
Abstract
Theileria annulata (T. annulata) is a tick-borne apicomplexan parasite that affects bovine. It is endemic in many tropical and subtropics areas, including Odisha, India. The objective of this study is to identify T. annulata infection in the peripheral blood of cattle as a biological sample by conventional PCR (cPCR) and quantitative PCR (qPCR). The phylogenetic analysis was done using the T. annulata merozoite surface antigen (Tams 1) gene. Out of 552 samples of examined blood smears by microscopy, 454 (82.24%) animals were positive for Theileria species. Out of 454 samples, 96 samples were further examined by both cPCR and qPCR, 52 samples (54.16%) were found positive for T. annulata in both PCR methodologies. Phylogenetic analysis revealed that T. annulata Odisha isolate was closely related to T. annulata Uttarakhand, India isolate (KM061799) and Hyderabad, India isolate (MK034702) with Nucleotide sequence identity 95.36%, 95.25%, respectively. This is the first study to detect T. annulata by qPCR in Odisha and supported that both PCR techniques were equally effective for the detection of Tams 1 gene of T. annulata in cattle’s blood.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Theileriosis is a tick-borne parasitic disease caused by hemoprotozoan parasites, which commonly infect ruminants especially cattle in tropical and subtropical regions (Nazifi et al. 2009; Demessie and Derso 2015). Theileria is an intracellular protozoan parasite transmitted by ticks and has complex life cycles in vertebrate and invertebrate hosts together (Sitotaw et al. 2014). The presence of infective ticks around the year and transplacental transmission of T. annulata from the dam to the fetus causes peracute infection and fatal disease in the neonates (Godara et al. 2009). The clinical signs of tropical theileriosis mainly depend on the destructive outcome of the parasite on the host lymphatic tissues and immune system. The clinical signs of naturally affected animals are fever, lymph nodes enlargement, anaemia, anorexia, weakness, difficult respiration, petechial haemorrhages in the conjunctiva, nasal and oral mucous membranes and corneal opacity of the eye (Sengupta et al. 1993; Branco et al. 2010; Sudan et al. 2012).
Molecular techniques have been developed for accurate and rapid identification of Theileria spp in animals having negative serological tests that can still infect ticks (Zaeemi et al. 2011; Ghaemi et al. 2012). However, conventional PCR assays don’t give any information on parasite load and quantity in blood making it impossible to differentiate between clinically affected animals and carrier animals. To solve these problems, qPCR had been used for the detection of Theileria species (Bogema et al. 2015; Perera et al. 2015; Gebrekidan et al. 2018).
The qPCR had not been yet used to detect T. annulata in Odisha. Although the qPCR technique is very expensive, which may prevent using it in poor economic situations; its rapid and accurate results guarantee its use as a very hopeful technique for effective diagnosis of parasitic infections. The aim of this study is to detect T. annulata infection in cattle blood by cPCR and qPCR. The phylogenetic analysis was done using the Tams 1 gene. To the best of the authors’ knowledge this first study to make identification of T. annulata infection in cattle blood by cPCR and qPCR in Odisha..
Materials and methods
Blood samples collection, DNA extraction and microscopic examination
A total of 552 blood samples were collected from Odisha, India in EDTA tubes for DNA extraction and blood smears preparation. Thin blood smear preparation and staining was performed according to Jain (1993). The DNA was extracted using the QIAamp® DNA Extraction Kit (QIAGEN, USA) according to the manufacturers’ protocols. By the aid of Nanodrop, the quantity and purity of the DNA samples were checked then, DNA samples were kept at − 20 °C until further analysis.
Conventional PCR (cPCR), cloning and nucleotide sequencing of Tams 1 gene
T. annulata specific Tams 1 gene primers (Tams 1-F 5′ CCAGGACCACCCTCAAGTTC-3′ and Tams 1-R 5′ GCATCTAGTTCCTTGGCGGA-3′) were used in conventional PCR protocol for the amplification of a fragment (441 bp) (Kundave et al. 2014) PCR was performed in volume of 25 µl containing 2.5 µl of 10X PCR buffer, 1.25 U of Taq DNA polymerase (Himedia, India), 2.0 µl of dNTP (2.5 mM each), 1.0 µl of each primer (10 pmol), 2.0 µl of DNA template and 16.25 µl of nucleus free water. Thermocycling conditions were standardized as initial denaturation (one-step) at 95 °C for 5 min, after that 37 cycles of denaturation at 95 °C for 30 s, 55 °C for 30 s, and extension at 72 °C for 30 s, with a final extension at 72 °C for 5 min. The reactions were done by the Thermal Cycler (T100 TM Thermal Cycler, Bio-Rad, USA). Electrophoresis of the PCR products along with 100 bp DNA ladder marker was performed on 1.5% agarose gel stained with ethidium bromide dye (Himedia, India) and visualized under UV light gel documentation system (Bio-Rad, Hercules, CA, USA). A negative control (nucleus free water), and a positive control DNA from an animal infected with T. annulata, were incorporated in each amplification.
The amplified Tams 1 gene from each positive sample was selected and sent for sequencing. Purification of PCR amplicons were performed with Illustra DNA purification Kit (GE Healthcare, UK), cloned into a TOPO® TA Cloning® Kit vector (Invitrogen, USA), and followed by transformation into Escherichia coli DH5α competent cells with ampicillin. Selection of positive colonies were performed by PCR and digested by EcoR1 enzyme (Thermofisher Scientific, USA). Plasmids were isolated from positive colonies using the GeneJET™ Plasmid Miniprep Kit (Thermofisher Scientific, USA). Two clones were selected from each sample and sequenced using universal primers of the M13 gene. The Tams 1 gene nucleotide sequence was determined by DNA sequencing through Sangers sequencing using the BigDye™ Terminator v3.1 cycle sequencing kit (Thermofisher Scientific, USA) in the platform of ABI Genetic Analyser model 3100 (Applied Biosystems, Germany). Sequencing was performed in the Department of Biotechnology, Institute of Life Sciences, Odisha, India.
Sequences alignments were performed using MEGA X program. The sequence of the partial Tams 1 gene analyzed and compared with other gene sequences available in GenBank through ClustalX software. The phylogenetic analysis was performed through nucleotide alignments using Neighbor-Joining method (Saitou and Nei 1987). Bootstrap analysis was performed with 1000 replicates by the aid of MEGA 6 program (Tamura et al. 2013).
Quantitative real-time PCR (qPCR)
Primer design was performed from the same sequence through primer BLAST NCBI. The forward primer used in cPCR was used in qPCR. Quantitative real-time PCR was done in 96-well plates on a Bio-Rad CFX96 qPCR thermal cycler (Bio-Rad Laboratories, Hercules, CA, USA). The qPCR assays specific for T. annulata was performed in 10 µL reaction volume consisting of 5 µL SYBR green qPCR master mix (Sigma- Aldrich), 1.5 µL of DNA, 2.5 µL of nucleus free water and 5 pM each of primer (Tams 1 F 5′-CCAGGACCACCCTCAAGTTC-3′ and Tams 1 R 5′-CTTGAAACGGTAGCCATCCG-3′). Thermal cycling conditions were started with initial denaturation step at 95 °C for 2 min, after that 45 cycles of 95 °C for 15 s (denaturation) and 60 °C for 30 s (annealing/extension). The melt curve analysis was done from 72 °C to 95 °C with 0.2 °C increments for 5 s. In each run, negative and positive control samples that had been previously diagnosed by PCR for Tams 1 gene were used to determine the accuracy of the run. Three replicates for each concentration were run to prove the stability of the qPCR. Additionally, electrophoresis of the qPCR products along with 100 bp DNA ladder marker was performed on 1.5% agarose gel and visualized in a gel documentation system.
Statistical analysis
Statistical analysis was performed using IBM SPSS 26 windows version program.
Results
Positive blood smears for Theileria spp infections revealed intra-erythrocytic bodies that appeared dot and comma-shaped in the case of T. annulata. Out of 552 samples of examined blood smears, 454 (82.24%) animals were positive for Theileria species (Fig. 1).

Blood smear of an animal infected with T. annulata previously diagnosed by PCR. There is evidence of intracellular piroplasms within erythrocytes
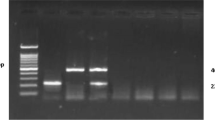
Amplifications of the isolated DNA from randomly collected blood samples of cattle showed an amplicon size of 441 bp which considered positive for T. annulata (Fig. 2). Out of the 96 cows examined by PCR in this study, T. annulata DNA was detected in 52 animals (54.16%). Nucleotide sequence of T. annulata Tams1 gene demonstrated a similarity of 95.36%, 95.25%, with Uttarakhand India isolate (KM061799) and Hyderabad India isolate (MK034702), respectively (Fig. 3).

Amplification of the T. annulata Tams 1 gene showing a target of 441 bp. M-100 bp size DNA marker. Lane with sample number 2, 3 and 5 are positive samples while, lane with sample 1, 4 and 6 are negative samples

Phylogenetic tree of constructed using the nucleotide sequence of the Tams 1 gene. The sequence obtained in the present study is indicated by a black triangle. Bootstrap values are provided at the beginning of each branch
A Cq (quantification cycle) value was calculated, equivalent to the PCR cycle at which the concentration of the target DNA crossed the threshold line established by the device Software (Bio-Rad CFX96 analysis software). The average of Cq values were 27.35 ± 0.79. After evaluating 96 submitted samples, 54.16% of cases were detected positive by qPCR assay procedures. Amplification plot and the melt curve for T. annulata were generated. Melting curve analysis was found to be a specific protocol and no detectable peaks were observed in no template control (NTC) which proved the specificity of the protocol. Electrophoresis of the qPCR products on gel revealed the target amplicons size 200 bp (Fig. 4).

Amplification of the T. annulata Tams 1 gene showing a target of 200 bp
Discussion
Rapid and accurate diagnosis has a potential effect on the establishment of strict measures to control the spread of disease in endemic regions. The qPCR become an important assay for the detection of vector born diseases. It provides rapid and accurate results which are crucial for screening large numbers of samples without faults (Kearns et al. 2001; Azimi et al. 2011).
In blood smear examination, T. annulata appeared dot and comma shaped intra-erythrocytic bodies. Out of 552 samples of examined blood smears, 454 (82.24%) animals were positive for Theileria species. The high infection rate is similar to a previous study in Odisha with a prevalence rate of 74% (Acharya et al. 2017). Blood smear examination is more probable to give false positive diagnosis of theileriosis due to the high pleomorphism among the piroplasms between Koch’s Blue Bodies and azurophilic granules (d’Oliveira et al. 1995). Consequently, it is recommended that the bovine theileriosis must be regularly screened by PCR technique (Mohammed et al. 2010; Sivakumar et al. 2012).
Several molecular techniques based on PCR are present for the detection of T. annulata DNA (Bilgic et al. 2010; Ros-García et al. 2012). Real-time quantitative PCR has a great outcome for analytical applications in quantitative DNA analysis, but giving quantitative data needs a more problematic data analysis than qualitative PCR (Ros-García et al. 2012). In the present study, the whole blood PCR examinations revealed a high rate of 54.16% (52/96) of animals were positive with T. annulata. The samples were collected from different districts of Odisha. In India, little studies performed molecular diagnosis of T. annulata for example study conducted in Odisha where 34 samples were found positive out of 50 (Acharya et al. 2017), in Gujarat where 74 samples were found positive out of 113 (Kundave et al. 2014). Additionally, a study conducted in Bangalore where 41 cases were found positive out of 132 (Ananda et al. 2009). This result will provide information about the infection rate of bovine tropical theileriosis in Odisha which will help in the establishment of strict control Measures.
Phylogenetic analysis of the Tams 1 gene sequence isolated from Odisha revealed that there was a relation between T. annulata Odisha sequence and other isolates available in GenBank. In the phylogenetic tree, T. annulata Odisha isolate was closely related to T. annulata Uttarakhand, India isolate (KM061799) and Hyderabad, India isolate (MK034702) with Nucleotide sequence identity 95.36%, 95.25%, respectively. Tams 1 gene has phylogenetically instructive variable regions that are species specific and useful in differentiation between species (Santos et al. 2013).
The current study confirmed the agreement of results between cPCR and qPCR in 100% tested samples. These results support that both methodologies were similarly effective for the detection of the Tams1 gene of T. annulata in the cattle’s blood, with the need for crucial adjustments according to the conditions inside the laboratory, to maintain the sensitivity and accuracy. This result agreed with a previous study performed to detect Leishmania in human blood (Pereira et al. 2014). It is crucial to mention that the extracted DNA for amplification when it was examined in diluted ten times in sterilized nucleus free water or in full concentration (data not showed) therefore, this study advised that all DNA of the blood samples should be examined in full concentrations and diluted ten times in sterilized nucleus free water, regardless the quantity of DNA according to the study made by (Pereira et al. 2014). This study completely supports some studies that revealed the need for detailed standardization to improve the quality of PCR reaction (Ros-García et al. 2012; Dandasena et al. 2018).
The advantage of qPCR is its ability to. make amplification of the target sequence and analysis of the products which exclude the need for subsequent gel electrophoresis and staining with carcinogenic dyes like Ethidium bromide. The size of the Real-time PCR amplicons is less than 200 bp, also annealing/extension steps combined in one step consequently, the chance of dissociation of the target sequence through the reaction reduced (Zarlenga and Higgins 2001; Niedbalski et al. 2010). Finally, our findings indicated that applying of qPCR can be sufficient for the rapid diagnosis of T. annulata as a helpful technique in conjunction with traditional procedures.
Conclusions
In summary, Our study has led to the identification of T. annulata infection in cattle through microscopic examination, cPCR and qPCR. The present study also revealed that qPCR is an accurate technique for the detection of T. annulata infection in cattle with less time as compared to conventional PCR. Furthermore, this study support that both methodologies were similarly effective for the detection of the Tams1 gene of T. annulata in the cattle’s blood.
References
Acharya AP, Panda SK, Prusty BK (2017) Diagnosis and confirmation of Theileria annulata infection in cattle in Odisha, India. J Entomol Zool Stud 5(4):1543–1546
Ananda KJ, D’Souza P, Puttalakshmamma GC (2009) Prevalence of Haemoprotozoan diseases in crossbred cattle in Bangalore north. Vet World 2:5–16
Azimi S, Mahravani H, Jeirani F, Shoshtari A (2011) Appling real time RT-PCR for bluetongue virus detection in Iran. Archi Razi 66:75–80
Bilgic HB, Karagenc T, Shiels B, Tait A, Eren H, Weir W (2010) Evaluation of cytochrome b as a sensitive target for PCR based detection of T. annulata carrier animals. Vet Parasitol 174:341–347
Bogema DR, Deutscher AT, Fell S, Collins D, Eamens GJ, Jenkins C (2015) Development and validation of a quantitative PCR assay using multiplexed hydrolysis probes for detection and quantification of Theileria orientalis isolates and differentiation of clinically relevant subtypes. J Clin Microbiol 53:941–950
Branco S, Orvalho J, Leitao A, Pereira I, Malta M, Mariano I, Carvalho T, Baptista R, Shiels BR, Peleteiro MC (2010) Fatal cases of Theileria annulata infection in calves in Portugal associated with neoplastic-like lymphoid cell proliferation. J Vet Sci 11:27–34
d’Oliveira C, Van Der Weide M, Habela P, Jacquiet P, Jongejan F (1995) Detection of Theileria annulata in blood samples of carrier cattle by PCR. J Clin Microbiol 13:2665–2669
Dandasena D, Bhandari V, Sreenivasamurthy GS, Murthy S, Roy S, Bhanot V, Singh S, Sharma P (2018) A Real-Time PCR based assay for determining parasite to host ratio and parasitaemia in the clinical samples of Bovine Theileriosis. Sci Rep 8(1):15441
Demessie Y, Derso S (2015) Tick Borne Hemoparasitic Diseases of Ruminants: A Review. Adv Bio Res 9:210–224
Gebrekidan H, Gasser RB, Stevenson MA, Jabbar A (2018) Multiplexed tandem PCR (MT-PCR) assay using the major piroplasm surface protein gene for the diagnosis of Theileria orientalis infection in cattle. J Clin Microbiol 56:1661–1671
Ghaemi P, Hoghooghi-Rad N, Shayan P, Eckert B (2012) Detection of Theileria orientalis in Iran by semi-nested PCR. Parasitol Res 110:527–531
Godara R, Sharma RL, Sharma CS (2009) Bovine Tropical Theileriosis in a neonate calf. Trop Anim Health Prod 42:551–553
Jain K (1993) Essentials of Veterinary Hematology. Blackwell Publishing, U.S.A
Kearns A, Guiver M, James V, King J (2001) Development and evaluation of a real-time quantitative PCR for the detection of human cytomegalovirus. J Virol Methods 95:121–131
Kundave VR, Patel AK, Patel PV, Hasnani JJ, Joshi CG (2014) Qualitative and quantitative assessment of Theileria annulata in cattle and buffaloes by Polymerase Chain Reaction. Trop Biomed 31(4):728–735
Mohammad Al-Saeed AT, Omer LT, Abdo J, Habibi G, Salih DA, Seitzer U, Ahmed J (2010) Epidemiological studies on tropical theileriosis (Theileria annulata infection of cattle) in Kurdistan Region, Iraq. Parasitol Res 106(2):403–407
Nazifi S, Razavi SM, Esmailnejad Z, Gheisari H (2009) Study on acute phase proteins (haptoglobin, serum amyloid A, fibrinogen, and ceruloplasmin) changes and their diagnostic values in bovine tropical theileriosis. Parasitol Res 105:41–46
Niedbalski W, Ksy A, Procajo Z, Szweda W, Mikulska SE, Platt SA, Guo-Wen L, Zhi-Gang Z, Wen-Yan Y, Lian-Yu Y (2010) Rapid detection and quantification of foot-and-mouth disease virus by a real-time reverse transcription PCR. B Vet I Pulawy 54
Pereira MR, Rocha-Silva F, Graciele-Melo C, Lafuente CR, Magalhães T, Caligiorne RB (2014) Comparison between conventional and real-time PCR assays for diagnosis of visceral leishmaniasis. Biomed Res Int 63:93–100
Perera PK, Gasser RB, Firestone SM, Smith L, Roeber F, Jabbar A (2015) A semiquantitative multiplexed tandem PCR for detection and differentiation of four Theileria orientalis genotypes in cattle. J Clin Microbiol 53:79–87
Ros-García A, Nicolás A, García-Pérez AL, Juste RA, Hurtado A (2012) Development and evaluation of a real-time PCR assay for the quantitative detection of Theileria annulata in cattle. Parasit Vectors 5:171
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Santos M, Soares R, Costa P (2013) Revisiting the Tams1-encoding gene as a species-specific target for the molecular detection of Theileria annulata in bovine blood samples. Ticks Tick Borne Dis 4:72–77
Sengupta PP, Bansal GC, Ray D (1993) Ocular lesions in experimental theileriosis. J Vet Parasitol 7:127–129
Sitotaw T, Regassa F, Zeru F, Kahsay AG (2014) Epidemiological significance of major hemoparasites of ruminants in and around Debre-Zeit, Central Ethiopia. J Parasitol Vector Bio 6:16–22
Sivakumar T, Kothalawala H, Abeyratne SA, Vimalakumar SC, Meewewa AS, Hadirampela DT, Puvirajan T, Sukumar S, Kuleswarakumar K, Chandrasiri AD, Igarashi I, Yokoyama N (2012) A PCR-based survey of selected Babesia and Theileria parasites in cattle in Sri Lanka.Vet Parasitol 190(1–2):263–267
Sudan V, Sharma RL, Yadav R, Borah MK (2012) Turning sickness in a cross bred cow naturally infected with Theileria annulata. J Parasit Dis 36:226–229
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA 6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729
Zaeemi M, Haddadzadeh H, Khazraiinia P, Kazemi B, Bandehpour M (2011) Identification of different Theileria species (Theileria lestoquardi, Theileria ovis, and Theileria annulata) in naturally infected sheep using nested PCR–RFLP. Parasitol Res 108:837–843
Zarlenga DS, Higgins J (2001) PCR as adiagnostic and quantitative technique in veterinary parasitology. Vet Parasitol 101:215–230
Author information
Authors and Affiliations
Contributions
SKS and IN conceived and designed the study; AS and GRG contributed to the writing and revision of the manuscript; MS performed laboratory testing; AS and CM contributed to the genome sequencing and phylogenetic analysis. SS and MD read and revised the manuscript. All authors’ read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors of this study stated that they have no conflicts of interest.
Ethical standards
There is no precise law for blood sample collection in India for clinical diagnosis and so no consent was obligatory. In this study 4 ml blood collected from the jugular vein under aseptic conditions from cattle after taking the permission of the farm owners.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Selim, A.M., Das, M., Senapati, S.K. et al. Molecular detection of Theileria annulata infection in cattle by conventional PCR and quantitative real time PCR in India. J Parasit Dis 45, 72–77 (2021). https://doi.org/10.1007/s12639-020-01278-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12639-020-01278-6