso group. Fukui function DFT calculations for nitrosobenzene and 2-methyl-2-nitrosopropane. J Mol Struct (theochem) 539:41–49. https://doi.org/10.1016/S0166-1280(00)00642-4
Ramanathan P (2017) Synthesis, Spectral Characterization and Biological Studies of 2-(4-Methoxynaphthalen-1-Yl)-1-(4-Methoxyphenyl)-1H-Phenanthro[9,10-d] imidazole. Mod Chem Appl. https://doi.org/10.4172/2329-6798.1000242
Ravindranath L, Reddy BV (2020) Theoretical and experimental study of torsional potentials, molecular structure (monomer and dimer), vibrational analysis and molecular characteristics of some dimethyl bipyridines. J Mol Struct 1200(36):127089. https://doi.org/10.1016/j.molstruc.2019.127089
RSCB PDB Protein Data Bank. https://www.rcsb.org/. Accessed 01 October 2021.
Saalem H, Krishnan AR, Erdogdu Y, Subashchandrabose S, Thanikachalam V, Manikandan G (2011) Density functional theory studies on 2,5-bis(4-hydroxy-3-methoxybenzylidene)-cyclopentanone. J Mol Struct 999:2–9. https://doi.org/10.1016/j.molstruc.2011.02.039
Saravanan RR, Seshadri S, Gunasekaran S, Mendoza RM, Granda SG (2015) Conformational analysis, X-ray crystallographic, FT-IR, FT-Raman, DFT, MEP and molecular docking studies on 1-(1-(3-methoxyphenyl) ethylidene) thiosemicarbazide. Spectrochim Acta a: Mol Biomol Spect 139:321–328. https://doi.org/10.1016/j.saa.2014.12.026
Sarıkaya EK, Dereli O (2013) Molecular structure and vibrational spectra of 7-Methoxy- 4-methylcoumarin by density functional method. J Mol Struct 1052:214–220. https://doi.org/10.1016/j.molstruc.2013.08.024
Senge MO, Fazekas M, Notaras EGA, Blau WJ, Zawadzka M, Locos OB, Mhuircheartaigh EMN (2007) Nonlinear optical properties of porphyrins. Adv Mater 19:2737–2774. https://doi.org/10.1002/adma.200601850
Sethi A, Singh RP, Shukla D, Singh P (2016) Synthesis of novel pregnane-diosgenin prodrugs via ring a and ring a connection: a combined experimental and theoretical studies. J Mol Struct 1125:616–623. https://doi.org/10.1016/j.molstruc.2016.07.020
Shalini K, Sharma PK, Kumar N (2010) Imidazole and its biological activities: a review. Pelagia Res Lib Der Chem Sinica 1(3):36–47
Sheeba BQ, Mary MSM, Amalanathan M, Job CB (2021) Structural and vibrational spectral investigation on the identification of Non-Linear Optical properties and wave function analyses (electrostatic potential, electron localisation function, localised orbital locator) of 3-Ethoxy Salicilaldehyde. Mol Simulat 47(15):1217–1233. https://doi.org/10.1080/08927022.2021.1962862
Sigma Aldrich Company (2006) Safety data sheet 1907. https://www.sigmaaldrich.com/TR/en/sds/aldrich/457612. Accessed 10 July 2021.
Singh P, Islam SS, Ahmad H, Prabaharan A (2018) Spectroscopic investigation (FT-IR, FT-Raman), HOMO-LUMO, NBO, and molecular docking analysis of N-ethyl-N-nitrosourea, a potential anticancer agent. J Mol Struct 1154:39–50. https://doi.org/10.1016/j.molstruc.2017.10.012
Spasov AR, Iezhitsa IN, Bugaeva LI, Anisimova VA (1999) Benzimidazole derivatives: Spectrum of pharmacological activity and toxicological properties (a review). Khim Farm Zhurn 33:6–17. https://doi.org/10.1007/BF02510042
Srivastava S, Gupta P, Sethi A, Pratap SR (2016) One pot synthesis of Curcumin-NSAIDs prodrug, spectroscopic characterization, conformational analysis, chemical reactivity, intramolecular interactions and first order hyperpolarizability by DFT method. J Mol Struct 1117:173–180. https://doi.org/10.1016/j.molstruc.2016.03.033
Stigliani JL, Genisson VB, Bernadou J, Pratviel G (2012) Cross-docking study on InhA inhibitors: a combination of Autodock Vina and PM6-DH2 simulations to retrieve bio-active conformations. Org Biomol Chem 10:6341–6349. https://doi.org/10.1039/c2ob25602a
Subashini K, Periandy S (2016) Spectroscopic (FT-IR, FT-Raman, UV, NMR, NBO) investigation and molecular docking study of (R)-2-Amino-1-PhenylEthanol. J Mol Struct 1117:240–256. https://doi.org/10.1016/j.molstruc.2016.03.063
Thomas R, Hossain M, Mary YS, Resmi KS, Armakovic S, Armakovic SJ, Nanda AK, Ranjan VK, Vijayakumar G, Alsenoy CV (2018) Spectroscopic analysis and molecular docking of imidazole derivatives and investigation of its reactive properties by DFT and molecular dynamics simulations. J Mol Struct 1158:156–175. https://doi.org/10.1016/j.molstruc.2018.01.021
Trott O, Olson AJ (2009) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461. https://doi.org/10.1002/jcc.21334
Uzun S, Esen Z, Koç E, Usta NC, Ceylan M (2019) Experimental and density functional theory (MEP, FMO, NLO, Fukui functions) and antibacterial activity studies on 2-amino-4-(4-nitrophenyl) -5,6-dihydrobenzo [h] quinoline-3-carbonitrile. J Mol Struct 1178:450–457. https://doi.org/10.1016/j.molstruc.2018.10.001
Verma A, Joshi S, Singh D (2013) Imidazole: Having Versatile Biological Activities. J Chemist. https://doi.org/10.1155/2013/329412
Vijesh AM, Isloor AM, Telkar S, Peethambar SK, Rai S, Isloor N (2011) Synthesis, characterization and antimicrobial studies of some new pyrazole incorporated imidazole derivatives. Eur J Med Chem 46:3531–3556. https://doi.org/10.1016/j.ejmech.2011.05.005
Yamijala SSRKC, Mukhopadhyay M, Pati SK (2015) Linear and nonlinear optical properties of graphene quantum dots: a computational study. J Phys Chem C 119:12079–12087. https://doi.org/10.1021/acs.jpcc.5b03531
Yazıcı S, Albayrak Ç, Gümrükçüoğlu İ, Şenel İ, Büyükgüngör O (2011) Experimental and density functional theory (DFT) studies on (E)-2-Acetyl-4-(4-nitrophenyldiazenyl) phenol. J Mol Struct 985(2–3):292–298. https://doi.org/10.1016/j.molstruc.2010.11.009
Yurdakul S, Temel E, Buyukgungor O (2019) Crystal structure, spectroscopic characterization, thermal properties and theoretical investigations on [Ag(methyl 4-pyridylketone)2NO3]. J Mol Struct 1191:301–313. https://doi.org/10.1016/j.molstruc.2019.04.071
Zampieri D, Mamolo MG, Laurini E, Vioa SG, L, (2008) Antifungal and antimycobacterial activity of 1-(3,5-diaryl-4,5- dihydro-1H-pyrazol-4-yl)-1H-imidazole derivatives. Bioorgan Med Chem 16:4516–4522. https://doi.org/10.1016/j.bmc.2008.02.055
Acknowledgements
We would like to thank to our reviewers for their comments that help improve the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kucuk, C., Yurdakul, S. & Erdem, B. Experimental and theoretical Fourier transform infrared and Raman spectroscopy, density functional theory, antibacterial activity and molecular docking studies on 1-(4-methoxyphenyl)-1H-imidazole. Chem. Pap. 76, 2833–2854 (2022). https://doi.org/10.1007/s11696-021-02017-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-021-02017-8