
Abstract
Microglia-involved neuroinflammation in the central nervous system (CNS) has been shown to aggravate brain damage and is associated with the pathogenesis of various neurodegenerative diseases. Thus, suppression of microglial activity has the potential to be a strategy for the treatment of neurodegenerative diseases. Pinitol, a methylated product of d-chiro-inositol, has been used as a treatment for blood-sugar metabolism and as an anti-tumor agent via its anti-inflammatory effects in cancer. However, whether or not pinitol can inhibit microglia-associated neuroinflammation is still unknown. This study aims to determine the effects of pinitol on inflammatory responses in BV2 microglia induced by LPS. Here, we found that the presence of pinitol ameliorates LPS-induced oxidative stress by reducing the production of ROS. Pinitol suppresses the expression and secretion of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6. Notably, pinitol prevents the production of NO and PGE2 by inhibiting the expression of iNOS and COX-2. Mechanistically, our findings demonstrate that pinitol inhibits the phosphorylation and degradation of IκBα and subsequent activation of NF-κB. Furthermore, we show that pinitol increases the expression of TREM2 in BV2 microglia, and silencing of TREM2 abolished the anti-inflammatory effects of pinitol. These findings suggest that TREM2 mediates the protective effects of pinitol against LPS in microglia. In summary, our results display that pinitol possesses a robust and beneficial effect against the LPS-induced inflammatory response in microglia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammation is an organism’s host defense response to harmful pathogens, stimulation, and injury (Aggarwal et al. 2009). It plays a vital role in innate immunity by repairing damaged tissue, protecting the organism, and initiating the healing process (Biber et al. 2016). Specifically, neuroinflammation is the inflammation that occurs in the central nervous system (CNS) (More et al. 2013). In neurodegenerative diseases, neuroinflammation can clear up infections to control disease development. However, persistent and excessive neuroinflammation in the CNS can accelerate many neurodegenerative diseases, including Alzheimer’s disease (AD) (Eikelenboom et al. 2002) and Parkinson’s disease (PD) (Marinova-Mutafchieva et al. 2009). Lipopolysaccharide (LPS), a central component of the membrane in Gram-negative bacteria, is a recognized endotoxin that induces inflammation. Microglia are essential immune cells in the brain and have a critical role in regulating immune system responses to brain injuries and immunological stimuli (Lee et al. 2012). In acute inflammation, microglia release substantial neurotrophic factors to rescue neurons. However, in chronic inflammation, overly activated microglia secrete various pro-inflammatory cytokines and cytotoxic mediators including interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), prostaglandin E2 (PGE2), and nitric oxide (NO) (Kumagai et al. 2007). These factors are related to the progression and development of neurodegenerative diseases (Block and Hong 2005a, b). Also, the production of reactive oxygen species (ROS) in the inflammatory process leads to oxidative stress and takes on an essential role in neuron damage (Shi et al. 2017). The release of these pro-inflammatory cytokines is closely related to the mobilization of the mitogen-activated protein kinase (MAPK) and nuclear factor-κB (NF-κB) signaling pathways (Fujioka et al. 2004; Ramachandiran et al. 2002). Triggering receptor expressed on myeloid cells 2 (TREM2) is expressed on the surface of microglia and mediates pivotal functions of microglia, including clearance of apoptotic neurons (Takahashi et al. 2005) and inhibition of pro-inflammatory responses (Hamerman et al. 2006; Zhong et al. 2015). The inhibition of abnormal microglial activation has become a therapeutic strategy for the treatment of neuroinflammation-related disorders.
Pinitol, first extracted from legumes, is a type of methylated product of d-chiro-inositol (Streeter 1980). Concerning pinitol transforming into d-chiro-inositol in the body, it has been shown to exert insulin-like effects (Bates et al. 2000). Previous research regarding pinitol has focused primarily on its regulative impact on blood-sugar metabolism (Joseph 2010), its protective effect on pancreatic tissue (Sivakumar and Subramanian 2009), and its use as an anti-tumor agent (Rengarajan et al. 2012). Moreover, pinitol has displayed diverse biological capacities such as suppression of the T helper cell-1 response, and anti-oxidant, anti-viral, larvicidal, anti-hyperlipidemic, and cardioprotective effects (Sethi et al. 2008; Sivakumar et al. 2010). Importantly, pinitol has a robust anti-inflammatory activity by blocking the activation of NF-κB abrogation of IKK activation and IκBα phosphorylation (Kumar et al. 2004). However, few studies have researched the effects of pinitol in neuroinflammation. The purpose of this study is to investigate the impacts of pinitol on inflammatory responses in BV2 microglia induced by lipopolysaccharide (LPS).
Materials and Methods
Cell Culture, siRNA Transfection, and Treatment
BV2 microglial cells were maintained in DMEM buffer supplemented with 15% FBS and 1% penicillin/streptomycin. For all experiments, the cells were pretreated with 10 μM pinitol for 3 h, followed by stimulation with 1 μg/mL LPS for 24 h. For the TREM2 silencing experiment, BV2 cells were transfected with TREM2 siRNA for 12 h, followed by the indicated treatment.
Dihydroethidium Staining
To determine the level of ROS production, we used the dye dihydroethidium (DHE) (Sigma-Aldrich, USA) dissolved in anhydrous dimethyl sulfoxide (DMSO). Briefly, the cells were probed with 5 μmol/L DHE in Krebs buffer for 20 min at a temperature of 37 °C in darkness. The cells were then washed 3 times with PBS buffer, and a fluorescence microscope (Olympus, Japan) was used to determine the fluorescence intensity. The intracellular levels of ROS were calculated using the software ImageJ. Briefly, we defined regions of interest (ROI) in the fluorescent image and counted the average number of cells presented in ROI: intracellular levels of ROS = the integrated density value (IDV)/the average number of cells.
Real-time Polymerase Chain Reaction Analysis
After the indicated treatment, RNA was extracted from BV2 cells using QIAzol reagent (Qiagen, USA). A NanoDrop spectrophotometer was used to assess the extracted RNA. cDNA was synthesized using a Bio-Rad cDNA synthesis kit following the manufacturer’s instructions. Real-time polymerase chain reaction (PCR) analysis was performed on an ABI 7500 platform using the SYBR Green–based method. Quantification of the target genes was performed using the 2-ΔΔCt method. The following primers were used in this study: TNF-α: For: 5′-CCAGACCCTCACACTCAGATC-3′, Rev: 5′-CACTTGGTGGTTTGCTACGAC-3′; IL-1β: For: 5′-TGGGCCTCAAAGGAAAGAAT-3′, Rev: 5′-CAGGCTTGTGCTCTGCTTGT-3′; IL-6 For: 5′-TGGAGTCACAGAAGGAGTGGCTAAG-3′, Rev: 5′-TCTGACCACAGTGAGGAATGTCCAC-3′; iNOS: For: 5′-CAGGAGGAGAGAGATCCGATTTA-3′, Rev: 5′-GCATTAGCATGGAAGCAAAGA-3′; COX-2: For: 5′-TGCATGTG-GCTGTGGATGTCATCAA-3′, Rev: 5′-CACTAAGACAGACCCGTCATCTCCA-3′; TREM2: For: 5′-TGTGGTCAGAGGGCTGGACT-3′, Rev: 5′-CTCCGGGTCCAGTGAGGA-3′; GAPDH: For: 5′-ACTCCCACTCTTCCACCTTC-3′, Rev: 5′-TCTTGCTCAGTGTCCTTGC-3′.
Western Blot Analysis
To determine the protein expression of the target genes, cell lysates were prepared with cell lysis buffer. To measure nuclear levels of NF-κB p65, nuclear fractions were prepared using a commercial Nuclear Protein Extraction Kit (Thermo Fisher Scientific, USA). Briefly, equal amounts of cell lysates or nuclear extractions (20 μg) were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then electronically transferred onto a polyvinylidene fluoride (PVDF) membrane. The membrane was then blocked with 5% non-fat dry milk followed by probing with primary antibodies overnight at 4 °C. The membrane was washed 3 times and then probed with HRP-linked secondary antibody for 1 h at room temperature (RT). The resulting bands were visualized using enhanced chemiluminescence (ECL) (Bio-Rad, USA). The following antibodies were used in this study: iNOS (1:2000, #ab15323, Abcam, USA); COX-2 (1:2000, #ab15191, Abcam, USA); IκBα (1:1000, #4812, Cell Signaling Technology, USA); p-IκBα (1:1000, #2859, Cell Signaling Technology, USA); p65 (1:2000, #8242, Cell Signaling Technology, USA); TREM2 (1:2000, #PA5-46980, Thermo Fisher Scientific, USA); Lamin B1 (1:5000, sc-374015, Santa Cruz Biotechnology, USA); β-actin (1:10,000, #3700, Cell Signaling Technology, USA); anti-rabbit IgG, HRP-linked antibody (1:2000, #7074, Cell Signaling Technology, USA); anti-mouse IgG, HRP-linked antibody (1:2000, #7076, Cell Signaling Technology, USA). The western blot bands were scanned, and the sum optical density of the bands was quantitatively analyzed using the Kodak Digital Science 1D software (Eastman Kodak Company, USA). Firstly, we subtracted the background. Then, we selected the bands and quantified signal intensities. Data were exported for statistical analysis.
Enzyme-Linked Immunosorbent Assay
Commercial enzyme-linked immunosorbent assay (ELISA) kits were used to measure the protein expression levels of TNF-α (#MTA00B, R&D Systems, USA), IL-6 (#M6000B, R&D Systems, USA), IL-1β (#MLB00C, R&D Systems, USA), and PGE2 (#ab133021, Abcam, USA). Briefly, the culture media were collected, and the analysis was performed following the manufacturer’s instructions. The results were collected using 96-plate reader spectrometry. A standardized 4-PL curve was used to obtain absolute values.
Luciferase Activity
The transcriptional activity of NF-κB was determined by visualizing the 3x NF-κB bind sites containing luciferase vector. BV2 microglia were co-transfected with NF-κB promoter and a firefly promoter using Lipofectamine 2000 from Invitrogen (Carlsbad, CA). At 48 h after transfection, the cells underwent the indicated treatment, and the lysates were collected. The dual-luciferase activity of NF-κB and renilla promoters was determined. The relative activity of NF-κB was calculated by normalizing to that of renilla luciferase.
Statistical Analysis
The data resulting from all experiments are presented as means ± standard error of measurement (S.E.M). Statistical analysis was performed using the software SPSS (Version 19.0). The one-way analysis of variance (ANOVA) method was used to determine statistical significance, followed by Turkey’s post hoc test. A value of P < 0.05 was considered to be of statistical significance.
Results
Pinitol Reduced LPS-Induced Generation of ROS in BV2 Microglia
Microglia were pretreated with 5 and 10 μM pinitol for 3 h, followed by exposure to 1 μg/mL LPS for 24 h. To reveal the anti-oxidant effect of pinitol against LPS-induced oxidative stress, the generation of intracellular ROS was examined through dihydroethidium (DHE) staining. The results in Fig. 1 reveal that ROS levels gradually increased upon exposure to LPS by 3.5-fold. Conversely, the levels of ROS decreased to 2.4- and 1.4-fold when exposed to the two concentrations of pinitol. Thus, the results show that oxidative stress was impeded by pinitol through the inhibition of ROS.

Pinitol reduced LPS-induced generation of ROS in BV2 microglia. Cells were pretreated with 5 and 10 μM pinitol for 3 h, followed by stimulation with 1 μg/mL LPS for 24 h. Intracellular ROS was measured by dihydroethidium (DHE) staining; scale bar, 100 μm (****P < 0.0001 vs. control group; $$, $$P < 0.01, 0.0001 vs. LPS treatment group)
Pinitol Prevented the Secretion of Pro-inflammatory Cytokines Induced by LPS
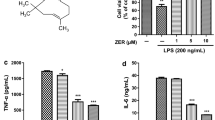
TNF-α, IL-1β, and IL-6 are important pro-inflammatory cytokines in the inflammatory response. Therefore, we investigated the impact of pinitol on the expression of these cytokines. The results in Fig. 2a showed that the secretions of TNF-α increased from 122.6 to 985.2 pg/mL with exposure to LPS. However, secretions of TNF-α decreased to 675.4 and 462.9 pg/mL after the addition of the two doses of pinitol. In Fig. 2b, we showed that treatment with LPS increased the secretions of IL-6 from 85.3 to 1344.1 pg/mL, while treatment with pinitol reduced it to 931.8 and 692.6 pg/mL. Similarly, the two doses of pinitol decreased the secretions of IL-1β induced by LPS from 485.3 to 322.7 and 189.7 pg/mL, as we showed in Fig. 2c.

Pinitol prevented LPS-induced secretion of pro-inflammatory cytokines. Cells were pretreated with 5 and 10 μM pinitol for 3 h, followed by stimulation with 1 μg/mL LPS for 24 h. a Secretions of TNF-α. b Secretions of IL-6. c Secretions of IL-1β (****P < 0.0001 vs. control group; $$, $$P < 0.01, 0.0001 vs. LPS treatment group)
Pinitol Suppressed iNOS Expression and NO Production Induced by LPS
iNOS is a critical regulatory enzyme in inflammation that acts through modulation of NO production. In Fig. 3a and b, we found that LPS stimulation increased iNOS at both the mRNA and protein levels, while the expression was decreased significantly by the two doses of pinitol. Notably, the DAF-FM staining results in Fig. 3c show that LPS treatment increased the production of NO to 4.3-fold, which was reduced to 2.6- and 1.8-fold by 5 and 10 μM pinitol, respectively. These results indicate that pinitol suppressed LPS-induced NO production via downregulation of iNOS.

Pinitol suppressed LPS-induced iNOS expression and NO production. Cells were pretreated with 5 and 10 μM pinitol for 3 h, followed by stimulation with 1 μg/mL LPS for 24 h. a mRNA of iNOS. b Protein of iNOS as measured by western blot analysis. c DAF-FM staining of NO; scale bar, 100 μm (****P < 0.0001 vs. control group; $$, $$P < 0.01, 0.0001 vs. LPS treatment group)
Pinitol Reduced COX-2 Expression and PGE2 Production Induced by LPS
COX-2 is another major inflammation-related enzyme involved in combining PGs, such as PGE2. Results in Fig. 4a and b demonstrate that LPS significantly increased the expression of COX-2, which was attenuated by the pinitol dose-dependently. Consistently, in Fig. 4c, we showed that LPS treatment increased the secretion of PGE2 from 337.8 to 1987.5 pg/mL, which was reduced to 1356.7 and 899.5 pg/mL by 5 and 10 μM of pinitol, respectively.

Pinitol reduced LPS-induced expression of COX-2 and the production of PGE2. Cells were pretreated with 5 and 10 μM pinitol for 3 h, followed by stimulation with 1 μg/mL LPS for 24 h. a mRNA of COX-2. b Protein of COX-2. c Secretions of PGE2 (****P < 0.0001 vs. control group; $$, $$P < 0.01, 0.0001 vs. LPS treatment group)
Pinitol Prevented LPS-Induced Activation of the IκBα/NF-κB Pathway in BV2 Microglia
It is well known that phosphorylation and degradation of IκBα is a trigger for NF-κB activation. Therefore, we studied the impact of pinitol on IκBα. Phosphorylated levels of IκBα increased to 2.8-fold by exposure to LPS alone, while it was reduced to 2.0- and 1.4-fold in the presence of the two concentrations of pinitol. Correspondingly, LPS treatment decreased the total level of IκBα by 45%, which was reduced by pinitol in a dose-dependent manner (Fig. 5).

Pinitol prevented LPS-induced phosphorylation and degradation of IκBα in BV2 microglia. Cells were pretreated with 5 and 10 μM pinitol for 3 h, followed by stimulation with 1 μg/mL LPS for 24 h. Phosphorylated and total levels of IκBα (****P < 0.0001 vs. control group; $$, $$P < 0.01, 0.0001 vs. LPS treatment group)
Since the NF-κB signaling pathway plays a vital role in the induction of various pro-inflammatory cytokines and mediators, determining whether pinitol influences the activation of NF-κB is essential. The results in Fig. 6a showed that LPS treatment increased nuclear translocation of NF-κB p65 to 3.6-fold, while the levels were reduced by the two concentrations of pinitol to 2.3- and 1.6-fold. We further tested the transcriptional activity of NF-κB. The luciferase activity assay revealed that pinitol had a powerful effect against the LPS-induced increase in NF-κB activity (Fig. 6b).

Pinitol prevented LPS-induced activation of NF-κB. Cells were pretreated with 5 and 10 μM pinitol for 3 h, followed by stimulation with 1 μg/mL LPS for 24 h. a Nuclear translocation of NF-κB p65. b Luciferase activity of NF-κB (****P < 0.0001 vs. control group; $$, $$P < 0.01, 0.0001 vs. LPS treatment group)
The Anti-inflammatory Effects of Pinitol in Microglia Are Mediated by TREM2
TREM2 is a vital receptor of microglia. Interestingly, we found that pinitol treatment dose-dependently increased the expression of TREM2 at both the mRNA and protein levels, as we showed in Fig. 7a and b, suggesting the possible involvement of TREM2 in the effects of pinitol. To further verify the role of TREM2 in this process, the expression of TREM2 was knocked down by transfection with TREM2 siRNA for 12 h. This successful knockdown of TREM2 was revealed by western blot analysis (Fig. 8a). Notably, the results show that knockdown of TREM2 almost completely abolished the inhibitory effects of pinitol on NF-κB activation (Fig. 8b) and the secretion of TNF-α (Fig. 8c). This data suggests that TREM2 mediates the anti-inflammatory effects of pinitol in microglia.

Pinitol increased TREM2 in LPS-treated microglia. Cells were pretreated with 5 and 10 μM pinitol for 3 h, followed by stimulation with 1 μg/mL LPS for 24 h. a mRNA of TREM2. b Protein of TREM2 (****P < 0.0001 vs. control group; $$, $$P < 0.01, 0.0001 vs. LPS treatment group)

Silencing of TREM2 abolished the protective effects of pinitol against LPS-induced inflammation in BV2 microglia. Cells were transfected with TREM2 siRNA for 12 h. Then, cells were pretreated with 10 μM pinitol for 3 h, followed by stimulation with 1 μg/mL LPS for 24 h. a Western blot analysis demonstrated successful silencing of TREM2. b Knockdown of TREM2 abolished the effects of pinitol in luciferase activity of NF-κB. c Knockdown of TREM2 abolished the inhibitory effects of pinitol in secretions of TNF-α (****P < 0.0001 vs. control group; $$P < 0.01, 0.0001 vs. LPS treatment group; ####P < 0.0001 vs. LPS + 10 μM pinitol)
Discussion
A 2012 report published by the World Health Organization (WHO) predicts that the number of patients with neurodegenerative diseases will reach over 70 million by 2030 and 106 million by 2050 (Word Health Organization 2012). Previous research has shown that such neurodegenerative diseases are associated with chronic neuroinflammation and primarily affect the elderly population (Eikelenboom et al. 2002; Marinova-Mutafchieva et al. 2009; Amor et al. 2010). Microglia, the primary cells in the brain, play a vital role in the progression and development of chronic inflammation in the CNS. Regulating over-activated microglia in neurodegenerative diseases could have a significant therapeutic impact. In this study, we demonstrated that pinitol has anti-neuroinflammatory properties in BV2 microglia stimulated with LPS. Our results show that pinitol reduces LPS-induced secretion of pro-inflammatory cytokines and mediators, suppresses the generation of ROS, and inhibits both IκBα phosphorylation and NF-κB activation. Furthermore, we proved that TREM2 mediates these effects.
Microglia are significant contributors to oxidative stress in the CNS as they induce the release of ROS and other cytokines to modulate the inflammatory response (Loane and Kumar 2016). Low levels of ROS serve as signaling molecules to regulate the immune response against harmful pathogens. However, overproduction of ROS in chronic inflammation leads to neurotoxicity, which is a cause of neuronal apoptosis (Von et al. 2015). Exacerbation of oxidative stress and increased ROS in brain tissues greatly contributes to the pathological progression of neurodegenerative diseases. Our research found that the presence of pinitol ameliorates the oxidative changes induced by LPS, suggesting an essential role of pinitol in the oxidative balance in microglia.
The endotoxin hypothesis of neurodegeneration has been studied over the past few decades. An example of this hypothesis is that LPS concentrations are elevated in the blood and brain cells of patients with AD. Increased levels of these endotoxins have a direct impact on neurodegeneration (Brown 2019). LPS-induced aberrant expression and production of pro-inflammatory mediators produced by microglia results in neuroinflammation and promotes the progression of CNS disorders. The blockage of abnormal responses mediated by microglia, could potentially prevent the development of neurodegenerative diseases (Teeling and Perry 2009). Upregulation of TNF-α, IL-6, IL-1β, and PGE2 are capable of initiating and amplifying the inflammatory response in brain cells (Lucas et al. 2006). Excessive generation of NO via over-activated microglia has been considered as another critical biomarker for neuroinflammation in neurodegenerative diseases. Our results demonstrate that pinitol strongly inhibits the production of TNF-α, IL-6, IL-1β, NO, and PGE2, as well as the expression of COX-2 and iNOS. Our mechanistic investigation revealed that the protective effects of pinitol in inflammatory responses are dependent on the inhibition of IκBα/NF-κB activation. NF-κB is one of the most important signaling pathways regulating the expression of pro-inflammatory cytokines and mediators (Kaltschmidt and Kaltschmidt 2009). The protein p65, one of the NF-κB subunits, has well-documented significance in the expression of inflammatory mediators in microglia (Erica and Mollie 2019). It is known that the IκB inhibitors, such as IκBα, can prevent nuclear translocation of NF-κB p65 (Kopitar 2015). However, the phosphorylation and degradation of IκBα also trigger the transference of NF-κB p65 into the nucleus. Abnormal activation of NF-κB has been widely associated with the development of CNS disorders (Sivandzade et al. 2019). The inhibitory effects of pinitol on NF-κB activity have broadened our understanding of its beneficial effects against neuroinflammation.
Finally, we focused on TREM2, the microglial surface receptor. Previous studies have shown that TREM2 can mediate the functions of microglia in the CNS, such as clearance of apoptotic neurons and secretion of pro-inflammatory cytokines (Takahashi et al. 2005; Hamerman et al. 2006). Our study has demonstrated that pinitol treatment increases the expression of TREM2 at both the mRNA and protein levels in BV2 microglia. On the other hand, silencing of TREM2 abolishes the effects of pinitol on NF-κB activity and secretions of TNF-α. These findings suggest that TREM2 is involved in the protective effects of pinitol against LPS-induced inflammation in microglia. TREM2 plays a critical role in transforming microglia from a homeostatic to a disease-associated state.
Additionally, TREM2 has been considered a principal regulator of microglial activity, including apoptosis, proliferation, phagocytosis, and inflammation, in various neurodegenerative diseases, such as AD and ischemia (Konishi and Kiyama 2018; Ulland and Colonna 2018). TREM2 has been proposed as a new therapeutic target for the treatment of these disorders. The involvement of TREM2 in the pharmacological functions of pinitol in microglial activity implicates a novel molecular mechanism.
Taken together, our findings suggest that pinitol has the potential to act as a therapeutic agent in neuroinflammation-related brain diseases by alleviating microglial activation.
Credit Author Statement
Conceptualization: Dr. Juan Kong and Dr. Li Dong; data curation: Dr. Juan Kong, Dr. Zhigang Du, and Dr. Li Dong; formal analysis: Dr. Li Dong; funding acquisition: Dr. Juan Kong; investigation: Dr. Juan Kong; methodology: Dr. Zhigang Du, Dr. Li Dong; project administration: Juan Kong; resources: Dr. Juan Kong and Dr. Zhigang Du; software: Dr. Juan Kong and Dr. Zhigang Du; supervision: Dr. Juan Kong; validation: Dr. Juan Kong; visualization: Dr. Zhigang Du and Dr. Li Dong; roles/writing—original draft: Dr. Juan Kong, Dr. Zhigang Du; writing—review and editing: Dr. Zhigang Du and Dr. Li Dong.
References
Aggarwal BB, Van Kuiken ME, Iyer LH, Harikumar KB, Sung B (2009) Molecular targets of nutraceuticals derived from dietary spices: potential role in suppression of inflammation and tumorigenesis. Exp Biol Med (Maywood) 234(8):825–849
Amor S, Puentes F, Baker D, van der Valk P (2010) Inflammation in neurodegenerative diseases. Immunology 129(2):154–169
Bates SH, Jones RB, Bailey CJ (2000) Insulin-like effect of pinitol. Br J Pharmacol 130(8):1944–1948
Biber K, Moller T, Boddeke E, Prinz M (2016) Central nervous system myeloid cells as drug targets: current status and translational challenges. Nat Rev Drug Discov 15(2):110–124
Block ML, Hong JS (2005a) Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76(2):77–98
Block ML, Hong JS (2005b) Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76:77–98
Brown GC (2019) The endotoxin hypothesis of neurodegeneration. J Neuroinflammation 16(1):180
Eikelenboom P, Bate C, VanGooletal WA (2002) Neuroinflammation in Alzheimer’s disease and prion disease. Glia 40(2):232–239
Erica CD, Mollie KM (2019) Cellular specificity of NF-κB function in the nervous system. Front Immunol 10:1043
Fujioka S, Niu J, Schmidt C, Sclabas GM, Peng B, Uwagawa T, Li Z, Evans DB, Abbruzzese JL, Chiao PJ (2004) NF-κB and AP-1 connection: mechanism of NF-κB-dependent regulation of AP-1 activity. Mol Cell Biol 24(17):7806–7819
Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL (2006) Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol 177(4):2051–2055
Joseph L (2010) D-Chiro-inositol glycans in insulin signaling and insulin resistance. Mol Med 16(11–12):543–551
Kaltschmidt B, Kaltschmidt C (2009) NF-κB in the nervous system. Cold Spring Harb Perspect Biol 1(3):a001271
Konishi H, Kiyama H (2018) Microglial TREM2/DAP12 signaling: a double-edged sword in neural diseases. Front Cell Neurosci:12–206
Kopitar JN (2015) Innate immune response in brain, NF-Kappa B signaling and cystatins. Front Mol Neurosci:8–73
Kumagai N, Chiba Y, Hosono M, Fujii M, Kawamura N, Keino H, Yoshikawa K, Ishii S, Saitoh Y, Satoh M, Shimada A, Hosokawa M (2007) Involvement of pro-inflammatory cytokines and microglia in an age-associated neurodegeneration model, the SAMP10 mouse. Brain Res 1185:75–85
Kumar A, Takada Y, Boriek AM, Aggarwal BB (2004) Nuclear factor-kB: its role in health and disease. J Mol Med 82:434–448
Lee KW, Jung SY, Choi SM, Yang EJ (2012) Effects of ginsenoside Re on LPS-induced inflammatory mediators in BV2 microglial cells. BMC Complement Altern Med:12–196
Loane DJ, Kumar A (2016) Microglia in the TBI brain: the good, the bad, and the dysregulated. Exp Neurol 275(3):316–327
Lucas SM, Rothwell NJ, Gibson RM (2006) The role of inflammation in CNS injury and disease. Br J Pharmacol 147(Suppl 1):S232–S240
Marinova-Mutafchieva L, Sadeghian M, Broom L, Davis JB, Medhurst AD, Dexter DT (2009) Relationship between microglial activation and dopaminergic neuronal loss in the substantia nigra: a time course study in a 6-hydroxydopamine model of Parkinson’s disease. J Neurochem 8:966–975
More SV, Kumar H, Kim IS, Song SY, Choi DK (2013) Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson's disease. Mediat Inflamm:2013–952375
Ramachandiran S, Huang Q, Dong J, Lau SS, Monks TJ (2002) Mitogen-activated protein kinases contribute to reactive oxygen species-induced cell death in renal proximal tubule epithelial cells. Chem Res Toxicol 15(12):1635–1642
Rengarajan T, Nandakumar N, Balasubramanian MP (2012) D-Pinitol a low-molecular cyclitol prevents 7, 12-Dimethylbenz anthracene induced experimental breast cancer through regulating anti-apoptotic protein Bcl-2, mitochondrial and carbohydrate key metabolizing enzymes. Biomed Preven Nutri 2:25–30
Sethi G, Ahn KS, Sung B, Aggarwal BB (2008) Pinitol targets nuclear factor-KB activation pathway leading to inhibition of gene products associated with proliferation, apoptosis, invasion, and angiogenesis. Mol Cancer Ther 7:1604–1614
Shi H, Deng HX, Gius D, Schumacker PT, Surmeier DJ, Ma YC (2017) Sirt3 protects dopaminergic neurons from mitochondrial oxidative stress. Hum Mol Genet 26(10):1915–1926
Sivakumar S, Subramanian SP (2009) Pancreatic tissue protective nature of D-Pinitol studied in streptozotocin-mediated oxidative stress in experimental diabetic rats. Eur J Pharmacol 622(1–3):65–70
Sivakumar S, Palsamy P, Subramanian S (2010) Impact of D-pinitol on the attenuation of proinflammatory cytokines, hyperglycemia-mediated oxidative stress and protection of kidney tissue ultrastructure in streptozotocin-induced diabetic rats. Chem Biol Interact 188:237–245
Sivandzade F, Prasad S, Bhalerao A, Cucullo L (2019) NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: molecular mechanisms and possible therapeutic approaches. Redox Biol:21–101059
Streeter JG (1980) Carbohydrates in soybean nodules: II. Distribution of compounds in seedlings during the onset of nitrogen fixation. Plant Physiol 66(3):471–476
Takahashi K, Rochford CDP, Neumann H (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 201(4):647–657
Teeling JL, Perry VH (2009) Systemic infection and inflammation in acute CNS injury and chronic neurodegeneration: underlying mechanisms. Neuroscience 158:1062–1073
Ulland TK, Colonna M (2018) TREM2 - a key player in microglial biology and Alzheimer disease. Nat Rev Neurol 4(11):667–675
Von BR, Eugenínvon BL, Eugenín J (2015) Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci 7:124
World Health Organization (2012) Dementia a public health priority. [Geneva]: World Health Organization http://site.ebrary.com/lib/ucmerced/Doc?id=10718026 [accessed 14 November 2013]
Zhong L, Chen XF, Zhang ZL, Wang Z, Shi XZ, Xu K, Zhang YW, Xu H, Bu G (2015) DAP12 stabilizes the C-terminal fragment of the triggering receptor expressed on myeloid cells-2 (TREM2) and protects against LPS-induced pro-inflammatory response. J Biol Chem 290(25):15866–15877
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that there is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kong, J., Du, Z. & Dong, L. Pinitol Prevents Lipopolysaccharide (LPS)-Induced Inflammatory Responses in BV2 Microglia Mediated by TREM2. Neurotox Res 38, 96–104 (2020). https://doi.org/10.1007/s12640-020-00187-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-020-00187-z