
Abstract
It is now well accepted that there is a close relationship between noradrenergic and dopaminergic neurons in the brain, especially referring to the modulation of the locus coeruleus–norepinephrine (LC-NE) system on dopamine transmission. The disturbance of this modulation may contribute to neurodegeneration of dopaminergic neurons in Parkinson’s disease. In this article, we briefly review evidence related to such modulation. Firstly, we illustrated the noradrenergic innervation and functional implication for the LC-NE system and nigra–striatum dopaminergic system. Furthermore, we depicted neuroprotective effects of the LC-NE on dopaminergic neurons in vivo and in vitro. Moreover, we present data implicating the potential mechanisms underlying the modulation of the LC-NE system on dopaminergic neurons, in particular the effects of NE as a neurotrophic factor and through its ability to stimulate the expression of other neurotrophic factors, such as the brain-derived neurotrophic factor. Finally, we discussed other mechanisms intrinsic to NE’s effects. A better understanding of the noradrenergic modulation on dopaminergic neurons may be rewarding by significant advances in etiologic study and promising treatment of Parkinson’s disease.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Noradrenergic and dopaminergic systems are the major neuronal circuits in the brain. Their corresponding neurotransmitters norepinephrine (NE) and dopamine (DA) are the main catecholamines involved in a variety of physiological processes. The major noradrenergic nucleus in the brain is the locus coeruleus (LC, A6) located in the pons (Vijayashankar and Brody 1979; Brodal 1981) with wide afferents to the cortex, cerebellum, thalamus, and spinal cord (Moore and Bloom 1979; Morrison et al. 1979; Segal and Bloom 1976; Swanson and Hartman 1975). In particular, the LC is the sole noradrenergic source to innervate the hippocampus (Swanson and Hartman 1975; Haring and Davis 1985) and frontal cortex (Morrison et al. 1979; Samuels and Szabadi 2008). LC-NE neurons and released NE throughout the brain are involved in a variety of physiological functions and behaviors such as cognition (Sterpenich et al. 2006; Sara 2009), attention (Robbins 2000), locomotor control, and contributing to the affective state (Robbins and Everitt 1995). The substantia nigra pars compacta (SNpc, A9) (Anden et al. 1966; Domesick 1988) and ventral tegmental area (VTA, A10) (Swanson 1982; Oades and Halliday 1987) represent two of the nine major dopaminergic neuron groups in the midbrain. Neurons from the SNpc and VTA are characterized by distinct but overlapping projection patterns. The majority of projections that originate from the SNpc innervate the dorsal striatum, and only some nigral fibers project to the ventral striatum and cortex. In contrast, neurons from the VTA mainly project to the ventral striatum as well as cortical areas, with significantly fewer projections innervating the dorsal striatum (Bjorklund and Dunnett 2007). While dopaminergic neurons in the SNpc are heavily involved in the control of movements, those in the VTA are responsible for the regulation of reward, emotional behavior and addiction (Satoh et al. 2003; Matsumoto and Hikosaka 2009; Bromberg-Martin et al. 2010). Nevertheless, although the LC-NE and DA systems have different characteristics, they have close relationship in anatomy, physiology, and functions, somewhat even overlapped. Therefore, their dysfunctions are also closely related to the pathogenesis of many neurodegenerative diseases.
It is a general knowledge that the degeneration in a clearly defined population of dopaminergic neurons in the brain is the main pathologic alteration in Parkinson’s disease (PD) (Hirsch et al. 1988; Fearnley and Lees 1991). However, significant neuronal loss also occurs in the LC in PD. Neurodegeneration of LC neurons even starts earlier than that seen in the SNpc and at a greater magnitude (Gesi et al. 2000; Zarow et al. 2003). Furthermore, there is a correlation between NE deficiency and degenerated dopaminergic neurons with the severity of PD neurological symptoms, indicating that the LC-NE system shares a critical role with the DA system in the progression of PD. Moreover, the neuroprotective effects of an intact LC-NE neuronal system on nigrostriatal dopaminergic neurons and the contribution of endogenous NE to the recovery of the dopaminergic neurons have been reported in many studies (Delaville et al. 2011; Isaias et al. 2011). Therefore, understanding of their interaction, especially for the modulatory effects of the LC-NE system on dopaminergic neurons, may benefit the treatment of PD.
In this paper, we put emphasis on the modulatory effects of noradrenergic neurons on dopaminergic neurons. First, the anatomical and functional relationships between the LC-NE and dopaminergic system are illustrated. Second, the neuroprotective role of noradrenergic neurons on dopaminergic neurons is depicted. Finally, the potential mechanisms underlying the modulation of the LC-NE system on dopaminergic neurons, in particular NE as a neurotrophic factor and its ability to stimulate the expression of other neurotrophic factors, such as the neurotrophin brain-derived neurotrophic factor (BDNF), are discussed. Other potential mechanisms are taken into account of as well.
Noradrenergic Innervation and Functional Implication for the LC-NE System and Nigra Dopaminergic System
An important characteristic between the noradrenergic and dopaminergic systems is their anatomical correlation, which is the basis for the functional modulation of the LC-NE system on midbrain dopaminergic neurons. Anatomical analysis demonstrated that noradrenergic neurons from the LC widely innervate dopaminergic neurons in the midbrain by sending projections to the SNpc, VTA, and striatum in the vicinity of dopaminergic neuronal cell bodies (Simon et al. 1979; Berridge et al. 1997; Schroeter et al. 2000; Liprando et al. 2004; Mejias-Aponte et al. 2009). Furthermore, dopamine β-hydroxylase (DBH) and NE transporter (NET), as well as NE and its receptors such as β- (β1 and β2) and α- (α1 and α2) adrenergic receptors (ARs), can be detected in the most midbrain dopaminergic neurons including the VTA and striatum (Ross and Reis 1974; Chen and Reith 1994; Liprando et al. 2004; Mejias-Aponte 2016) by autoradiographic and immunostaining approaches (Jones et al. 1985; Lee et al. 1998). Such closely anatomical innervation raises the possibility that noradrenergic inputs play a role in modulating DA neuronal activity. For example, electrical stimulation of the LC results an excitation action showing by burst firing of dopaminergic neurons in the SNpc (Grenhoff et al. 1993), which can be attenuated by administration of the α1-AR antagonist prazosin (Grenhoff and Svensson 1993). Furthermore, either lesion of LC neurons or chronic NE depletion has been shown to reduce basal and amphetamine-induced release of DA in the striatum and SNpc (Lategan et al. 1990; Lategan et al. 1992). Consistently, the systemic administration of the selective NE reuptake inhibitor reboxetine enhanced the burst firing activity of dopaminergic neurons in the VTA (Linner et al. 2001). Stimulation of the LC facilitates firing rate in midbrain dopaminergic neurons (Grenhoff et al. 1993), which can also be reduced or increased by application of antagonists of α1-ARs or α2-ARs (Grenhoff and Svensson 1993). However, there is a report that selective lesion of LC neurons increases the mean firing activity of dopaminergic neurons and their burst activity in the VTA (Guiard et al. 2008), suggesting a complexity in regulation of dopaminergic neuronal activity by noradrenergic inputs.
It is noteworthy that although NE terminals make close contacts onto midbrain DA neurons, the majority of contacts are not in direct synaptic apposition, as showing by an ultrastructural study (Liprando et al. 2004). That means, in the SNpc and VTA areas, there are predominant nonsynaptic appositions between NET-immunoreactive axons and dopaminergic neurons, which are separated by glia (Liprando et al. 2004). Although the real function of glia between these neurons is unknown, such glial separations may provide a selective physical barrier to transmission between some noradrenergic terminals and dopaminergic dendrites, allowing one subpopulation of dendrites to be modulated by NE, but not another. These ultrastructural observations suggest that their functional interactions between these two systems are more likely through extrasynaptic mechanisms and considered as a paracrine or volume transmission system (Mejias-Aponte 2016). Thus, NE released from noradrenergic terminals in this area seems to act as a neuromodulator on DA neurons more than a neurotransmitter (Gesi et al. 2000). The similar ultrastructural innervation between noradrenergic terminals and astrocytes is also found in the cortex and thymus (Vizi et al. 1995; Cohen et al. 1997). As NE induced expression of a range of growth factors such as BDNF, nerve growth factor (NGF), glial cell line-derived neurotrophic factor (GDNF), and fibroblast growth factor (FGF) in glial cells via an action on astrocytic β2-ARs (Juric et al. 2006; Day et al. 2014), such innervation may be related to the modulatory effects of NE on neuronal plasticity.
In combination, these results not only establish a physiological and functional connection between the noradrenergic and dopaminergic systems, but also form the basis for the influence of NE on dopaminergic neurons in the neuropathology of degenerative diseases.
The Neuroprotection of LC-NE System on the Nigra Dopaminergic System
It is now known that the LC-NE system exhibits a neuronal protection on the nigra dopaminergic system, which has been revealed by a variety of studies. First, a functional and intact LC-NE system facilitates the survival of the dopaminergic system, which was revealed by the studies that the disturbance and/or a functional enhancement of the LC-NE system influences both the onset and the progression of neuronal damage to the DA nigrostriatal tract (Delaville et al. 2011; Isaias et al. 2011). For example, in neurotoxin-induced animal models of PD, concomitant lesions of the LC-NE system with 6-hydroxy-DA resulted in more dopaminergic neuronal loss or activity reduction in the SNpc and VTA areas caused by subsequent exposure to MPTP (Mavridis et al. 1991; Bing et al. 1994; Fornai et al. 1995; Srinivasan and Schmidt 2003). In line with these observations, neurotoxin such as DSP-4-induced reductions in LC activities and functions worsens DA deficit caused by MPTP in animal models of PD (Marien et al. 1993). In addition, these treatments resulted in a significant reduction in striatal concentrations of DA and its metabolites (Mavridis et al. 1991; Fornai et al. 1996; Srinivasan and Schmidt 2003) and an alteration in DA-related behavior (Antelman and Caggiula 1977; Wang et al. 2010). In contrast, administration of β2-AR agonists protects the MPTP-induced degeneration of dopaminergic neurons in the SNpc of mouse model and a β2-AR antagonist correlated with increased risk of developing PD (Mittal et al. 2017).
Second, the neuroprotection of noradrenergic neurons on dopaminergic neurons can be found by administration of NE or increase of NE synthesis, which has been evidenced by in vitro and in vivo studies. For example, in vitro administration of NE (0.3–1 μM) confers substantial and long-term protection to dopaminergic neurons by reducing spontaneously occurring oxidative stress in primary cultured mesencephalic cells (Troadec et al. 2001). Similarly, elevation of extracellular NE levels by treatment with α2-AR antagonists (Martel et al. 1998) or by genetic methods (Kilbourn et al. 1998) protected dopaminergic neurons from neurotoxin-induced cell death. NE hyperinnervation of target areas or treatment of rats with NE has led to resistance to experimental parkinsonism (Marien et al. 1994; Rommelfanger et al. 2004; Rommelfanger et al. 2007). It is well known that methamphetamine can cause degeneration of dopaminergic axon in nigra-striatal regions (Hotchkiss and Gibb 1980; Hirata et al. 1996). Such methamphetamine-induced dopaminergic degeneration is exacerbated after experimental lesion of LC neurons with enhanced methamphetamine toxicity and damage to these neurons (Fornai et al. 1995; Fornai et al. 1998; Weinshenker et al. 2008). In addition, a postmortem survey from persons with PD revealed that the brain areas that were high in NE tended to be spared from DA loss, consistent to a neuroprotective role of NE, with an exception being the putamen (Tong et al. 2006). In clinic trials, to increase extracellular NE concentrations has been used for treatment of PD patients, including use of α2- and β2-AR antagonists, NET inhibitor, and increase NE synthesis (Espay et al. 2014; Mittal et al. 2017).
The Role of NE in the Modulation of the LC-NE on Dopaminergic Neurons
As a classic neurotransmitter of the LC-NE system, NE plays a critical role for the noradrenergic functions and behaviors in the homeostasis of body (Berridge and Waterhouse 2003). In addition, NE may act as a modulator for the modulation to other neuronal circuits especially for the dopaminergic neurons. It has been suggested that the neuronal beneficial effects of NE on other neurons can be direct and indirect. For the direct effect, NE is considered to act as a neurotrophic factor. For the indirect effect, it refers to its ability to facilitate the expression of other neurotrophic factors such as the BDNF (Aloyz et al. 1999; Chen et al. 2007; Counts and Mufson 2010), FGF-2 (Kajitani et al. 2012), Bcl-2 (Huang et al. 2007), and NGF (Culmsee et al. 1999; Counts and Mufson 2010). However, both mechanisms can be overlapped. This review focusses on the mediator role of BDNF for NE’s effects, although other growth factors are also often associated with an ability of NE to protect dopaminergic neurons from toxicity (Timmer et al. 2007; Xing et al. 2010; Dobolyi et al. 2012).
NE Operates as an Endogenous Neurotrophic Substance
Acting as a neurotrophic factor, NE can influence both development and adulthood. For these effects, NE has been considered to drive DNA synthesis in the mature and developing nervous system (Lauder 1993), which induces neurite outgrowth in primary cultured neurons (Day et al. 2014). For instance, stimulation of α2-ARs expressed within proliferative germinal zones decreases DNA synthesis in the developing forebrain (Lidow and Rakic 1994; Kreider et al. 2004). Such effect of NE to drive DNA synthesis has been suggested possibly as a common mechanism in the developing and adult brain.
During the development period, NE is thought to be required for the critical period plasticity in development of the cerebral cortex and olfactory. For instance, NE can participate in the formation and organization of neuronal circuits in the olfactory bulb, as injection of either NE or β-AR agonists into rat pups increased odor experience and the differentiation of olfactory bulb plasticity (Sullivan et al. 1989; Sullivan et al. 1991). In contrast, early NE depletion induced by cytotoxic lesion of the LC in rat pups impaired the development of the cerebral cortex (Felten et al. 1982; Siciliano et al. 1999). Furthermore, the use of dbh−/− mice verified NE’s necessary effect on the development of auditory cortex (Shepard et al. 2015). In addition, in vitro studies demonstrated that NE increases embryonic neuroepithelial cell division during early development and in cultured neurons from embryonic rodent brain via the α1-ARs (Popovik and Haynes 2000), promotes differentiation and neurite outgrowth, and increases expression of genes related to neuronal sprouting (Day et al. 2014) and differentiation (Laifenfeld et al. 2002). Similarly, the facilitative effects of NE on synaptic plasticity during development period were confirmed by ex vivo experiments (Hu et al. 2007; Liu et al. 2010).
Similarly, the LC-NE system also plays a prominent role to maintain its neurotrophic influence on adulthood period. This effect mainly exhibits as its action on the cellular plasticity through neurogenesis. For example, in adult rats, administration of NE markedly increased hippocampal synaptic plasticity in adult rats (Katsuki et al. 1997) and directly activated self-renewing and multipotent neuronal precursors, including stem cells from the hippocampus of adult mice (Jhaveri et al. 2010). BrdU-positive cells in the adult hippocampus were significantly enhanced by increased extracellular NE resulted from administration of reboxetine (Malberg et al. 2000). l-Dihydroxyphenylserine (L-DOPS), a NE prodrug, is reported to stimulate neuronal sprouting and synaptogenesis (Stroemer et al. 1998). Furthermore, NE could affect neurogenesis indirectly through α1-ARs on interneurons (Hillman et al. 2009). In contrast, a significant reduction in hippocampal neurogenesis was observed by neurotoxin DSP-4-induced depletion of NE (Kulkarni et al. 2002), by bilateral infusions of the anti-DBH-saporin into the LC (Coradazzi et al. 2016), or by administration of α2-AR agonists (Yanpallewar et al. 2010) which can be reversed by α2-AR antagonist yohimbine via increasing NE release (Yanpallewar et al. 2010). Similarly, noradrenergic activation resulted from LC stimulation increased plasticity in the auditory cortex and auditory thalamus neurons in the adult rats (Edeline et al. 2011). The neurotrophic benefit of the LC-NE system on dopaminergic neurons is supported from investigations related to PD patients and PD animal models. A plague of the DA replacement therapy for PD patients is the onset of abnormal involuntary movements (AIMs, also called l-DOPA-induced dyskinesia) (Blin et al. 1988). It appeared to be a result of an aberrant synaptic plasticity of dopaminergic neurons in the striatum (Calabresi et al. 2000; Picconi et al. 2005). However, it is considered to be associated with the loss of a trophic support of the NE system for the growth, differentiation, and repair of the nigrostriatal DA pathway, as the use of α2-AR antagonists yohimbine and idazoxan to increase NE availability can dramatically reduce the AIMs in PD patients and PD animal models (Henry et al. 1999; Rascol et al. 2001; Lundblad et al. 2002). Furthermore, a loss of LC-ascending NE axons forestalls the onset, worsens the severity, and decides the ultrastructural correlates of l-DOPA-induced AIMs (Fulceri et al. 2006; Fulceri et al. 2007). Together, these findings clearly demonstrated the robust impact of the LC-NE system on dopaminergic neuronal plasticity in the adult brain.
The neurotrophic effects of NE have been also explained as one of the mechanisms underlying NE’s neuroprotective role. Although ARs are involved in NE neurotrophic actions, several observations revealed that in some cases, NE’s effects are independent of ARs. Instead, the catechol moiety of NE plays an important role. For example, the neuroprotective effect of NE against oxidative stress in cultured cholinergic neurons and SK-N-SH cells appears to be related to its catechol moiety, as a compound that reproduces the diphenolic structure of NE without binding affinity for ARs (Vauquelin et al. 1979) mimicked NE’s neuroprotective effects (Traver et al. 2005; Jhang et al. 2014). The similar phenomenon was also found on the dopaminergic neurons (Troadec et al. 2001). The diphenolic structure in NE may be at the origin of the trophic effect on DA and responsible for the neuroprotective effect of NE, possibly via the production of corresponding metabolites by autoxidation (Troadec et al. 2001). These indicate that the precursors and metabolites of NE, which exhibited on their aromatic ring two free hydroxyl groups in the orthoposion, afford neuroprotection. It is such diphenolic structure that is responsible for the neuroprotective/antioxidant activity of NE. Consistent with this view, the compounds which possess a catechol moiety, such as the β- and β1-AR agonists isoproterenol and dobutamine, as well as o-catechol (pyrocatechol), mimicked the protective effects of NE in dopaminergic and other cultures (Ancerewicz et al. 1998; Noh et al. 1999; Troadec et al. 2001).
NE’s Effects May Be Mediated Through BDNF
BDNF is a member of the neurotrophin family (Barde et al. 1982; Leibrock et al. 1989). It is predominantly produced by neurons (Zafra et al. 1992; da Penha Berzaghi et al. 1993; Lindholm et al. 1994; Thoenen 1995) and astrocytes (Miklic et al. 2004). Binding BDNF and its preferred receptor, the tropomyosin receptor kinase B (TrkB) (Reichardt 2006), triggers the activation of diverse signaling cascades, further regulating neuronal development and survival, as well as neurogenesis in the central nervous system (Ghosh et al. 1994; Jones et al. 1994; Nawa et al. 1994; Cabelli et al. 1995; McAllister et al. 1995, 1996; Zuccato and Cattaneo 2009). Accumulating evidence shows that NE can facilitate the expression of several neurotrophic factors. However, BDNF can be a main mediator for the trophic signal derived from noradrenergic afferents (Fawcett et al. 1998), for which the extensive studies have well documented.
First, BDNF is synthesized by LC neurons (Castren et al. 1995; Smith et al. 1995; Conner et al. 1997; Numan et al. 1998). DBH and BDNF are co-localized in the noradrenergic axons and terminal nerve fibers in the brain (Castren et al. 1995; Fawcett et al. 1998). Second, BDNF is anterogradely transported (from the cell soma) by afferents to fibers in noradrenergic terminals and then secreted onto target neurons (Conner et al. 1997; Fawcett et al. 1998). Such anterogradely transported BDNF causes activation of TrkB in target regions such as the neocortex for neurons survival and differentiation (Fawcett et al. 1998). This presynaptic secretion of BDNF may provide a cellular mechanism for noradrenergic modulating on other neural circuitries, in either developing or mature nervous systems. An in vivo study demonstrated that mice overexpression BDNF in DBH-positive neurons exhibited a 52% increase of TH-positive neurons in the SNpc (Alonso-Vanegas et al. 1999), suggesting that an increased anterograde transport of BDNF through coeruleus-nigral projection could benefit the growth of mesencephalic DA neurons (Vitalis et al. 2005). Third, NE stimulates the synthesis of BDNF in neurons and astrocytes, which has been confirmed in vivo (Fawcett et al. 1998; Ivy et al. 2003) and in vitro (Zafra et al. 1992; Schwartz and Nishiyama 1994; Schwartz et al. 1994; Inoue et al. 1997; Juric et al. 2006; Chen et al. 2007; Musazzi et al. 2014). In contrast, the antagonists of α1- and β1/β2-ARs inhibited this stimulatory effect of NE (Juric et al. 2008). Fourth, the neuroprotective effects of NE require TrkB activation. NE triggers TrkB phosphorylation. It has been reported that the TrkB, but not ARs, is essential for the ability of NE to protect cultured neurons from rat cortex, hippocampus, and LC, as well as human hNT neurons following Aβ exposure (Counts and Mufson 2010; Liu et al. 2015). The ability of NE to prevent Aβ-induced cells death was fully prevented by the TrkB antagonist k252a. Fifth, NE may transactivate TrkB via Src family kinase activity, which has been indicated in the studies using adenosine, pituitary adenylate cyclase-activating peptide, and zinc (Lee and Chao 2001; Lee et al. 2002; Rajagopal et al. 2004; Huang et al. 2008), as NE and the small molecule TrkB agonist 7,8-dihydroxyphenylserine share structural similarities (a catechol ring).
Both NE and BDNF Act Through Survival Signaling Pathways
Neuronal viability is maintained through a complex interacting network of signaling pathways. While these diverse cascades are critical for the proper formation of the central nervous system (Yuan and Yankner 2000), they are associated with neuronal protections for affected neurons and called as the neuronal survival signaling pathways. For example, the phoshpatidyloinositol-3-kinase (PI3K)/Akt pathway is a major mediator of cell survival signaling leading to the transcription of many pro-survival genes (Datta et al. 1999; Brunet et al. 2001; Kang et al. 2004; Patel et al. 2010). The mitogen-activated protein kinase/extracellular signal regulated kinase 1/2 (MAPK/ERK) pathways promote neuronal growth and neuroplasticity and influence gene expression through activation of transcription factor such as CREB (Shaywitz and Greenberg 1999; Troadec et al. 2002; Einat et al. 2003; Chen et al. 2007; Cottingham et al. 2012). In addition to increasing cellular survival, the PI3K and MAPK pathways play principal roles in promoting neurite growth, synaptic strength and plasticity (Heerssen and Segal 2002). NE and BDNF are not transcriptional factors and cannot directly act on transactivation of related genes (Ruiz et al. 2014). As such, these pathways have been shown to mediate the neurotrophic effects of NE and BDNF. As one of the consequences, activation of these signaling pathways leads to the binding of transcription factors with cis-acting elements such as AP-1, CRE, and Egr1/SP1 on the TH promoter and transactivation of the TH gene (Nagamoto-Combs et al. 1997; Lim et al. 2000; Suzuki et al. 2004; Kalashnikova et al. 2006; Fukuchi et al. 2010).
Many lines of investigations have revealed these survival pathways as an important mean for NE to enhance neuronal growth or protections. For instance, administration of NE dose-dependently induces a robust activation of ERK1/2 (Cottingham et al. 2012), PI3K, and CREB proteins (Chen et al. 2007). Similarly, desipramine, which increases extracellular NE levels, not only can act as a signaling potentiator to selectively enhance NE-induced ERK1/2 signaling (Cottingham et al. 2012), but also activate the MAPK/ERK pathway (Huang et al. 2007). Cyclic adenosine monophosphate (cAMP) was described as a prosurvival molecule for several populations of catecholaminergic neurons. The neuroprotective effect of NE on dopaminergic neuronal cells is strongly enhanced by forskolin, a cAMP-elevating agent, which did not involve ARs (Troadec et al. 2002). However, this effect involves cAMP-dependent MAPK, as forskolin stimulated the phosphorylation of extracellular ERK1/2 in dopaminergic neurons (Troadec et al. 2002). Nevertheless, other studies showed that α2A-AR (Cottingham et al. 2012) or β-ARs (Counts and Mufson 2010) are involved in NE-induced neuroprotective properties through stimulation of cAMP production and pCREB signaling. The activation of these signal pathways is believed to promote cell survival both in vivo (Chen and Russo-Neustadt 2005) and in vitro (Chen and Russo-Neustadt 2007).
As mentioned above, activation of diverse signaling cascades trigged by binding of BDNF and TrkB also has critical roles in neuronal plasticity, survival, and neurogenesis (Zuccato and Cattaneo 2009). The three major pathways include the phospholipase C-γ (PLC-γ), PI3K)/Akt, and MAPK/ERK pathway (Baydyuk and Xu 2014). By activating these diverse signaling cascades in neurons, BDNF can regulate neuronal development and progenitor cell survival, proliferation, initiation of neurite outgrowth, and path-finding (Bonni et al. 1999; Encinas et al. 1999; Yamada et al. 1999). It can mediate various structural plasticities of neurons, including dendrite formation and maintenance (Widmer et al. 1993; McAllister et al. 1999; Berghuis et al. 2006; Sciarretta et al. 2010; Orefice et al. 2013). The MAPK/ERK pathway also activates regulators of protein translation (Segal 2003). Deletion of either the TrkB or Bdnf gene leads to cell atrophy, dendritic degeneration, and neuronal loss, as shown in the excitatory neurons of the dorsal forebrain (Gorski et al. 2003). BDNF-activated ERK1/2 protects cultured rat cortical neurons against apoptosis induced by DNA damage, which is necessary and sufficient for the anti-apoptotic action of BDNF (Hetman et al. 1999; Gozdz et al. 2003).
A foundation of previous studies as described above has illustrated the potential mechanisms underlying effects of NE on dopaminergic neurons through its neurotrophic action and through other neurotrophic factors such as BDNF. The LC-NE system can affect neuronal growth, survival and plasticity of dopaminergic neurons, indicating that NE must activate the translation of pro-growth or plasticity-related protein synthesis. Therefore, activation of signaling pathways as listed above is a necessary step and thus leads to transcription of the TH gene acting on some cis-elements in the TH promoter (Kim et al. 1993; Tinti et al. 1996; Suzuki et al. 2004).
Other Potential Mechanisms Underlying Effects of NE on Dopaminergic System
NE Has Anti-inflammatory Properties: Suppress Mediators of Inflammation
Accumulating evidence suggests that inflammation contributes to the onset and evolution of degeneration in dopaminergic neurons (Mosley et al. 2012). Therefore, the neuroprotective effects of NE on dopaminergic neurons may also be related to its ability to maintain the immunosuppressive environment in the brain as a repression of proinflammatory mediators (Feinstein 1998; Heneka et al. 2002). In vivo studies show that increasing NE levels reduce inflammation (Kalinin et al. 2006) and provide neuroprotection (Troadec et al. 2002). First, NE can reduce damage during neuroinflammatory and neurodegenerative conditions by inducing expression of neurotrophic factors as mentioned in the above section. Second, NE or stimulation of ARs promotes anti-inflammatory phenotypes. For example, the β2-AR agonist protects neurons from kainic acid-induced inflammatory damage (Gleeson et al. 2010), as activation of the β2-ARs promotes the M2 macrophage phenotypes (Grailer et al. 2014) and Th2-type immune responses by reduced cytokine secretion (Anderson and Mosser 2002). Third, NE can suppress inflammatory gene expression from astrocytes. For example, in vitro studies have shown that NE or β-AR agonists suppresses glial expression of proinflammatory cytokines such as IL-1β (Thastrup et al. 1985; Dello Russo et al. 2004), MIP1-α and TNF-α from macrophages (Spengler et al. 1994; Hasko et al. 1998), and the inducible nitric oxide synthase (iNOS) (Feinstein et al. 1993; Gleeson et al. 2010). Similarly, these treatments reduce system levels of TNF-α, IL-6, and nitric oxide during endotoxemia (Elenkov et al. 1995; Szabo et al. 1997). Also, NE reuptake inhibitors desipramine and atomoxetine suppress expression of inflammation-related chemokine and cell adhesion molecule in vivo and in vitro (O’Sullivan et al. 2009, 2010). Other in vivo studies demonstrated that experimental LC destruction resulted in a robust increase and prolonged expression of both IL-1β and iNOS (Heneka et al. 2002).
NE’s Protection May Be Related to the Reduction of Oxidative Stress-Induced Damage
Oxidative stress has been considered as a causative factor for degeneration of dopaminergic neurons in PD. One protective role of NE on dopaminergic neurons may be related to its ability to reduce oxidative stress (Noh et al. 1999). For example, treatment with NE markedly reduced the production of free radical species, which can be mimicked by treatment with catalase (Troadec et al. 2001), an enzyme that blocks the conversion of H2O2 into the highly reactive hydroxyl radicals (Takahashi and Niki 1998). Further, NE and its putative active metabolites are hydrophilic, and they can exert their protective action at the level of the outer plasma membrane by preventing the propagation of lipid peroxidation (Andorn and Pappolla 2001). Moreover, NE was shown to act as both a scavenger of hydroxyl radicals and an inhibitor of lipid peroxidation in cell-free system (Liu and Mori 1993). Finally, trolox is a vitamin E analogue known to protect cell membranes from hydroxyl radical-mediated lipid peroxidation and showed its neuronal protection (Buettner 1993). NE has been showed as potent as trolox in reducing the levels of reactive oxygen species produced in degenerating neurons (Troadec et al. 2001; Alvarez-Diduk and Galano 2015). Therefore, NE exerts its protective action at the levels of the outer plasma membrane by preventing the propagation of lipid peroxidation.
Conclusions
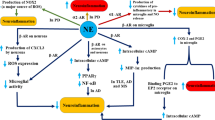
It is well known that the LC-NE system plays a critical role in homeostatic control of brain functions and is involved in a variety of physiological process and behaviors. Furthermore, there are a number of lines of studies revealed the crosstalk between the LC-NE and dopaminergic neurons. However, the neuroprotective effects of the LC-NE system on dopaminergic neurons, especially the involved mechanisms, have not been discussed in detail. Based on the evidence reviewed above, a functional and activity deficiency in the LC-NE system has been taken as a critical factor in determining the evolution of progressive degeneration in dopaminergic neurons in the brain. An intact LC-NE system provides an important role for the survival and normal function of dopaminergic neurons. Among these effects, NE as a potential modulator for dopaminergic neurons plays a central role through its neurotrophic factor and by means of other neuronal growth factors especially BDNF (Fig. 1). Extensive and appropriate studies will be necessary to elucidate the mechanisms involved in NE’s neurotrophic effects, which will improve our understanding the modulation of the LC-NE on dopaminergic neurons and benefit for the strategy in paving a path to drug development and treatment of neurodegenerative diseases.

Schematic representation of potential mechanisms underlying the protective role of the LC-NE system on dopaminergic neurons through trophic influence and positive signaling
Abbreviations
- ARs:
-
Adrenergic receptors
- BDNF:
-
Brain-derived neurotrophic factor
- DA:
-
Dopamine
- CREB:
-
cAMP response element binding
- DBH:
-
Dopamine β-hydroxylase
- DSP4:
-
N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine hydrochloride
- LC:
-
Locus coeruleus
- NE:
-
Norepinephrine
- NET:
-
NE transporter
- PD:
-
Parkinson’s disease
- SNpc:
-
Substantia nigra pars compacta
- TrkB:
-
Tropomyosin receptor kinase B
- VTA:
-
Ventral tegmental area
References
Alonso-Vanegas MA, Fawcett JP, Causing CG, Miller FD, Sadikot AF (1999) Characterization of dopaminergic midbrain neurons in a DBH:BDNF transgenic mouse. J Comp Neurol 413:449–462
Aloyz R, Fawcett JP, Kaplan DR, Murphy RA, Miller FD (1999) Activity-dependent activation of TrkB neurotrophin receptors in the adult CNS. Learn Mem 6:216–231
Alvarez-Diduk R, Galano A (2015) Adrenaline and noradrenaline: protectors against oxidative stress or molecular targets? J Phys Chem B 119:3479–3491
Ancerewicz J, Migliavacca E, Carrupt PA et al (1998) Structure-property relationships of trimetazidine derivatives and model compounds as potential antioxidants. Free Radic Biol Med 25:113–120
Anden NE, Hfuxe K, Hamberger B, Hokfelt T (1966) A quantitative study on the nigro-neostriatal dopamine neuron system in the rat. Acta Physiol Scand 67:306–312
Anderson CF, Mosser DM (2002) A novel phenotype for an activated macrophage: the type 2 activated macrophage. J Leukoc Biol 72:101–106
Andorn AC, Pappolla MA (2001) Catecholamines inhibit lipid peroxidation in young, aged, and Alzheimer’s disease brain. Free Radic Biol Med 31:315–320
Antelman SM, Caggiula AR (1977) Norepinephrine-dopamine interactions and behavior. Science 195:646–653
Barde YA, Edgar D, Thoenen H (1982) Purification of a new neurotrophic factor from mammalian brain. EMBO J 1:549–553
Baydyuk M, Xu B (2014) BDNF signaling and survival of striatal neurons. Front Cell Neurosci 8:254
Berghuis P, Agerman K, Dobszay MB, Minichiello L, Harkany T, Ernfors P (2006) Brain-derived neurotrophic factor selectively regulates dendritogenesis of parvalbumin-containing interneurons in the main olfactory bulb through the PLCgamma pathway. J Neurobiol 66:1437–1451
Berridge CW, Waterhouse BD (2003) The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev 42:33–84
Berridge CW, Stratford TL, Foote SL, Kelley AE (1997) Distribution of dopamine beta-hydroxylase-like immunoreactive fibers within the shell subregion of the nucleus accumbens. Synapse 27:230–241
Bing G, Zhang Y, Watanabe Y, McEwen BS, Stone EA (1994) Locus coeruleus lesions potentiate neurotoxic effects of MPTP in dopaminergic neurons of the substantia nigra. Brain Res 668:261–265
Bjorklund A, Dunnett SB (2007) Dopamine neuron systems in the brain: an update. Trends Neurosci 30:194–202
Blin J, Bonnet AM, Agid Y (1988) Does levodopa aggravate Parkinson’s disease? Neurology 38:1410–1416
Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286:1358–1362
Brodal A (1981) Neurological anatomy in relation to clinical medicine. Oxford University Press, New York, pp 416–419
Bromberg-Martin ES, Matsumoto M, Hikosaka O (2010) Dopamine in motivational control: rewarding, aversive, and alerting. Neuron 68:815–834
Brunet A, Datta SR, Greenberg ME (2001) Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 11:297–305
Buettner GR (1993) The pecking order of free radicals and antioxidants: lipid peroxidation, alpha-tocopherol, and ascorbate. Arch Biochem Biophys 300:535–543
Cabelli RJ, Hohn A, Shatz CJ (1995) Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science 267:1662–1666
Calabresi P, Giacomini P, Centonze D, Bernardi G (2000) Levodopa-induced dyskinesia: a pathological form of striatal synaptic plasticity? Ann Neurol 47:S60–S68 discussion S68-69
Castren E, Thoenen H, Lindholm D (1995) Brain-derived neurotrophic factor messenger RNA is expressed in the septum, hypothalamus and in adrenergic brain stem nuclei of adult rat brain and is increased by osmotic stimulation in the paraventricular nucleus. Neuroscience 64:71–80
Chen NH, Reith ME (1994) Autoregulation and monoamine interactions in the ventral tegmental area in the absence and presence of cocaine: a microdialysis study in freely moving rats. J Pharmacol Exp Ther 271:1597–1610
Chen MJ, Russo-Neustadt AA (2005) Exercise activates the phosphatidylinositol 3-kinase pathway. Brain Res Mol Brain Res 135:181–193
Chen MJ, Russo-Neustadt AA (2007) Nitric oxide signaling participates in norepinephrine-induced activity of neuronal intracellular survival pathways. Life Sci 81:1280–1290
Chen MJ, Nguyen TV, Pike CJ, Russo-Neustadt AA (2007) Norepinephrine induces BDNF and activates the PI-3K and MAPK cascades in embryonic hippocampal neurons. Cell Signal 19:114–128
Cohen Z, Molinatti G, Hamel E (1997) Astroglial and vascular interactions of noradrenaline terminals in the rat cerebral cortex. J Cereb Blood Flow Metab 17:894–904
Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S (1997) Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci 17:2295–2313
Coradazzi M, Gulino R, Fieramosca F, Falzacappa LV, Riggi M, Leanza G (2016) Selective noradrenaline depletion impairs working memory and hippocampal neurogenesis. Neurobiol Aging 48:93–102
Cottingham C, Jones A, Wang Q (2012) Desipramine selectively potentiates norepinephrine-elicited ERK1/2 activation through the alpha2A adrenergic receptor. Biochem Biophys Res Commun 420:161–165
Counts SE, Mufson EJ (2010) Noradrenaline activation of neurotrophic pathways protects against neuronal amyloid toxicity. J Neurochem 113:649–660
Culmsee C, Semkova I, Krieglstein J (1999) NGF mediates the neuroprotective effect of the beta2-adrenoceptor agonist clenbuterol in vitro and in vivo: evidence from an NGF-antisense study. Neurochem Int 35:47–57
da Penha Berzaghi M, Cooper J, Castren E, Zafra F, Sofroniew M, Thoenen H, Lindholm D (1993) Cholinergic regulation of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) but not neurotrophin-3 (NT-3) mRNA levels in the developing rat hippocampus. J Neurosci 13:3818–3826
Datta SR, Brunet A, Greenberg ME (1999) Cellular survival: a play in three Akts. Genes Dev 13:2905–2927
Day JS, O’Neill E, Cawley C, Aretz NK, Kilroy D, Gibney SM, Harkin A, Connor TJ (2014) Noradrenaline acting on astrocytic beta(2)-adrenoceptors induces neurite outgrowth in primary cortical neurons. Neuropharmacology 77:234–248
Delaville C, Deurwaerdere PD, Benazzouz A (2011) Noradrenaline and Parkinson’s disease. Front Syst Neurosci 5:31
Dello Russo C, Boullerne AI, Gavrilyuk V, Feinstein DL (2004) Inhibition of microglial inflammatory responses by norepinephrine: effects on nitric oxide and interleukin-1beta production. J Neuroinflammation 1:9
Dobolyi A, Vincze C, Pal G, Lovas G (2012) The neuroprotective functions of transforming growth factor beta proteins. Int J Mol Sci 13:8219–8258
Domesick VB (1988) Neuroanatomical organization of dopamine neurons in the ventral tegmental area. Ann N Y Acad Sci 537:10–26
Edeline JM, Manunta Y, Hennevin E (2011) Induction of selective plasticity in the frequency tuning of auditory cortex and auditory thalamus neurons by locus coeruleus stimulation. Hear Res 274:75–84
Einat H, Yuan P, Gould TD, Li J, Du J, Zhang L, Manji HK, Chen G (2003) The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci 23:7311–7316
Elenkov IJ, Hasko G, Kovacs KJ, Vizi ES (1995) Modulation of lipopolysaccharide-induced tumor necrosis factor-alpha production by selective alpha- and beta-adrenergic drugs in mice. J Neuroimmunol 61:123–131
Encinas M, Iglesias M, Llecha N, Comella JX (1999) Extracellular-regulated kinases and phosphatidylinositol 3-kinase are involved in brain-derived neurotrophic factor-mediated survival and neuritogenesis of the neuroblastoma cell line SH-SY5Y. J Neurochem 73:1409–1421
Espay AJ, LeWitt PA, Kaufmann H (2014) Norepinephrine deficiency in Parkinson’s disease: the case for noradrenergic enhancement. Mov Disord 29:1710–1719
Fawcett JP, Bamji SX, Causing CG, Aloyz R, Ase AR, Reader TA, McLean JH, Miller FD (1998) Functional evidence that BDNF is an anterograde neuronal trophic factor in the CNS. J Neurosci 18:2808–2821
Fearnley JM, Lees AJ (1991) Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114(Pt 5):2283–2301
Feinstein DL (1998) Suppression of astroglial nitric oxide synthase expression by norepinephrine results from decreased NOS-2 promoter activity. J Neurochem 70:1484–1496
Feinstein DL, Galea E, Reis DJ (1993) Norepinephrine suppresses inducible nitric oxide synthase activity in rat astroglial cultures. J Neurochem 60:1945–1948
Felten DL, Hallman H, Jonsson G (1982) Evidence for a neurotropic role of noradrenaline neurons in the postnatal development of rat cerebral cortex. J Neurocytol 11:119–135
Fornai F, Bassi L, Torracca MT, Scalori V, Corsini GU (1995) Norepinephrine loss exacerbates methamphetamine-induced striatal dopamine depletion in mice. Eur J Pharmacol 283:99–102
Fornai F, Torracca MT, Bassi L, D’Errigo DA, Scalori V, Corsini GU (1996) Norepinephrine loss selectively enhances chronic nigrostriatal dopamine depletion in mice and rats. Brain Res 735:349–353
Fornai F, Alessandri MG, Torracca MT, Bassi L, Scalori V, Corsini GU (1998) Noradrenergic modulation of methamphetamine-induced striatal dopamine depletion. Ann N Y Acad Sci 844:166–177
Fukuchi M, Fujii H, Takachi H, Ichinose H, Kuwana Y, Tabuchi A, Tsuda M (2010) Activation of tyrosine hydroxylase (TH) gene transcription induced by brain-derived neurotrophic factor (BDNF) and its selective inhibition through Ca(2+) signals evoked via the N-methyl-D-aspartate (NMDA) receptor. Brain Res 1366:18–26
Fulceri F, Biagioni F, Lenzi P, Falleni A, Gesi M, Ruggieri S, Fornai F (2006) Nigrostriatal damage with 6-OHDA: validation of routinely applied procedures. Ann N Y Acad Sci 1074:344–348
Fulceri F, Biagioni F, Ferrucci M, Lazzeri G, Bartalucci A, Galli V, Ruggieri S, Paparelli A, Fornai F (2007) Abnormal involuntary movements (AIMs) following pulsatile dopaminergic stimulation: severe deterioration and morphological correlates following the loss of locus coeruleus neurons. Brain Res 1135:219–229
Gesi M, Soldani P, Giorgi FS, Santinami A, Bonaccorsi I, Fornai F (2000) The role of the locus coeruleus in the development of Parkinson’s disease. Neurosci Biobehav Rev 24:655–668
Ghosh A, Carnahan J, Greenberg ME (1994) Requirement for BDNF in activity-dependent survival of cortical neurons. Science 263:1618–1623
Gleeson LC, Ryan KJ, Griffin EW, Connor TJ, Harkin A (2010) The beta2-adrenoceptor agonist clenbuterol elicits neuroprotective, anti-inflammatory and neurotrophic actions in the kainic acid model of excitotoxicity. Brain Behav Immun 24:1354–1361
Gorski JA, Zeiler SR, Tamowski S, Jones KR (2003) Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J Neurosci 23:6856–6865
Gozdz A, Habas A, Jaworski J, Zielinska M, Albrecht J, Chlystun M, Jalili A, Hetman M (2003) Role of N-methyl-D-aspartate receptors in the neuroprotective activation of extracellular signal-regulated kinase 1/2 by cisplatin. J Biol Chem 278:43663–43671
Grailer JJ, Haggadone MD, Sarma JV, Zetoune FS, Ward PA (2014) Induction of M2 regulatory macrophages through the beta2-adrenergic receptor with protection during endotoxemia and acute lung injury. J Innate Immun 6:607–618
Grenhoff J, Svensson TH (1993) Prazosin modulates the firing pattern of dopamine neurons in rat ventral tegmental area. Eur J Pharmacol 233:79–84
Grenhoff J, Nisell M, Ferre S, Aston-Jones G, Svensson TH (1993) Noradrenergic modulation of midbrain dopamine cell firing elicited by stimulation of the locus coeruleus in the rat. J Neural Transm Gen Sect 93:11–25
Guiard BP, El Mansari M, Merali Z, Blier P (2008) Functional interactions between dopamine, serotonin and norepinephrine neurons: an in-vivo electrophysiological study in rats with monoaminergic lesions. Int J Neuropsychopharmacol 11:625–639
Haring JH, Davis JN (1985) Differential distribution of locus coeruleus projections to the hippocampal formation: anatomical and biochemical evidence. Brain Res 325:366–369
Hasko G, Shanley TP, Egnaczyk G, Nemeth ZH, Salzman AL, Vizi ES, Szabo C (1998) Exogenous and endogenous catecholamines inhibit the production of macrophage inflammatory protein (MIP) 1 alpha via a beta adrenoceptor mediated mechanism. Br J Pharmacol 125:1297–1303
Heerssen HM, Segal RA (2002) Location, location, location: a spatial view of neurotrophin signal transduction. Trends Neurosci 25:160–165
Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O’Banion MK, Weinberg G, Klockgether T, Feinstein DL (2002) Noradrenergic depletion potentiates beta-amyloid-induced cortical inflammation: implications for Alzheimer’s disease. J Neurosci 22:2434–2442
Henry B, Fox SH, Peggs D, Crossman AR, Brotchie JM (1999) The alpha2-adrenergic receptor antagonist idazoxan reduces dyskinesia and enhances anti-parkinsonian actions of L-dopa in the MPTP-lesioned primate model of Parkinson’s disease. Mov Disord 14:744–753
Hetman M, Kanning K, Cavanaugh JE, Xia Z (1999) Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J Biol Chem 274:22569–22580
Hillman KL, Lei S, Doze VA, Porter JE (2009) Alpha-1A adrenergic receptor activation increases inhibitory tone in CA1 hippocampus. Epilepsy Res 84:97–109
Hirata H, Ladenheim B, Carlson E, Epstein C, Cadet JL (1996) Autoradiographic evidence for methamphetamine-induced striatal dopaminergic loss in mouse brain: attenuation in CuZn-superoxide dismutase transgenic mice. Brain Res 714:95–103
Hirsch E, Graybiel AM, Agid YA (1988) Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 334:345–348
Hotchkiss AJ, Gibb JW (1980) Long-term effects of multiple doses of methamphetamine on tryptophan hydroxylase and tyrosine hydroxylase activity in rat brain. J Pharmacol Exp Ther 214:257–262
Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R (2007) Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell 131:160–173
Huang YY, Peng CH, Yang YP et al (2007) Desipramine activated Bcl-2 expression and inhibited lipopolysaccharide-induced apoptosis in hippocampus-derived adult neural stem cells. J Pharmacol Sci 104:61–72
Huang YZ, Pan E, Xiong ZQ, McNamara JO (2008) Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron 57:546–558
Inoue S, Susukida M, Ikeda K, Murase K, Hayashi K (1997) Dopaminergic transmitter up-regulation of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) synthesis in mouse astrocytes in culture. Biochem Biophys Res Commun 238:468–472
Isaias IU, Marotta G, Pezzoli G, Sabri O, Schwarz J, Crenna P, Classen J, Cavallari P (2011) Enhanced catecholamine transporter binding in the locus coeruleus of patients with early Parkinson disease. BMC Neurol 11:88
Ivy AS, Rodriguez FG, Garcia C, Chen MJ, Russo-Neustadt AA (2003) Noradrenergic and serotonergic blockade inhibits BDNF mRNA activation following exercise and antidepressant. Pharmacol Biochem Behav 75:81–88
Jhang KA, Lee EO, Kim HS, Chong YH (2014) Norepinephrine provides short-term neuroprotection against Abeta1-42 by reducing oxidative stress independent of Nrf2 activation. Neurobiol Aging 35:2465–2473
Jhaveri DJ, Mackay EW, Hamlin AS, Marathe SV, Nandam LS, Vaidya VA, Bartlett PF (2010) Norepinephrine directly activates adult hippocampal precursors via beta3-adrenergic receptors. J Neurosci 30:2795–2806
Jones LS, Gauger LL, Davis JN (1985) Anatomy of brain alpha 1-adrenergic receptors: in vitro autoradiography with [125I]-heat. J Comp Neurol 231:190–208
Jones KR, Farinas I, Backus C, Reichardt LF (1994) Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell 76:989–999
Juric DM, Miklic S, Carman-Krzan M (2006) Monoaminergic neuronal activity up-regulates BDNF synthesis in cultured neonatal rat astrocytes. Brain Res 1108:54–62
Juric DM, Loncar D, Carman-Krzan M (2008) Noradrenergic stimulation of BDNF synthesis in astrocytes: mediation via alpha1- and beta1/beta2-adrenergic receptors. Neurochem Int 52:297–306
Kajitani N, Hisaoka-Nakashima K, Morioka N, Okada-Tsuchioka M, Kaneko M, Kasai M, Shibasaki C, Nakata Y, Takebayashi M (2012) Antidepressant acts on astrocytes leading to an increase in the expression of neurotrophic/growth factors: differential regulation of FGF-2 by noradrenaline. PLoS One 7:e51197
Kalashnikova EV, Dymshits G, Kolpakov FA (2006) Signal transduction pathways involved in transcriptional regulation of tyrosine hydroxylase. In: Fight International Conference on Bioinformatics of Genome Regulation and Structure-BGRS’2006, pp. 128–131. Novosibirsk
Kalinin S, Feinstein DL, Xu HL, Huesa G, Pelligrino DA, Galea E (2006) Degeneration of noradrenergic fibres from the locus coeruleus causes tight-junction disorganisation in the rat brain. Eur J Neurosci 24:3393–3400
Kang YC, Kim PK, Choi BM, Chung HT, Ha KS, Kwon YG, Kim YM (2004) Regulation of programmed cell death in neuronal cells by nitric oxide. In Vivo 18:367–376
Katsuki H, Izumi Y, Zorumski CF (1997) Noradrenergic regulation of synaptic plasticity in the hippocampal CA1 region. J Neurophysiol 77:3013–3020
Kilbourn MR, Sherman P, Abbott LC (1998) Reduced MPTP neurotoxicity in striatum of the mutant mouse tottering. Synapse 30:205–210
Kim KS, Lee MK, Carroll J, Joh TH (1993) Both the basal and inducible transcription of the tyrosine hydroxylase gene are dependent upon a cAMP response element. J Biol Chem 268:15689–15695
Kreider ML, Seidler FJ, Cousins MM, Tate CA, Slotkin TA (2004) Transiently overexpressed alpha2-adrenoceptors and their control of DNA synthesis in the developing brain. Brain Res Dev Brain Res 152:233–239
Kulkarni VA, Jha S, Vaidya VA (2002) Depletion of norepinephrine decreases the proliferation, but does not influence the survival and differentiation, of granule cell progenitors in the adult rat hippocampus. Eur J Neurosci 16:2008–2012
Laifenfeld D, Klein E, Ben-Shachar D (2002) Norepinephrine alters the expression of genes involved in neuronal sprouting and differentiation: relevance for major depression and antidepressant mechanisms. J Neurochem 83:1054–1064
Lategan AJ, Marien MR, Colpaert FC (1990) Effects of locus coeruleus lesions on the release of endogenous dopamine in the rat nucleus accumbens and caudate nucleus as determined by intracerebral microdialysis. Brain Res 523:134–138
Lategan AJ, Marien MR, Colpaert FC (1992) Suppression of nigrostriatal and mesolimbic dopamine release in vivo following noradrenaline depletion by DSP-4: a microdialysis study. Life Sci 50:995–999
Lauder JM (1993) Neurotransmitters as growth regulatory signals: role of receptors and second messengers. Trends Neurosci 16:233–240
Lee FS, Chao MV (2001) Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc Natl Acad Sci U S A 98:3555–3560
Lee A, Wissekerke AE, Rosin DL, Lynch KR (1998) Localization of alpha2C-adrenergic receptor immunoreactivity in catecholaminergic neurons in the rat central nervous system. Neuroscience 84:1085–1096
Lee FS, Rajagopal R, Kim AH, Chang PC, Chao MV (2002) Activation of Trk neurotrophin receptor signaling by pituitary adenylate cyclase-activating polypeptides. J Biol Chem 277:9096–9102
Leibrock J, Lottspeich F, Hohn A, Hofer M, Hengerer B, Masiakowski P, Thoenen H, Barde YA (1989) Molecular cloning and expression of brain-derived neurotrophic factor. Nature 341:149–152
Lidow MS, Rakic P (1994) Unique profiles of the alpha 1-, alpha 2-, and beta-adrenergic receptors in the developing cortical plate and transient embryonic zones of the rhesus monkey. J Neurosci 14:4064–4078
Lim J, Yang C, Hong SJ, Kim KS (2000) Regulation of tyrosine hydroxylase gene transcription by the cAMP-signaling pathway: involvement of multiple transcription factors. Mol Cell Biochem 212:51–60
Lindholm D, Castren E, Berzaghi M, Blochl A, Thoenen H (1994) Activity-dependent and hormonal regulation of neurotrophin mRNA levels in the brain—implications for neuronal plasticity. J Neurobiol 25:1362–1372
Linner L, Endersz H, Ohman D, Bengtsson F, Schalling M, Svensson TH (2001) Reboxetine modulates the firing pattern of dopamine cells in the ventral tegmental area and selectively increases dopamine availability in the prefrontal cortex. J Pharmacol Exp Ther 297:540–546
Liprando LA, Miner LH, Blakely RD, Lewis DA, Sesack SR (2004) Ultrastructural interactions between terminals expressing the norepinephrine transporter and dopamine neurons in the rat and monkey ventral tegmental area. Synapse 52:233–244
Liu J, Mori A (1993) Monoamine metabolism provides an antioxidant defense in the brain against oxidant- and free radical-induced damage. Arch Biochem Biophys 302:118–127
Liu Y, Formisano L, Savtchouk I, Takayasu Y, Szabo G, Zukin RS, Liu SJ (2010) A single fear-inducing stimulus induces a transcription-dependent switch in synaptic AMPAR phenotype. Nat Neurosci 13:223–231
Liu X, Ye K, Weinshenker D (2015) Norepinephrine protects against amyloid-beta toxicity via TrkB. J Alzheimers Dis 44:251–260
Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci MA (2002) Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson’s disease. Eur J Neurosci 15:120–132
Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000) Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20:9104–9110
Marien M, Briley M, Colpaert F (1993) Noradrenaline depletion exacerbates MPTP-induced striatal dopamine loss in mice. Eur J Pharmacol 236:487–489
Marien M, Lategan A, Colpaert F (1994) Noradrenergic control of striatal dopamine. In: Briley M, Marien M (eds) Noradrenergic mechanisms in Parkinson’s disease. CRC Press, Boca Raton, pp 127–138
Martel J, Chopin P, Colpaert F, Marien M (1998) Neuroprotective effects of the alpha2-adrenoceptor antagonists, (+)-efaroxan and (+/−)-idazoxan, against quinolinic acid-induced lesions of the rat striatum. Exp Neurol 154:595–601
Matsumoto M, Hikosaka O (2009) Two types of dopamine neuron distinctly convey positive and negative motivational signals. Nature 459:837–841
Mavridis M, Degryse AD, Lategan AJ, Marien MR, Colpaert FC (1991) Effects of locus coeruleus lesions on parkinsonian signs, striatal dopamine and substantia nigra cell loss after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in monkeys: a possible role for the locus coeruleus in the progression of Parkinson’s disease. Neuroscience 41:507–523
McAllister AK, Lo DC, Katz LC (1995) Neurotrophins regulate dendritic growth in developing visual cortex. Neuron 15:791–803
McAllister AK, Katz LC, Lo DC (1996) Neurotrophin regulation of cortical dendritic growth requires activity. Neuron 17:1057–1064
McAllister AK, Katz LC, Lo DC (1999) Neurotrophins and synaptic plasticity. Annu Rev Neurosci 22:295–318
Mejias-Aponte CA (2016) Specificity and impact of adrenergic projections to the midbrain dopamine system. Brain Res 1641:258–273
Mejias-Aponte CA, Drouin C, Aston-Jones G (2009) Adrenergic and noradrenergic innervation of the midbrain ventral tegmental area and retrorubral field: prominent inputs from medullary homeostatic centers. J Neurosci 29:3613–3626
Miklic S, Juric DM, Carman-Krzan M (2004) Differences in the regulation of BDNF and NGF synthesis in cultured neonatal rat astrocytes. Int J Dev Neurosci 22:119–130
Mittal S, Bjornevik K, Im DS et al (2017) beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science 357:891–898
Moore RY, Bloom FE (1979) Central catecholamine neuron systems: anatomy and physiology of the norepinephrine and epinephrine systems. Annu Rev Neurosci 2:113–168
Morrison JH, Molliver ME, Grzanna R (1979) Noradrenergic innervation of cerebral cortex: widespread effects of local cortical lesions. Science 205:313–316
Mosley RL, Hutter-Saunders JA, Stone DK, Gendelman HE (2012) Inflammation and adaptive immunity in Parkinson’s disease. Cold Spring Harb Perspect Med 2:a009381
Musazzi L, Rimland JM, Ieraci A, Racagni G, Domenici E, Popoli M (2014) Pharmacological characterization of BDNF promoters I, II and IV reveals that serotonin and norepinephrine input is sufficient for transcription activation. Int J Neuropsychopharmacol 17:779–791
Nagamoto-Combs K, Piech KM, Best JA, Sun B, Tank AW (1997) Tyrosine hydroxylase gene promoter activity is regulated by both cyclic AMP-responsive element and AP1 sites following calcium influx. Evidence for cyclic amp-responsive element binding protein-independent regulation. J Biol Chem 272:6051–6058
Nawa H, Pelleymounter MA, Carnahan J (1994) Intraventricular administration of BDNF increases neuropeptide expression in newborn rat brain. J Neurosci 14:3751–3765
Noh JS, Kim EY, Kang JS, Kim HR, Oh YJ, Gwag BJ (1999) Neurotoxic and neuroprotective actions of catecholamines in cortical neurons. Exp Neurol 159:217–224
Numan S, Lane-Ladd SB, Zhang L, Lundgren KH, Russell DS, Seroogy KB, Nestler EJ (1998) Differential regulation of neurotrophin and trk receptor mRNAs in catecholaminergic nuclei during chronic opiate treatment and withdrawal. J Neurosci 18:10700–10708
O’Sullivan JB, Ryan KM, Curtin NM, Harkin A, Connor TJ (2009) Noradrenaline reuptake inhibitors limit neuroinflammation in rat cortex following a systemic inflammatory challenge: implications for depression and neurodegeneration. Int J Neuropsychopharmacol 12:687–699
O’Sullivan JB, Ryan KM, Harkin A, Connor TJ (2010) Noradrenaline reuptake inhibitors inhibit expression of chemokines IP-10 and RANTES and cell adhesion molecules VCAM-1 and ICAM-1 in the CNS following a systemic inflammatory challenge. J Neuroimmunol 220:34–42
Oades RD, Halliday GM (1987) Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Res 434:117–165
Orefice LL, Waterhouse EG, Partridge JG, Lalchandani RR, Vicini S, Xu B (2013) Distinct roles for somatically and dendritically synthesized brain-derived neurotrophic factor in morphogenesis of dendritic spines. J Neurosci 33:11618–11632
Patel NJ, Chen MJ, Russo-Neustadt AA (2010) Norepinephrine and nitric oxide promote cell survival signaling in hippocampal neurons. Eur J Pharmacol 633:1–9
Picconi B, Pisani A, Barone I, Bonsi P, Centonze D, Bernardi G, Calabresi P (2005) Pathological synaptic plasticity in the striatum: implications for Parkinson’s disease. Neurotoxicology 26:779–783
Popovik E, Haynes LW (2000) Survival and mitogenesis of neuroepithelial cells are influenced by noradrenergic but not cholinergic innervation in cultured embryonic rat neopallium. Brain Res 853:227–235
Rajagopal R, Chen ZY, Lee FS, Chao MV (2004) Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J Neurosci 24:6650–6658
Rascol O, Arnulf I, Peyro-Saint Paul H et al (2001) Idazoxan, an alpha-2 antagonist, and L-DOPA-induced dyskinesias in patients with Parkinson’s disease. Mov Disord 16:708–713
Reichardt LF (2006) Neurotrophin-regulated signalling pathways. Philos Trans R Soc London Ser B Biol Sci 361:1545–1564
Robbins TW (2000) From arousal to cognition: the integrative position of the prefrontal cortex. Prog Brain Res 126:469–483
Robbins T, Everitt B (1995) Central norepinephrine neurons and behavior. In: Bloom F, Kupfer D (eds) Neuropsychopharmacology: the fourth generation of progress. Raven Press, New York
Rommelfanger KS, Weinshenker D, Miller GW (2004) Reduced MPTP toxicity in noradrenaline transporter knockout mice. J Neurochem 91:1116–1124
Rommelfanger KS, Edwards GL, Freeman KG, Liles LC, Miller GW, Weinshenker D (2007) Norepinephrine loss produces more profound motor deficits than MPTP treatment in mice. Proc Natl Acad Sci U S A 104:13804–13809
Ross RA, Reis DJ (1974) Effects of lesions of locus coeruleus on regional distribution of dopamine-beta-hydroxylase activity in rat brain. Brain Res 73:161–166
Ruiz CR, Shi J, Meffert MK (2014) Transcript specificity in BDNF-regulated protein synthesis. Neuropharmacology 76(Pt C):657–663
Samuels ER, Szabadi E (2008) Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part I: principles of functional organisation. Curr Neuropharmacol 6:235–253
Sara SJ (2009) The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci 10:211–223
Satoh T, Nakai S, Sato T, Kimura M (2003) Correlated coding of motivation and outcome of decision by dopamine neurons. J Neurosci 23:9913–9923
Schroeter S, Apparsundaram S, Wiley RG, Miner LH, Sesack SR, Blakely RD (2000) Immunolocalization of the cocaine- and antidepressant-sensitive l-norepinephrine transporter. J Comp Neurol 420:211–232
Schwartz JP, Nishiyama N (1994) Neurotrophic factor gene expression in astrocytes during development and following injury. Brain Res Bull 35:403–407
Schwartz JP, Nishiyama N, Wilson D, Taniwaki T (1994) Receptor-mediated regulation of neuropeptide gene expression in astrocytes. Glia 11:185–190
Sciarretta C, Fritzsch B, Beisel K, Rocha-Sanchez SM, Buniello A, Horn JM, Minichiello L (2010) PLCgamma-activated signalling is essential for TrkB mediated sensory neuron structural plasticity. BMC Dev Biol 10:103
Segal M, Bloom FE (1976) The action of norepinephrine in the rat hippocampus. III. Hippocampal cellular responses to locus coeruleus stimulation in the awake rat. Brain Res 107:499–511
Segal RA (2003) Selectivity in neurotrophin signaling: theme and variations. Annu Rev Neurosci 26:299–330
Shaywitz AJ, Greenberg ME (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68:821–861
Shepard KN, Liles LC, Weinshenker D, Liu RC (2015) Norepinephrine is necessary for experience-dependent plasticity in the developing mouse auditory cortex. J Neurosci 35:2432–2437
Siciliano R, Fornai F, Bonaccorsi I, Domenici L, Bagnoli P (1999) Cholinergic and noradrenergic afferents influence the functional properties of the postnatal visual cortex in rats. Vis Neurosci 16:1015–1028
Simon H, Le Moal M, Stinus L, Calas A (1979) Anatomical relationships between the ventral mesencephalic tegmentum—a 10 region and the locus coeruleus as demonstrated by anterograde and retrograde tracing techniques. J Neural Transm 44:77–86
Smith MA, Makino S, Altemus M, Michelson D, Hong SK, Kvetnansky R, Post RM (1995) Stress and antidepressants differentially regulate neurotrophin 3 mRNA expression in the locus coeruleus. Proc Natl Acad Sci U S A 92:8788–8792
Spengler RN, Chensue SW, Giacherio DA, Blenk N, Kunkel SL (1994) Endogenous norepinephrine regulates tumor necrosis factor-alpha production from macrophages in vitro. J Immunol 152:3024–3031
Srinivasan J, Schmidt WJ (2003) Potentiation of parkinsonian symptoms by depletion of locus coeruleus noradrenaline in 6-hydroxydopamine-induced partial degeneration of substantia nigra in rats. Eur J Neurosci 17:2586–2592
Sterpenich V, D’Argembeau A, Desseilles M et al (2006) The locus ceruleus is involved in the successful retrieval of emotional memories in humans. J Neurosci 26:7416–7423
Stroemer RP, Kent TA, Hulsebosch CE (1998) Enhanced neocortical neural sprouting, synaptogenesis, and behavioral recovery with D-amphetamine therapy after neocortical infarction in rats. Stroke 29:2381–2393 discussion 2393-2385
Sullivan RM, Wilson DA, Leon M (1989) Norepinephrine and learning-induced plasticity in infant rat olfactory system. J Neurosci 9:3998–4006
Sullivan RM, McGaugh JL, Leon M (1991) Norepinephrine-induced plasticity and one-trial olfactory learning in neonatal rats. Brain Res Dev Brain Res 60:219–228
Suzuki T, Kurahashi H, Ichinose H (2004) Ras/MEK pathway is required for NGF-induced expression of tyrosine hydroxylase gene. Biochem Biophys Res Commun 315:389–396
Swanson LW (1982) The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull 9:321–353
Swanson LW, Hartman BK (1975) The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-beta-hydroxylase as a marker. J Comp Neurol 163:467–505
Szabo C, Hasko G, Zingarelli B, Nemeth ZH, Salzman AL, Kvetan V, Pastores SM, Vizi ES (1997) Isoproterenol regulates tumour necrosis factor, interleukin-10, interleukin-6 and nitric oxide production and protects against the development of vascular hyporeactivity in endotoxaemia. Immunology 90:95–100
Takahashi M, Niki E (1998) The effect of oxidative stress on cells by oxygen radicals and its inhibition by antioxidants. In: Montagnier L, Olivier R, Pasquier F (eds) Oxidative stress in cancer, AIDA and neurodegenerative diseases. Marcel Decker, New York, pp 9–14
Thastrup O, Knudsen JB, Lemmich J, Winther K (1985) Inhibition of human platelet aggregation by dihydropyrano- and dihydrofuranocoumarins, a new class of cAMP-phosphodiesterase inhibitors. Biochem Pharmacol 34:2137–2140
Thoenen H (1995) Neurotrophins and neuronal plasticity. Science 270:593–598
Timmer M, Cesnulevicius K, Winkler C, Kolb J, Lipokatic-Takacs E, Jungnickel J, Grothe C (2007) Fibroblast growth factor (FGF)-2 and FGF receptor 3 are required for the development of the substantia nigra, and FGF-2 plays a crucial role for the rescue of dopaminergic neurons after 6-hydroxydopamine lesion. J Neurosci 27:459–471
Tinti C, Conti B, Cubells JF, Kim KS, Baker H, Joh TH (1996) Inducible cAMP early repressor can modulate tyrosine hydroxylase gene expression after stimulation of cAMP synthesis. J Biol Chem 271:25375–25381
Tong J, Hornykiewicz O, Kish SJ (2006) Inverse relationship between brain noradrenaline level and dopamine loss in Parkinson disease: a possible neuroprotective role for noradrenaline. Arch Neurol 63:1724–1728
Traver S, Salthun-Lassalle B, Marien M, Hirsch EC, Colpaert F, Michel PP (2005) The neurotransmitter noradrenaline rescues septal cholinergic neurons in culture from degeneration caused by low-level oxidative stress. Mol Pharmacol 67:1882–1891
Troadec JD, Marien M, Darios F, Hartmann A, Ruberg M, Colpaert F, Michel PP (2001) Noradrenaline provides long-term protection to dopaminergic neurons by reducing oxidative stress. J Neurochem 79:200–210
Troadec JD, Marien M, Mourlevat S, Debeir T, Ruberg M, Colpaert F, Michel PP (2002) Activation of the mitogen-activated protein kinase (ERK(1/2)) signaling pathway by cyclic AMP potentiates the neuroprotective effect of the neurotransmitter noradrenaline on dopaminergic neurons. Mol Pharmacol 62:1043–1052
Vauquelin G, Geynet P, Hanoune J, Strosberg AD (1979) Affinity chromatography of the beta-adrenergic receptor from Turkey erythrocytes. Eur J Biochem 98:543–556
Vijayashankar N, Brody H (1979) A quantitative study of the pigmented neurons in the nuclei locus coeruleus and subcoeruleus in man as related to aging. J Neuropathol Exp Neurol 38:490–497
Vitalis T, Cases O, Parnavelas JG (2005) Development of the dopaminergic neurons in the rodent brainstem. Exp Neurol 191(Suppl 1):S104–S112
Vizi ES, Orso E, Osipenko ON, Hasko G, Elenkov IJ (1995) Neurochemical, electrophysiological and immunocytochemical evidence for a noradrenergic link between the sympathetic nervous system and thymocytes. Neuroscience 68:1263–1276
Wang Y, Zhang QJ, Liu J, Ali U, Gui ZH, Hui YP, Chen L, Wu ZH, Li Q (2010) Noradrenergic lesion of the locus coeruleus increases apomorphine-induced circling behavior and the firing activity of substantia nigra pars reticulata neurons in a rat model of Parkinson’s disease. Brain Res 1310:189–199
Weinshenker D, Ferrucci M, Busceti CL et al (2008) Genetic or pharmacological blockade of noradrenaline synthesis enhances the neurochemical, behavioral, and neurotoxic effects of methamphetamine. J Neurochem 105:471–483
Widmer HR, Kaplan DR, Rabin SJ, Beck KD, Hefti F, Knusel B (1993) Rapid phosphorylation of phospholipase C gamma 1 by brain-derived neurotrophic factor and neurotrophin-3 in cultures of embryonic rat cortical neurons. J Neurochem 60:2111–2123
Xing B, Xin T, Zhao L, Hunter RL, Chen Y, Bing G (2010) Glial cell line-derived neurotrophic factor protects midbrain dopaminergic neurons against lipopolysaccharide neurotoxicity. J Neuroimmunol 225:43–51
Yamada M, Ohnishi H, Sano S, Araki T, Nakatani A, Ikeuchi T, Hatanaka H (1999) Brain-derived neurotrophic factor stimulates interactions of Shp2 with phosphatidylinositol 3-kinase and Grb2 in cultured cerebral cortical neurons. J Neurochem 73:41–49
Yanpallewar SU, Fernandes K, Marathe SV et al (2010) Alpha2-adrenoceptor blockade accelerates the neurogenic, neurotrophic, and behavioral effects of chronic antidepressant treatment. J Neurosci 30:1096–1109
Yuan J, Yankner BA (2000) Apoptosis in the nervous system. Nature 407:802–809
Zafra F, Lindholm D, Castren E, Hartikka J, Thoenen H (1992) Regulation of brain-derived neurotrophic factor and nerve growth factor mRNA in primary cultures of hippocampal neurons and astrocytes. J Neurosci 12:4793–4799
Zarow C, Lyness SA, Mortimer JA, Chui HC (2003) Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol 60:337–341
Zuccato C, Cattaneo E (2009) Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol 5:311–322
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Zhu, MY. Noradrenergic Modulation on Dopaminergic Neurons. Neurotox Res 34, 848–859 (2018). https://doi.org/10.1007/s12640-018-9889-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-018-9889-z