
Abstract
The survival of probiotic microorganisms during their exposure to harsh environments plays a critical role in the fulfillment of their functional properties. In particular, transit through the human gastrointestinal tract (GIT) is considered one of the most challenging habitats that probiotics must endure, because of the particularly stressful conditions (e.g., oxygen level, pH variations, nutrient limitations, high osmolarity, oxidation, peristalsis) prevailing in the different sections of the GIT, which in turn can affect the growth, viability, physiological status, and functionality of microbial cells. Consequently, probiotics have developed a series of strategies, called “mechanisms of stress response,” to protect themselves from these adverse conditions. Such mechanisms may include but are not limited to the induction of new metabolic pathways, formation/production of particular metabolites, and changes of transcription rates. It should be highlighted that some of such mechanisms can be conserved across several different strains or can be unique for specific genera. Hence, this review attempts to review the state-of-the-art knowledge of mechanisms of stress response displayed by potential probiotic strains during their transit through the GIT. In addition, evidence whether stress responses can compromise the biosafety of such strains is also discussed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The human gastrointestinal tract (GIT) is a complex system involving tissues, glands, and organs which synchronously work for the proper digestion and absorption of food components/nutrients [1]. During digestion, a large number of substances, including electrolytes, hydrolytic enzymes, hydrochloric acid, bile, antimicrobial peptides, among others are secreted into the GIT [2], creating a unique environment with a plethora of harsh physicochemical conditions that affect microbial survival [3, 4]. Additionally, physical components (epithelial and mucus layers) and immunological factors (antibodies and epithelia-associated immune cells) also contribute to the highly stressful conditions affecting the microbial community dynamics in the gut [5, 6].
Under this scenario, autochthonous microorganisms (resident or native) found in the GIT have adapted to the different biochemical niches available in the gut by a selective reciprocal co-evolutionary process between human and their gut symbionts [7]. Moreover, allochthonous microorganisms (transient or foreing), such as probiotics, which are termed as “live microorganisms that when administered in adequate amounts confer health benefits on the host” [8], are also capable of temporarily integrating into the gut, where they may directly impact the host health, the later through modification of the composition and activity of the resident gut communities or indirectly by stimulating, regulating, and modulating different functions, including digestion, metabolism, epithelial innate immunity, and brain-gut communication [9].
In order to adapt and survive to the GIT conditions, probiotics have developed a robust and conserved “general mechanisms of stress response” to either maintain structural integrity, regulate, inhibit, or fortify physiology and metabolic activities, as well as to protect or repair damaged macromolecules [10,11,12,13]. Furthermore, novel evidence has suggested that probiotics are also capable of activating a notable class of coordinated cellular processes to increase the stress tolerance, namely “specific-associated stress response” [14], which includes a cascade of cellular events aimed to reprogram themselves to induce or repress specific/particular regulators (i.e., genes, proteins, lipids, and/or metabolites), associated to the improvement of cell fitness, robustness, and attain a level of adaptation not observed before [5, 15].
Despite that these responses allow probiotics not only to withstand specific GIT stressful conditions but also enhance their functionality, it is necessary to consider that such intricate mechanisms can be subjected to alterations in their expression and regulation that could critically jeopardize the probiotic biosafety. In this latter context, it has been reported that stress responses lead to the expression of pathogenic-associated molecules required for microbial survival and growth and which can potentially have negative or collateral activities in the host’s well-being [16]. Hence, the aim of this review is to describe the various stresses encountered by potential probiotic strains through the GIT transit, plus compile the most recent progress in the study of general and specific defense mechanisms by which they respond to stressful conditions, including new metabolic pathways. Finally, evidence on whether stress responses can compromise the biosafety of such strains is also discussed.
Methods
A comprehensive literature search was conducted using seven electronic databases, namely Scopus, PubMed, Science Direct, Web of Science, ResearchGate, Scientific Electronic Library Online (SciELO), and Google Scholar. The initial search was carried out in May 2021 without limiting the period of publication. Combination of the following keywords or terms was used as scoping search strategy: “gastrointestinal tract (GIT),” “probiotics,” “GIT compartments,” “stress survival,” “mechanisms of response,” “acidic, saline, osmotic, and oxidative environments,” “omics approaches,” “molecular characterization,” “biosafety,” and “gut-brain axis.” The titles and abstracts of the selected articles were examined; then, full-text articles were retrieved and used for data abstraction. Inclusion criteria were (1) accessible full articles; (2) articles published in journals with impact index; (3) recent and/or relevant articles, mainly over the previous 5 to 10 years; (4) studies carried out with potential probiotic strains belonging to core genera most often used, i.e., Lactobacillus, Bifidobacterium, Lactococcus, or Pediococcus; and (5) studies that characterized the stress response at any genomic, transcriptional, proteomic, or metabolomic level. Exclusion criteria were (1) unpublished articles; (2) articles published in languages other than English, (3) editorial material, letters to the editor, and abstracts without significant data; and (4) studies that combined a GIT stress condition with other compounds or substances not proper of the GIT.
On the Way to…
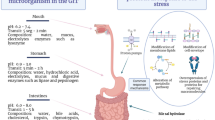
Overall, probiotics are orally administered, and their delivery is achieved through diverse dosage forms, including functional foods, beverages, and dietary supplements (e.g., tablets, capsules, powders, among others). However, consumers prefer food products over supplements considering the hedonic aspects of food intake. Hence, probiotics have been incorporated directly and in cell-free form into these products [17, 18]. In this regard, viability and stability of free probiotic cells are the main concerns for targeted delivery to the human gut when ingested orally. During their journey through the GIT, probiotics face a series of adverse conditions that may compromise not only their survival and colonization capacity, but also their functionality and efficacy [6, 12, 19]. Therefore, the compartments and/or accessory organs involved, as well as the harsh conditions prevalent in the GIT (i.e., physicochemical, enzymatic, and microbial parameters) are depicted in Fig. 1, and will be described in the following section.

The hazardous passage of probiotics through the GIT. Figure adapted from images created with BioRender.com
Mouth
The mouth is the opening entry of the GIT and the first barrier to overcome. When probiotics are orally ingested, they are initially exposed to saliva, which is an extracellular secretion produced by salivary glands [20]. This secretion is a complex fluid (pH ranging between 6.2 and 7.4) mainly composed by water (99%), mucus (proteoglycans and glycoproteins), electrolytes (sodium, potassium, chloride, and bicarbonate ions), and enzymes, including amylase, lipase, lactoperoxidase, lactoferrin, and lysozyme; the latter plays an important role in saliva’s natural antimicrobial properties [21, 22]. Additionally, saliva contains hydrogen peroxide and immunologic components which include secretory immunoglobulin A (IgA), immunoglobulin G (IgG), and immunoglobulin M (IgM) [12]. Furthermore, it has been described that saliva is an underestimated component of the non-specific immune defense which can exert direct antimicrobial activity through pore-forming peptides such as defensins, cathelicidins, and histatin, which are produced by salivary glands and/or immune cell populations. Additionally, saliva plays a dual role in modulating the attachment and colonization in the oral tissues, by binding microbial cells to soluble-phase saliva receptors (proline-rich proteins), which promotes agglutination and blocking of microbial surface adhesins [22]. Hence, all these components may collectively influence the viability and cell morphological structure of probiotics, further altering their adhesive and metabolic properties [23]. Although most microorganisms recognized to date as probiotics may be particularly sensitive to antimicrobial compounds in saliva [20], several in vitro studies have demonstrated that when probiotics are exposed to saliva, it does not significantly affect the cell viability or survival, even after 24 h of incubation [24,25,26]. Moreover, specific-strain response to saliva proteolytic enzymes has been reported as adhesive properties of some strains remain unaffected after lysozyme pretreatment [27]. Thus, these findings suggest that the impact of saliva (and its main components) on the survival rates and colonization properties of probiotics appears to be minimal [12].
Esophagus
After their passage through the mouth, probiotics move through the esophagus. The primary function of this compartment is to serve as a conduit for the passage of swallowed material to the stomach [28]. This seemingly simple basic function has made the esophagus an undervalued structure; hence, limited information is available on the difficulties that probiotics experience during their transit through this organ. However, it is recognized that there may be multiple barriers that compromise the probiotic efficiency [3, 29].
For instance, the esophageal transit of probiotics is accomplished by periodic and coordinated contractions and relaxation movements (peristalsis) that transport down microorganisms to be pulled towards the stomach [30]. These peristaltic movements imply the exposure of microbial cells to propulsive forces that travel at 3–4 cm s−1 and reach a peak pressure amplitude of 60–140 mmHg along the esophagus [31]. All these tensile (lengthening), compressive (shortening), and/or shear (shape-changing) mechanical forces could impose stress on probiotics during their journey. Equally important, it has been reported that the esophagus also contains a large number of widely distributed sub-mucosal glands that have the ability to secrete several products including bicarbonate (HCO3−), acid mucins (e.g., glycoproteins, sialomucins, and sulfomucins), epidermal growth factor (EGF), and prostaglandins [32]. Although research on the effects of both esophageal stresses on morphology, locomotion, and survival of probiotics are rare, some revealing studies have elucidated that mechanical pressure and bicarbonate secretion (which establish a surface pH gradient) may affect or interrupt the structure and function of the cell envelope assembly in microorganisms [33, 34].
Stomach
The stomach is the muscular organ that assists in the early stages of digestion and prepare the bolus for further processing in the small intestine. In the stomach, probiotics face severe mechanical and chemical conditions creating the most difficult phase for microbial survival [1]. Despite the gastric motility, a complex system of peristaltic waves and segmentation (mixing) contractions that may cause a deleterious environment to microorganisms by the mechanical forces during the normal stomach function [35], the most harmful condition for them is created by the acidic gastric fluid [12]. Gastric glands produce ca. 1.5–2.0 L gastric juice per day. This secretion is comprised of a variable mixture of water (99%), hydrochloric acid (0.4–0.5%), electrolytes (sodium, potassium, calcium, phosphate, sulfate, and bicarbonate ions), mucus, enzymes (lipase, rennin, and pepsinogen), hormones (gastrin and serotonin), and the intrinsic factor [1, 36]. As noted above, gastric juice is highly acidic reaching a pH that dynamically oscillates from 0.9 to 1.5–3.0. This unique composition and pH allow gastric juice to exert a rapid antimicrobial effect, in which allochthonous microorganisms, primarily originating from dietary intake, are usually destroyed in minutes [36, 37].
Therefore, the transit through the stomach represents a remarkable challenge for probiotic survival, since the acidic prevalent condition may induce serious morphological and phenotypic modifications at the cellular level such as (i) changes in the composition of the microbial cell membrane (embedded proteins and lipids, diffusion of other molecules), (ii) damage to the DNA (gene expression), and (iii) alteration of the peptidoglycan components (molecular length, saturation, and branching) [34, 38, 39].
Small Intestine
After gastric digestion, the chyme, and remaining microorganisms, released from the stomach enter the small intestine, where both most digestion occurs and practically all absorption proceeds. To accomplish these functions, the small intestine uses auxiliary secretions produced by the intestinal epithelia, liver, gallbladder, and pancreas [40]. Hence, probiotics first face the neutralized effect of bicarbonate, which suddenly raises the pH from 2.0 to 6.2. This abrupt change, from highly acidic to neutral environment, affects the structure of several microbial macromolecules (e.g., protein unfolding, membrane and DNA damage, among others), thus compromising their viability [41]. On the other hand, gut-on-a-chip analysis, utilizing motility-induced luminal fluid flow, but without physiological peristalsis-like mechanical motions, has indicated that microorganisms would overgrow without the peristaltic strain, which evidenced that mechanical forces arising from intestinal contraction may modulate the number of colonizing microorganisms [42].
Additionally, cells are exposed to the main degradation process conducted by the action of two potent actors: (a) the pancreatic juice, which comprises a series of enzymes (ribonuclease and deoxyribonuclease), pro-enzymes (protrypsin, prochymotrypsin, proelastase, procarboxypeptidases, pancreatic lipase, and α-amylase), protease inhibitors, and electrolytes (sodium, potassium, calcium, and bicarbonate ions) [1, 43], and (b) the bile secretion, mainly constituted by water (95%), bile salts (cholic acid and chenodeoxycholic acid), phospholipids, bilirubin, cholesterol, electrolytes (sodium, potassium, chlorine, calcium, magnesium, phosphate, sulfate, and bicarbonate ions), peptides and amino acids, steroids, enzymes, vitamins, and heavy metals [37]. Altogether, these secretions keep the total solution alkaline with a pH ranging between 7 and 8 [44, 45]. It has been reported that the pancreatic juice and bile constituents can not only severely affect the viability of probiotics, but can also significantly reduce their binding properties by inducing alterations in cell membrane conformation, deforming or denaturing proteins, inducing oxidative DNA damage, degrading nucleic acids, and disrupting phospholipids/fatty acids integrity [46, 47]. Finally, the intestinal epithelial cells harbor several distinctive immune effector molecules that play a key role in providing a barrier against microbial invasion and maintain homeostasis. Markedly, the secretion of antimicrobial peptides (AMPs) (e.g., defensins, cathelicidins, C-type lectins, cytokines, and mucins) and immunoglobulins (secretory immunoglobulin A (sIgA) and immunoglobulin G (IgG)) can seriously compromise the probiotic cell integrity. On the one hand, the AMPs can elicit bactericidal activity by (a) the formation of pores on the surface of microbial cell walls which cause nutrient leakage, depolarization, and impaired cell integrity or (b) by binding to, and cleavage of, peptidoglycans and phospholipids, resulting in the neutralization of their activity and eventually making microorganisms susceptible to lysis. Meanwhile, the immunoglobulins can enhance antibody-mediated microbial phagocytosis, exert immune exclusion by blocking microbial adhesins, and inhibit microbial motility by facilitated microorganisms entrapment in mucus [4, 48].
Large Intestine
The large intestine or large bowel, also known as the colon, is part of the final stages of digestion, most responsible for the absorption of water, electrolytes, and other key nutrients. Given the luminal pH value (ranging from 5.5 to 7.5), lower bile salt concentration and peristaltic activity, as well as minor components of the adaptive system, the colon is highly conducive to microbial survival and colonization, crucial for degradation of indigestible food material through the process of fermentation [3, 49, 50]. During colonic fermentation, a considerable number of metabolic by-products (e.g., enzymes, fatty acids, alcohols, phenols, indoles, amino acids, and co-factors) are released. Such metabolites can change the environmental conditions (pH, redox potential, oxygen availability) or act as antimicrobial substances, which may in turn (i) create a more inhospitable milieu or (ii) alter the cell surface morphology, metabolism, and regulation of gene expression of probiotics [51, 52].
Role of Gut-Brain Axis
The gut-brain axis (GBA) is a bidirectional communication network of signaling pathways between the GIT, the microorganisms which inhabit it, and the peripheral and central nervous systems. Different studies have shown that GBA has a critical role in maintaining homeostatic and cognitive processes [53]; thus, the brain has an influence on the gut through the gut-brain axis, and vice versa. Under normal conditions, the brain ensures proper gastrointestinal functions, such as motility, secretion of acid, bicarbonates, mucus, and signal molecules, intestinal fluid handling, mucosal immune response, and intestinal permeability [54]. Hence, although these conditions impose already a challenge for probiotics, the disruption of gastrointestinal functions can directly or indirectly affect probiotic survival. Different types of physical and psychological stressors (e.g., acoustic, mental, or social stresses, sleep disorders) can disrupt the GBA. It has been described that the presence of stress induces variation in quantity and quality of mucus secretion, affects gastric and intestinal postprandial motility, alters intestinal permeability, and induces overproduction of proinflammatory cytokines (IL-1β, IL-6, TNF-α, and interferon-γ), hormones (corticosterone), and antimicrobial compounds (α-defensin) [55, 56]. All these anomalies can contribute to limit the survival and proliferation of probiotics.
Deploying Well-Known Stress Responses to Survive
In light of the above, it is clear that the GIT exhibits numerous extreme environmental conditions which can determine the degree of probiotic survival. Under this context, probiotics respond adaptively by altering or deploying intrinsic phenotypes to overcome or resist these stressful conditions, at least long enough to reach a more conducive habitat [57]. Consequently, in recent years, research have focused on probiotic adaptive or stress responses [58].
To further explore these stress responses, first, it is necessary to define the concept of “GIT stressor.” Here, we use this concept to refer any physiochemical factor within the steady GIT environment which can exert an adverse effect on the physiological well-being of microbial cells, either by killing or slow and prevent their growth [59]. Thus, GIT stressors may include agents of a very different nature (e.g., pH, oxygen, high concentration of substances, starvation), and probiotics must respond appropriately to them in order to survive [11]. In this sense, the mechanisms of microbial resistance to any stressor are essentially classified as (1) innate/intrinsic or (2) adaptive. The innate responses comprise all those structures and functional pathways naturally occurring and active in the microbial cells, which allow tolerance to multiple stressing agents; meanwhile, adaptive responses encompass induced genotypic and phenotypic modifications, with or without mutations, arising as a consequence of the exposure of cells to a particular stressor, thus increasing the ability of microorganisms to survive [60, 61].
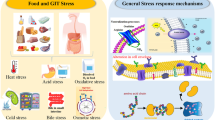
Although microbial resistance is highly dependent on the microorganism and the GIT stressor, there are conserved and well-characterized defense mechanisms shared among probiotic genera and species [4]. Particularly, conventional biochemical analyses and molecular techniques have provided an outline of the presence and activity of functional features that take part in these complex systems [5]. Some of the most reported innate and adaptive stress mechanisms in the literature include (1) alterations in specific stress-sensing/signaling and export systems (e.g., sensor molecules such as nucleic acids, polypeptides, proteins, lipids, proton pumps), (2) accumulation of compatible solutes (sugars, polyols, amino acids) to restore turgescent pressure and enable cell growth and division, (3) regulation of energy production and storage to dispose of an intracellular carbon stock, (4) perturbations of metabolic pathways (e.g., alternative fates of pyruvate, utilization of other carbon sources, activation of the proteolytic system, usage of the catabolism of free amino acids) to stimulate energy fluxes, (5) modifications in the cell envelope (e.g., regulation of membrane fluidity and cell wall composition, overexpression of exopolysaccharides and S-layer proteins) to maintain its integrity, cell shape, and counteract the extracellular effectors, (6) overproduction of proteins (e.g., chaperones, proteases, special shock proteins, miscellaneous enzymes) to protect or repair damaged macromolecules (DNA, denatured or misfolded proteins), and (7) production of antimicrobial substances (peptidic or proteinacious bacteriocins‒nisin, plantaricin, lacticin, bifidocin, mutacin, pediocin, etc., organic acids‒butyric, acetic, propionic, formic, and lactic acids, and other small molecules‒diacetyl, hydrogen peroxide, acetaldehyde, acetoine, reuterin, and reutericyclin) to gain a competitive advantage within the intestinal microbiota [2, 5, 34, 62, 63]. A summary of these defense mechanisms, induced by different stressing conditions, as well as the site where they are deployed during the descent of the probiotic through the GIT, is represented in Figs. 2 and 3, respectively.

Overview of key innate and adaptive mechanisms identified in the enhanced GIT tolerance of potential probiotic microorganisms. ArgG, argininosuccinate synthase; GapA, glyceraldehyde-3-phosphate dehydrogenase; GroES, heat shock protein 10-chaperonin; GroEL, heat shock protein 60-chaperonin; DnaK, heat shock protein 70; TurfB, translation elongation factor; Ppk, polyphosphate kinase; ADI system, arginine deaminase system. Figure adapted from images created with BioRender.com

Main stress resistance mechanisms and place where they are deployed during the descent of the probiotic through the GIT. Figure adapted from images created with BioRender.com
Under this context, it is undeniable that the innate or adaptive mechanisms can share the action of interconnected molecules to form a stress response network, which is common among different microorganisms; however, there could also be unique components at the genetic, transcriptional, protein, or metabolic level, involved in specific stress responses that are present only in some probiotic strains. Considering this, the identification of specific defense mechanisms has become a dynamic field of research in recent years [5].
Novel Players in Adaptation to a Hostile World
In order to overcome the GIT-associated host defense, probiotic microorganisms deploy stress response mechanisms to ensure their survival and persistence in the human gut [16]. Current research has evidenced that probiotics are able to implement novel and sophisticated cellular resilient strategies to sense and adapt to environmental conditions [5, 11]. As summarized in Table 1, a number of advanced experimental approaches, mainly based on multi-omic platforms, i.e., genomics, transcriptomics, proteomics, and metabolomics, have revealed specific molecular mechanisms (e.g., regulation of gene expression, post-translational protein regulation) that enhance the survival of potential probiotic strains in the GIT [5, 15, 64]. In the following sections, the current state of knowledge for the main stress responses mentioned above and how these new and specific defenses contribute to microbial survival is discussed.
Acid Stress
To understand the particular elements of microbial adaptation under acidic conditions, Jung and Lee [65] propagated Lactiplantibacillus plantarum WiKim18 in media with different pH values (5.0–5.5) and evaluated the transcriptional changes associated with survival. The authors observed that acidic conditions affected the expression by upregulation of genes located in the functional categories of the alanine-aspartate metabolism (pyrAA) and amino acid metabolism (cblB, cbs, cysE). Besides, transport-related genes involved in the distribution of essential nutrients, vital for bacterial survival, were also significantly upregulated, particularly, those associated with ABC transport: PTS system gene (pts4ABC), extracellular transglycosylase genes (lp_0302, lp_0304, lp_3014, lp_3050), oligo-peptide ABC transporter genes (lp_0018, lp_0783), nicotinamide nucleotide transporter gene (pnuC1), copper transporting ATPase genes (copA, copB), and carbamoyl phosphatase genes (pyrAB, pyrAA, pyrC).
In a similar work, Wei et al. [66] evidenced the transcriptional alterations in Bifidobacterium longum JDM301AR when cultured in a modified acid medium (sub-lethal batch cultures = pH 3.5). Data showed that the bacterial cells displayed an acquisition of acid-tolerant phenotype, mainly through the modification of cell wall and cell membrane. Such response was attributed to the upregulation of genes encoding for cystathionine (cystathionine gamma-synthase, MetC3), pantothenate, coenzyme A (CoA), and peptidoglycan (clusters BLJ_0525-531 and BLJ_1300-1303, respectively) biosynthesis. These molecules are implicated in the production of ammonia (NH3), fatty acids (FAs), and phospholipids, which are essential to neutralize H+ ions and modify the rigidity of bacterial cell wall. Also, the membrane composition profile revealed an increase production of C14:0 (tetradecanoic, myristic acid), suggesting the key role of this compound on bacterial survival under acid stress.
The metabolic changes and transcriptional/phenotypic adherence response of Lactiplantibacillus plantarum ATCC 14,917, under acid stress (pH 5.5) of initial growth, were also reported by Wang et al. [67]. The authors reported that the intracellular metabolites of bacteria were significantly influenced by the pH stress, compared with the control group. The differential metabolites were dominated by 16 compounds: 1 fatty acid (trans-vaccenic acid), 1 amino acid (L-histidine), and 14 metabolites involved in carbohydrate metabolism (e.g., uridine 5′-triphosphate (UTP), cytosine, adenosine, 2-hydroxyadenine, uracil, nicotinate, glycerophosphocholine, among others). Furthermore, stressed cells were richer in unsaturated (tetradecanoic/myristic acid, C14:0; cis-9-octadecenoic, C18:1 n-9; octadecadienoic acid, C18:2 n-7) and cyclopropane (methyleneoctadecenoic/dihydrosterculic acid, ΔC19:0 n-9) fatty acid content in the cell membranes. Finally, the results of the gene expression of adhesion-related proteins revealed that genes msa, mub1, mub2, mub3, mub4, lspA, and tuf were upregulated after acid stress. Therefore, the changes in bacteria metabolite profile were positive, and their effects on the adhesion ability of L. plantarum ATCC 14,917 evidence the impact of bacterial stress response on its interaction with their host.
In a related work, it was demonstrated that Bifidobacterium longum sub. longum BBMN68 was able to change its protein profile and physiology after subjecting the cells to a medium with sub-lethal pH value (4.5) [68]. Such condition of acid stress increased the abundance of proteins involved in (a) amino acid metabolism (aspartate aminotransferase-AspC; argininosuccinate synthase-ArgG; glutamine synthetase-Glna1; selenocysteinelyase-CsdB), (b) carbohydrate metabolism (phosphoketolase-Xfp; glyceraldehyde-3-phosphate dehydrogenase-GapA; glucokinase-GalK; ADP-glucose pyrophosphorylase-GlgC; fructokinase-KdgK; acetate kinase-AckA; transaldolase-MipB, enolase-Eno), and (c) protein protection (chaperones DnaK, DnaJ1, and GroEL; translation elongation factor-TurfB; aminoacyl-tRNA synthetases GatA, GlyS, ProS, PheS, TyrS). The change in protein profile observed suggests that B. longum BBMN68, under sub-lethal pH environment, is capable to prioritize meeting energy requirements and maintain protein structural integrity. Moreover, a significant shift in the ATP (> 60%), NH3 (> 65%), and peptidoglycan (> 35%) content, as well as an improvement in the H+-ATPase activity (> 50%) and maintenance of the intracellular pH (pHin), was observed in B. longum BBMN68. These are response mechanisms that lead to the H+ discharge/neutralization and cell wall strengthening.
Bile Stress
In a recent report, Bagon et al. [69] employed a proteomic approach to determine protein differentially expressed or modified after exposure of Lactobacillus johnsonii PF01 and C1-10 strains to bile stress in growth media (0.1 and 0.3% w/v). Clearly, in both strains, bile significantly stimulated the number of secreted proteins (up to 100 new proteins), which were classified into four main functional categories: (1) cellular processes and signaling (e.g., MetK, FtsK, AtpG, ClpP), (2) information storage and processing (e.g., Adk, COG3613, Hpt, NusB, RpsA-S1), (3) metabolism (e.g., Fba, L7/L12-RplL, GapA, PepC, ackA, PPX1, GalE), and (4) miscellaneous (e.g., Rv3717, 4-FlgJ, Spr, FrnE, ThrS, HisS). Additionally, a transcriptional analysis revealed that putative proteins with bile response roles (enolase-Eno, phosphoglycerate kinase-Pgk, pyridoxamine 5′-phosphate oxidase-Pyr, 50S ribosomal protein L7/L12-RplL, L-lactate dehydrogenase-Mdh, triosephosphate isomerase-TpiA) were highly upregulated in L. johnsonii strains. It has been reported that all this set of proteins may serve to extracellular nutrient breakdown and to create a matrix of proteins outside the cell, which may have individual or collective functions that promote survival [69].
Accordingly, a transcriptomic analysis showed an altered expression of genes and abundance of proteins in Ligilactobacillus salivarius Ren when exposed to different concentrations of bile salts (0.25 to 1.0 g L−1) [70]. The bacterium was able to shift the regulation (enhancement) of genes involved in (a) amino acid transport and metabolism (gshA, ilvE, serA, araT, pepX), (b) carbohydrate transport and metabolism (glpK, malB, malP, dhaK, malT), and (c) cell envelope biogenesis (betT, dacC-1). Besides, the abundance of several proteins was increased under bile stress, including pyruvate oxidase-poxB, branched-chain-amino-acid aminotransferase-ilvE, aspartate-semialdehyde dehydrogenase-asd, and ATP-dependent protease sub-unit hslV, among others. The potential involvements of these genes and proteins in bile resistance suggest that L. salivarius Ren expands its profile of carbon sources including utilization of maltose and glycerol for energy production; moreover, the presence of enzymes involved in modification of cell surface charge (e.g., araT, Asd, HlyIII) is supposed to hinder the penetration of bile. Finally, the existence of ABC transporters (lsr_RS00945, lsr_RS01160, lsr_RS01170) could contribute to expel bile accumulated in the cytoplasm [70].
In a comparable work, Ma et al. [71] reported that the gene expression, malolactic enzyme (MLE) pathway, and other physiological features were influenced in Lacticaseibacillus paracasei L9 when exposed to a bile-rich medium (0.1 to 0.2% w/v). On one side, the transcriptomic analysis showed that ca. 50 differential expressed genes were upregulated. The recognized functions of these genes are mainly associated with (a) carbon source utilization (LPL9_0432, LPL9_2760, LPL9_1931, etc.), (b) amino acids and peptide metabolism (opp operon, LysX, LysY, etc.), (c) transmembrane transport (FtsE family, LPL9_1281, LPL9_1668, etc.), (d) transcription factors (TetR family, LPL9_0056, LPL9_1280, etc.), and (e) membrane proteins (PspC, LPL9_0968, LPL9_0969). Some of these genes have not been reported to be involved in the bile-related stress in other bacteria. On the other hand, the stimulation of L-malic acid metabolism, which is governed by the MLE pathway (upregulation of mleS and mleT genes), demonstrated to play a crucial role in the alkalinization of the cytoplasm and maintenance of the integrity of the cell membrane. Finally, bacteria grown while in bile stress displayed a rougher and more shrunken appearance, with little variation in length. However, the most surprising finding was the formation of membrane vesicles on the surface of cells and the significant difference in hydrophobicity (threefold higher) when compared with the control treatment.
Similarly, it was found that conjugated (glycodeoxycholic acid-GDCA-) and free (deoxycholic acid-DCA-) bile acids (0.05% w/w) induce a deep metabolic reorganization in Limosilactobacillus reuteri CRL1098 (strain with a health benefit supported by a positive human clinical trial) [72]. Novel tolerance biomarkers were identified, primarily by differential expression of several proteins. The L. reuteri CRL1098 proteome was assigned to distinctive functional categories, namely nucleotide (iunh, ctps, AdSS, fhs) and glycerolipid metabolism (Fe-ADH), transcription and translation (tsf, fusA), pH homeostasis and stress responses (groEL, Otc, TypA), and amino acid biosynthesis (cth, gpt). Among all these proteins, cytosine triphosphate (CTP) synthetase, an enzyme related to the repair of oxidative DNA injuries, was remarkably over-expressed. Additionally, a bile salt hydrolase enzyme (bsh) was characterized as a protein of 325 amino acids with a calculated mass of 36,098.1 Da and predicted pI of ca. 4.81. A significant upregulation of the bsh gene in response to bile stress was also observed. Such enzyme catalyzes the bile acid deconjugation, which appears to be one of the most common detoxification strategy that mediate bacterial bile resistance.
Osmotic Stress
Different studies have been carried out to acquire genomic knowledge about possible new molecular mechanisms of microbial osmotic tolerance. For instance, Yao et al. [73] determined the salt tolerance-related genes of Lactiplantibacillus plantarum D31 and T9 strains by exposing bacteria to a NaCl-rich medium (5.0–15.0% w/v). Both strains were able to grow at high osmotic pressure caused by up to 8.0% NaCl. Then, draft genome sequences of both strains revealed that ca. 170 genes encoded hypothetical functions related to possible strain-specific mechanisms for stress tolerance and/or niche adaptation. These genes encompass at least four distinct categories: (a) recovery of intracellular ion balance (Na+/H+ reverse transport and K+ transport systems = kdp cluster, kup), (b) absorption or synthesis of compatible solutes (nitrate/sulfonate/bicarbonate ABC transporter and proline synthesis opuABCD, choSQ, proABC cluster, D7Y65_10050), (c) stress response (DnaK-DnaJ and GroES-GroEL regulatory systems = D7Y65_09835, D7Y65_09830, D7Y65_06915, D31_D7Y65_06920, etc.), and (d) transcriptional or response regulators (GntR, TetR, Crp/Fnr, and LysR families, RNA polymerase sigma factor RpoD = D7Y65_13295, D7Y66_11330, D7Y65_04790, D7Y66_09275). The particular presence of such genes supported the stress resistance phenotype observed in both strains.
On the other hand, some Lactiplantibacillus plantarum strains (ATCC14917, FS5-5, and 208) have shown the ability to differentially express proteins in response to exposure to osmotic stress (240 g L−1 NaCl) [74]. After proteomic analysis, 40 to 110 proteins with a molecular mass ranging between 6.9 kDa and 135.4 kDa and pI values between 4.41 and 11.34 were identified. Particularly, 26 proteins were found to be key enzymes involved in cell response to osmotic stress in different metabolic pathways. Overall, the proteins that were over-expressed within the L. plantarum strains are mainly involved in (1) sugar and energy metabolism (pyk, gnd, adh2, ldh, pfkA, gck, eno), (2) amino acid metabolism (gadA, cysK, glmS), (3) nucleotide metabolism (rpoA, deaD, mutS, purA, adk), (4) fatty acid metabolism and peptide polysaccharide biosynthesis (MurA, murB, FabI), (5) protein biosynthesis (rplD, rplE, rplM, rplO, rpsC, rpsM), and (6) oxidative phosphorylation (atpA, atpD). These metabolic perturbations suggest that L. plantarum strains focus primarily on the utilization of alternative carbon sources to assure their growth in salt-rich media.
In this same context, Qi et al. [75] characterized the intracellular metabolic response of Ligilactobacillus salivarius FDB89 when subjected to hyperosmotic growth conditions (0.8 mol L−1 NaCl). The metabolomic profile exhibited 44 new characteristic compounds including betaine, carnitine, proline, methionine, malonate, aspartate, cyclopentanecarboxylic acid, isoleucine, pyrimidine, and phenylalanine, as well as choline and their derivatives, sn-glycero-3-phosphocholine, phosphocholine, and acetylcholine, which may serve as potential biomarkers for osmotic stress response, since they were consistently accumulated and abundant in L. salivarius FDB89. Furthermore, it was described that such characteristic compounds could not only play a key role in protecting macromolecular structures, but also serve as compatible solutes, act as electron acceptors, or be used as unique carbon, nitrogen, and energy source.
Induced alterations in the expression of surface layer (S-layer) proteins, as adaptive mechanism to osmotic stress, was also observed when Lactobacillus acidophilus ATCC 4356 (strain with a health benefit supported by a positive human clinical trial) was subjected to elevated salt concentrations (0.3 to 0.8 M NaCl) [76]. In this sense, two distinctive S-layer bands which correspond to proteins with a molecular weight of 45.9 and 49.4 kDa and a pI value of 9.49 were found. The protein content of both bands significantly increased (40–60%) as the salt concentration of the culture medium augmented. This behavior was confirmed with the high transcription (expression) level of slpA and slpX genes. Moreover, the increase in the S-layer proteins conducted to the modification of the cell wall. Mass recovery of the complete cell wall, peptidoglycan, and lipoteichoic acids of cells grown in high-salt conditions was up to threefold lower when compared to the control condition. Such modification suggests that a bacterium carrying S-layers depends on these proteins to maintain cell wall stability.
Oxidative Stress
Several studies have been conducted to understand the physiological response of microorganisms to oxidative stress. For instance, the global intracellular metabolic profile of Pediococcus pentosaceus R1, exposed to sub-lethal concentrations (1 to 4 mM) of H2O2, was studied [77]. Data showed that P. pentosaceus R1 mobilized plenty of metabolites under oxidative stress. Specifically, 74 compounds were identified as critical biomarkers. These metabolites can be classified into eleven main categories, being the most abundant (a) amino acids (glycylproline, L-lysine, L-glutamine, 3-aminoisobutanoic acid, 2-hydroxybutyric acid, alpha-ketoisovaleric acid, L-alpha-aminobutyric, L-tyrosine, etc.), (b) carbohydrates (D-galactose, D-glucose, D-maltose, D-arabinose 5-phosphate), (c) organic acids (4-hydroxyphenylpyruvic acid, pyrophosphate, L-pipecolic acid, phenylpyruvic acid), (d) nucleotides (deoxyadenosine, deoxyguanosine, 5-thymidylic acid, uridine 5′-monophosphate), (e) fatty acids (myristic, caproic, dodecanoic acids), f) lipids (MG160, hexanoylcarnitine, glycerol 3-phosphate), and (g) vitamins (niacinamide, pantothenic acid). Such complex metabolite composition indicated that P. pentosaceus R1 redirected its physiology to satisfy various important priorities in order to survive and grow; these include energy conservation, reparation of cellular damage, regulation of membrane fluidity, and scavenging of reactive oxygen species (ROS).
Genes involved in detoxification and redox homeostasis (grxC1, grxC2, trxB1, nfnB1, nfnB2), amino acid transport and metabolism (leuABCD operon, ilvC1, ilvE, livKHMGF operon), nucleotide metabolism (uvrD1, uvrA1, dinp1, recN, mutT3, nrdGDIEF operon), and protein modification and repair (groEL, groES, DnaJ, DnaK, ClpB, GrpE, ibpA, clpP1, thiJ, pepO, etc.) increased their expression in Bifidobacterium longum subsp. longum BBMN68 as a part of its response to oxygen exposure (3% v/v). Conversely, those implicated in carbohydrate transport and metabolism (mglA3, xylH, MalE, MalF, BBMN68_1170, etc.) and translation, ribosomal structure, and biogenesis (ddpA1, tag, tagH, irp, etc.) were repressed after oxygen exposure. These findings suggest that B. longum BBMN68 mainly employs mechanisms of oxygen reduction and ROS detoxification, repair of damaged biomacromolecules, and adaptive modulation of several metabolic processes (e.g., utilization of other complex carbon sources) to effectively cope with oxygen-driven stresses [78].
Calderini et al. [79] simulated an oxidative environment (0.4, 0.8, or 1.2 mM H2O2) to analyze the protein profile of the oxidative stress response in Lactobacillus acidophilus NCFM (strain with a health benefit supported by a positive human clinical trial). The proteomic approach allowed the authors to identify 19 unique proteins (including their isozymes), which changed in abundance caused by H2O2. They were typically associated with four functional categories: (i) energy metabolism (gapdh, pk); (ii) nucleotide biosynthesis (nrdD, prpps); (iii) general stress (GrpE, DnaK, ClpP); and (iv) oxidative stress (cysK, abm). Such biomolecules help to enhance cell fitness by acting as redox sensors eliciting DNA repair mechanisms, satisfying energy requirements, or acting as coenzymes involved in the regeneration of antioxidant enzymes. The most important finding was the presence of enzymes with cysteine synthase activity (i.e., cysK, gapdh, pk), which are relevant for protein stability, enzyme catalysis, and disulfide-reducing pathway in overcoming oxygen stress.
The transcriptional response in Bifidobacterium longum NCC2705 (strain with a health benefit supported by a positive human clinical trial) and D2957, following a sub-lethal level of H2O2 (0.65 to 10 mM) exposure, showed the presence of ca. 90 to 110 genes that were differentially expressed after treatment. The genes that had more upregulation code for enzymes involved in the functional category of oxidative stress: thioredoxin (trx), thioredoxin reductase (trxR), peroxiredoxin (prdx), ferredoxin (fdx), glutaredoxin (grx), exodeoxyribonuclease VII small sub-unit (xseB), ribonucleotide reductase alpha sub-unit (rnr1), and oxygen-sensitive ribonucleoside-triphosphate reductase (nrdD), among others. Additionally, a complementary analysis revealed that B. longum strains were capable to shift their cell membrane fatty acid composition to positively affect the intrinsic resistance to H2O2 exposure. There was a significant presence of tetradecanoic/myristic acid (C14:0), hexadecenoic/palmitic acid (C16:0), cis-oleic acid (C18:1 n9 cis), trans-oleic acid (C18:1 n9 trans), and plasmalogens (C18:1 plas). These fatty acids might help to prevent the propagation of free radicals and decrease the amount of lipid peroxidation [80].
Other Stressors
As mentioned above, peristaltic movements are correlated with the prevalence of high-pressure forces, which can lead to cell disruption or alterations in microbial metabolic/physiological activities. In this sense, the study conducted by Siroli et al. [81] showed the effects of sub-lethal high-pressure treatments (0.1 to 200 MPa) on both the membrane fatty acid (FA) composition and the transcriptomic profile of Lacticaseibacillus paracasei A13. Data revealed an increased concentration of both saturated fatty acids, such as dodecanoic/lauric acid (C12:0), tridecanoic/tridecylic acid (C13:0), tetradecanoic/myristic acid (C14:0), hexadecenoic/palmitic acid (C16:0), and octadecanoic/stearic acid (C18:0), and unsaturated fatty acids, including hexadecenoic/palmitoleic acid (C16:1), octadecenoic/oleic acid (C18:1 cis), and octadecenoic/elaidic acid (C18:1 trans). Furthermore, significant perturbation in the expression (upregulation) of several genes involved in fatty acid biosynthesis pathway was observed; these include accA, accB, accC, fabD, fabH, fabG, fabZ, fabK, and fabF. It has been proposed that the presence of these particular FAs may lead to a quick rigidification of the cell membrane, whereas the regulated genes may be involved in FA initiation and elongation, as well as in the introduction of double bonds in the carbon chain, a mechanism employed by bacteria to control membrane fluidity and entrance of toxic molecules.
During colonic fermentation, the host-microbiota release different metabolic by-products (e.g., alcohols like ethanol) that tend to accumulate in the intestinal lumen. Some of these by-products may have the capacity to inhibit the cell growth and functionality of probiotics. Guo et al. [82] performed a transcriptome analysis in Lacticaseibacillus paracasei SMN-LBK to determine key tolerance genes expressed as a response to culture in an ethanol-rich media (5 to 10% v/v). It was found that ca. 300 differential genes were upregulated in L. paracasei SMN-LBK; however, only certain genes were remarkably expressed under ethanol stress. The first group includes the genes encoding for phosphofructokinase (pfk) and l-lactate dehydrogenase (ldh), which could be crucial regulators of the glycolytic pathway (improvement of utilization of glucose and satisfaction of energy requirements). The second group includes genes that codify for glycerol-3-phosphate dehydrogenase (gpdh), and glycerol kinase (gk), which may enhance glycerol production and maintain cellular redox homeostasis. This gene regulation can be closely connected to prioritize cell membrane maintenance.
Nutrient limitation or starvation can also induce metabolic stress in microbial cells, thus affecting their growth and survival. When Lactiplantibacillus plantarum B21 was grown in the absence of glucose (0 g L−1, carbohydrate starvation), specific metabolic and morphological changes were evidenced [83]. The metabolomic profile showed that a wide number of metabolites involved in amino acid metabolism were present in high quantities; these include glycine, lysine, norleucine, proline, valine, alanine, and serine, among others. Such composition indicates that L. plantarum B21 uses proteins as the main source of energy. Also, the bacterium was capable of altering its metabolic pathways to conserve energy. This response was supported by the elevated accumulation of β-hydroxypyruvic acid, aspartic acid, thiocyanic acid, and other organic acids, which suggests an inhibition of the fermentation and Krebs cycles. In addition, the absence of glucose resulted in an unusual shorter shape (coccoid-like cells), with bristly cell surface. This morphological alteration was reflected by a change in the membrane fatty acid composition. Specifically, the ratio of unsaturated fatty acids to saturated fatty acids (1:1) was dominated by the presence of 9-octadecenoic acid (C18:1 cis-9), 7-hexadecenoic acid (C16:1 n-9), and tetracosanoic acid (C24:0). These fatty acids can improve the liquidity, flexibility, and elasticity of cell membranes [83].
The dynamics of metabolomic biomarkers in Lacticaseibacillus casei Zhang (strain with a health benefit supported by a positive human clinical trial) subjected to starvation stress over 4000 generations (glucose-restricted medium, 0.02% v/v) was also studied [84]. The accumulating intracellular and extracellular metabolites profile displayed ca. 10,000 substances deferred between generations 0 and 4000. Only 66 metabolites were differentially expressed and considered as key biomarkers in starvation stress response. They were grouped into three main classes: amino acid metabolism (phenylacetaldehyde, indoxyl, urocanic acid, alanine, aspartate, glutamate, glycine, serine, threonine, histidine, cysteine, methionine, L-homoserine, etc.), nucleotide metabolism (thymine, hypoxanthine, purine uridine, guanosine, etc.), and vitamin and cofactors metabolism (9-cis-retinoic acid, pyridoxine, pyridoxamine, pantothenate, biotin, ascorbate, nicotinate, riboflavin, etc.). The abundance of these specific compounds indicates that, under a glucose-restricted environment, L. casei Zhang enters in a transition to metabolically diversify its carbon sources and supply the cellular energy requirements.
Beyond Survival: Is Adaptation Always for the Best?
The adaptation of probiotics to the harsh conditions prevailing in the GIT may enable their survival, thus increasing the chances of providing health benefits to the host. However, there could also be deleterious relationships between exposure to such stressful conditions and the functional properties of probiotic microorganisms [10]. As revised above, this behavior is generated by the modulation of a myriad of molecular features within the microbial cell; however, this molecular reprogramming may cause collateral activities that could negatively impact the host’s well-being. Thus, in this section, we aimed to review the possible biosafety implications of alterations in the stress response mechanisms of probiotics.
On one side, it is well known that the main factors of stress in the GIT are acid juices and bile. As previously described, in order to survive, microorganisms can reprogram their gene expression to adapt to the new environment. Thus, it is not surprising that under an acidic environment, an increase in the production of ammonia and CO2, as well as proteins and compounds related to intracellular repair caused by the stress, is observed. The released ammonia, produced through the arginine deiminase pathway, allows raising the pH of the environment [85, 86]. The same has been observed under stress caused by bile. Whitehead et al. [87] observed the induction of the arginine deiminase pathway genes during the adaptation of Limosilactobacillus reuteri ATCC 55,730 to bile. This pathway is responsible for providing energy and converting arginine into ornithine, ammonia, and CO2 which, so far, does not seem to be harmful, but a closer approach allows us to glimpse how biosafety can be compromised when bacteria try to survive and adapt to harsh environments. For instance, ornithine is the precursor amino acid of the biogenic amine putrescine, which despite having low toxicological activity, has been related to increased cardiac output, dilatation of the vascular system, hypotension, and bradycardia. Besides, they may have indirect toxic effects via potentiating the toxicity of histamine and tyramine (other biogenic amines), and by acting as precursor of carcinogenic N-nitrosamines [88]. In addition, the stress of acidic environments, and other factors that stress the cell such as O2 and NaCl, favor the production of biogenic amines by certain microorganisms (e.g., lactic acid bacteria) through the activity of the enzyme amino acid decarboxylase, since decarboxylation allows the consumption of protons and the production of amines and CO2, restoring the internal pH and consequently increasing survival [89, 90]. Therefore, the presence of biogenic amines represents a potential health problem for the host. For this reason, it is a necessity to evaluate the ability to generate biogenic amines in probiotics.
Furthermore, it has been reported that ammonia production, from arginine catalysis, is a widely distributed mechanism among microorganisms to resist acidity and osmotic stress produced by NaCl. The enzymatic mechanism depends on three enzymes and the activation their respective coding genes: arginine deiminase (arcA), ornithine carbamoyl transferase (arcB), and carbamate kinase (arcC) [91]. However, ammonia is known to activate cofactors of cellular damage and chronic liver damage; additionally, elevated concentrations of ammonia could generate hepatic encephalopathy, which has been associated with Alzheimer’s disease [92]. As an alternative mechanism, some microorganisms can use the arginine deiminase system to produce ammonia, carbon dioxide, and ATP, using agmatine and urea as substrate. During the process, agmatine is hydrolyzed to form putrescine [93]. Therefore, in addition to biogenic amines, the ammonia production capacity must be evaluated to prevent its formation and avoid intoxication [89].
In a previous study, screening of 200 strains of Levilactobacillus brevis showed that at least 36 strains contained AgDI genes, including agmatine/putrescine antiporter [94]. However, the agmatine deiminase pathway is induced by the presence of exogenous agmatine and is regulated by the carbon catabolic repression that acts through the CcpA protein [95].
As previously pointed out, the production of bile salt hydrolase (BSH) is one of the enzymes used as the main strategy of probiotics to survive the exposure to bile during digestion. The bsh gene encodes BSH, and its expression levels increase during growth in the presence of bile salts [96], although the intestinal factor could also trigger its expression [97]. BSH enzymes catalyze the conjugation of conjugated acids glycodeoxycholic and taurodeoxycholic (products of cholesterol metabolism), important for the emulsification of fat in circulation [98]. This conversion capacity has been controversial, as although it has shown positive effects, such as the reduction of serum cholesterol in the host [99], negative effects have also been reported, as the free or secondary bile acids formed could act as mutagens and play an important role in the development of gastrointestinal cancer [100].
Changes in carbohydrate metabolism have also been regarded as a modulation response during protein expression generated by stress factors, such as bile salts. Specifically, Bifidobacterium species have evidenced changes in glycosidic activity, which at first sight could improve the assimilation of indigestible carbohydrates [101]. However, these changes could also increase the expression of enzymes such as N-acetyl-β-D-glucosaminidase and α-D-galactosidase, which are involved in the degradation of mucin [102, 103]. An increase in this activity may cause boosted intestinal permeability and consequent contribute to sepsis by microbial translocation [104].
In another scenario, lactate (D- and L-stereoisomers) is a minor fermentation product in the gut; however, it plays an important role as electron sink in the colonic lumen. Specific microorganisms produce either L-lactate or D-lactate, while others may produce a racemic mix. L-lactic acid can be metabolized and used as energy source, while D-lactic acid requires the enzyme D-lactate dehydrogenase, which humans tend to lack in sufficient levels. Small amounts of D-lactic acid are not a concern; however, this can become a problem when those D-lactic acid-producing microorganisms outnumber the other type, because when large amounts of D-lactate are present, individuals can experience metabolic acidosis. Despite that studies have shown no evidence of the increase of D-lactate in the blood circulation after probiotic consumption, it has been hypothesized that an imbalance of stereoisomers may arise due to the differential modulation of L-lactate at the expense of D-lactate dehydrogenase, since this enzyme is upregulated during metabolic adaptation of some microorganisms (e.g., Lactobacillus kefiranofaciens M1 and L. reuteri strains) to bile salts and acidic environment [105, 106].
The journey that microorganisms take through the GIT after being ingested is full of environments with extreme conditions, which could lead probiotics to regulate an arsenal of molecules that allow it to survive, but which in turn puts its biosecurity in question. Hence, the list of contentious characteristics of probiotics could go on. Nonetheless, there is a lack of enough information about how their biosafety could be compromised by the stressors to which probiotics are subjected to during its passage through the GIT.
Concluding Remarks and Prospects
The GIT is a unique environment in which probiotics have to face several physiological challenges; however, these microorganisms have evolved sophisticated mechanisms to overcome such difficult stresses. The common resistance mechanisms include alteration of cell membranes, regulation of metabolism, repair of macromolecules, and pH homeostasis. However, as described in this review, probiotic strains may also employ a variety of specific elements directed to cope with GIT stress. Recent advances in omic techniques have provided valuable knowledge regarding the physiological and molecular networks involved in these particular processes. Research has revealed detailed insights into key players involved in gene expression and regulation, activation of specialized metabolic pathways, and promotion of unique biosynthetic capabilities, which may control the new evolutionary mechanisms in probiotic defense.
Under this scenario, the knowledge of probiotic adaptation or defense mechanisms continues to expand; there still will be some gaps that future research needs to focus on. Figure 4 proposes the integration of in silico, in vitro, and in vivo approaches that can aid in addressing the gaps in this area. We previously reported in-depth information about such integration and its contribution to gaining clearer insights into how probiotics adapt to the surrounding environment [107]. Consequently, investigations need to differentiate between the impact of individual stressors and their combinations on adaptive patterns (e.g., cell growth, survival), particularly because exposure to multiple stress conditions could induce a cross-protection response against unrelated agents. Similarly, further studies on pre-exposure to specific sub-lethal stress conditions and their influence on the induction of adaptation responses are needed. These studies can serve as a model for the design of industrial pre-adaptation systems leading to more robust probiotics with better performance (e.g., greater viability/stability, better functionality). Finally, further studies are also needed to understand if the stress response mechanisms might compromise the positive health effects and safety concerns of probiotic microorganisms. Thus, it is imperative to utilize in vivo trials, either in animal or human studies, in order to analyze the actual response dynamics of probiotics under the GIT challenge with respect to their desirable physiological characteristics.

Basic and clinical research to further study the mechanisms of probiotic stress response. Figure adapted from images created with BioRender.com
References
Sensoy I (2021) A review on the food digestion in the digestive tract and the used in vitro models. Curr Res Food Sci 4:308–319. https://doi.org/10.1016/j.crfs.2021.04.004
Gaucher F, Bonnassie S, Rabah H, Marchand P, Blanc P, Jeantet R, Jan G (2019) Review: adaptation of beneficial Propionibacteria, Lactobacilli, and Bifidobacteria improves tolerance toward technological and digestive stresses. Front Microbiol 10:841. https://doi.org/10.3389/fmicb.2019.00841
Hillman ET, Lu H, Yao T, Nakatsu CH (2017) Microbial ecology along the gastrointestinal tract. Microbes Environ 32(4):300–313. https://doi.org/10.1264/jsme2.me17017
Panwar H, Rokana N, Sudhakaran VA, Kaur J, Singh A, Singh J, Singh KS, Chaudhary V, Puniya AK (2021) Gastrointestinal stress as innate defense against microbial attack. J Appl Microbiol 130(4):1035–1061. https://doi.org/10.1111/jam.14836
Gandhi A, Shah NP (2017) Integrating omics to unravel the stress-response mechanisms in probiotic bacteria: approaches, challenges, and prospects. Crit Rev Food Sci Nutr 57(16):3464–3471. https://doi.org/10.1080/10408398.2015.1136805
George Kerry R, Patra JK, Gouda S, Park Y, Shin HS, Das G (2018) Benefaction of probiotics for human health: a review. J Food Drug Anal 26(3):927–939. https://doi.org/10.1016/j.jfda.2018.01.002
Groussin M, Mazel F, Alm EJ (2020) Co-evolution and co-speciation of host-gut bacteria systems. Cell Host Microbe 28(1):12–22. https://doi.org/10.1016/j.chom.2020.06.013
Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, Morelli L, Canani RB, Flint HJ, Salminen S, Calder PC, Sanders ME (2014) The international scientific association for probiotics and prebiotics consensus on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol 11:506–514. https://doi.org/10.1038/nrgastro.2014.66
Yan F, Polk DB (2020) Probiotics and probiotic-derived functional factors—mechanistic insights into applications for intestinal homeostasis. Front Immunol 11:1428. https://doi.org/10.3389/fimmu.2020.01428
Amund OD (2016) Exploring the relationship between exposure to technological and gastrointestinal stress and probiotic functional properties of lactobacilli and bifidobacteria. Can J Microbiol 62(9):715–725. https://doi.org/10.1139/cjm-2016-0186
Guan N, Li J, Shin H, Du G, Chen J, Liu L (2017) Microbial response to environmental stresses: from fundamental mechanisms to practical applications. Appl Microbiol Biotechnol 101(10):3991–4008. https://doi.org/10.1007/s00253-017-8264-y
Han S, Lu Y, Xie J, Fei Y, Zheng G, Wang Z, Liu J, Lv L, Ling Z, Berglund B, Yao M, Li L (2021) Probiotic gastrointestinal transit and colonization after oral administration: a long journey. Front Cell Infect Microbiol 11:609722. https://doi.org/10.3389/fcimb.2021.609722
Hosseini Nezhad M, Hussain MA, Britz ML (2015) Stress responses in probiotic Lactobacillus casei. Crit Rev Food Sci Nutr 55(6):740–749. https://doi.org/10.1080/10408398.2012.675601
Gottesman S (2019) Trouble is coming: signaling pathways that regulate general stress responses in bacteria. J Biol Chem 294(31):11685–11700. https://doi.org/10.1074/jbc.REV119.005593
Mbye M, Baig MA, AbuQamar SF, El-Tarabily KA, Obaid RS, Osaili TM, Al-Nabulsi AA, Turner MS, Shah NP, Ayyash MM (2020) Updates on understanding of probiotic lactic acid bacteria responses to environmental stresses and highlights on proteomic analyses. Compr Rev Food Sci Food Saf 19(3):1110–1124. https://doi.org/10.1111/1541-4337.12554
Fang FC, Frawley ER, Tapscott T, Vázquez-Torres A (2016) Bacterial stress responses during host infection. Cell Host Microbe 20(2):133–143. https://doi.org/10.1016/j.chom.2016.07.009
Yoha KS, Nida S, Dutta S, Moses JA, Anandharamakrishnan C (2022) Targeted delivery of probiotics: perspectives on research and commercialization. Probiotics Antimicrob Proteins 14:15–48. https://doi.org/10.1007/s12602-021-09791-7
Baral KC, Bajracharya R, Lee SH, Han HK (2021) Advancements in the pharmaceutical applications of probiotics: dosage forms and formulation technology. Int J Nanomed 16:7535–7556. https://doi.org/10.2147/IJN.S337427
Cassani L, Gomez-Zavaglia A, Simal-Gandara J (2019) Technological strategies ensuring the safe arrival of beneficial microorganisms to the gut: from food processing and storage to their passage through the gastrointestinal tract. Food Res Int 129:108852. https://doi.org/10.1016/j.foodres.2019.108852
Lynge Pedersena AM, Belstrøm D (2019) The role of natural salivary defences in maintaining a healthy oral microbiota. J Dent 80:S3–S12. https://doi.org/10.1016/j.jdent.2018.08.010
Mosca AC, Chen J (2016) Food oral management: physiology and objective assessment. Curr Opin Food Sci 9:11–20. https://doi.org/10.1016/j.cofs.2016.03.003
Vila T, Rizk AM, Sultan AS, Jabra-Rizk MA (2019) The power of saliva: antimicrobial and beyond. PLoS Pathog 15(11):e1008058. https://doi.org/10.1371/journal.ppat.1008058
Martínez B, Rodríguez A, Kulakauskas S, Chapot-Chartier M-P (2020) Cell wall homeostasis in lactic acid bacteria: threats and defences. FEMS Microbiol Rev 44(5):538–564. https://doi.org/10.1093/femsre/fuaa021
Hoffmann A, Daniels R (2017) Lactobacilli for the treatment of oral diseases. J Probiotics Health 5(3):181. https://doi.org/10.4172/2329-8901.1000181
Rolim FRL, dos Santos KMO, de Barcelos SC, do Egito AS, Ribeiro TS, da Conceição ML, Magnani M, Gomes de Oliveira ME, de Queiroga RCRE (2015) Survival of Lactobacillus rhamnosus EM1107 in simulated gastrointestinal conditions and its inhibitory effect against pathogenic bacteria in semi-hard goat cheese. LWT - Food Sci Technol 63(2):807–813. https://doi.org/10.1016/j.lwt.2015.05.004
Vemuri R, Shinde T, Shastri MD, Perera AP, Tristram S, Martoni CJ, Gundamaraju R, Ahuja KDK, Ball M, Eri R (2018) A human origin strain Lactobacillus acidophilus DDS-1 exhibits superior in vitro probiotic efficacy in comparison to plant or dairy origin probiotics. Int J Med Sci 15(9):840–848. https://doi.org/10.7150/ijms.25004
Haukioja A, Yli-Knuuttila H, Loimaranta V, Kari K, Ouwehand AC, Meurman JH, Tenovuo J (2006) Oral adhesion and survival of probiotic and other lactobacilli and bifidobacteria in vitro. Oral Microbiol Immunol 21(5):326–332. https://doi.org/10.1111/j.1399-302x.2006.00299.x
Su A, Parker CH, Conklin JL (2020) Esophageal anatomy and physiology. In: Rao SSC, Yeh Lee Y, Ghoshal UC (eds) Clinical and Basic Neurogastroenterology and Motility. Academic Press & Elsevier B.V, Amsterdam, Netherlands, pp 79–88
Zhang X, Patil D, Odze RD, Zhao L, Lisovsky M, Guindi M, Riddell R, Bellizzi A, Yantiss RK, Nalbantoglu I, Appelman HD (2018) The microscopic anatomy of the esophagus including the individual layers, specialized tissues, and unique components and their responses to injury. Ann NY Acad Sci 1434(1):304–318. https://doi.org/10.1111/nyas.13705
Nikaki K, Sawada A, Ustaoglu A, Sifrim D (2019) Neuronal control of esophageal peristalsis and its role in esophageal disease. Curr Gastroenterol Rep 21(11):59. https://doi.org/10.1007/s11894-019-0728-z
Mir M, Ali MN, Ansari U, Sami J (2016) Structure and motility of the esophagus from a mechanical perspective. Esophagus 13:8–16. https://doi.org/10.1007/s10388-015-0497-1
Nie L, Li W, Xue L, Wang L, Shen Y, Fan X (2020) Submucosal gland neoplasms of the esophagus: an update and review. Esophagus 17:376–384. https://doi.org/10.1007/s10388-020-00758-1
Genova LA, Roberts MF, Wong Y-C, Harper CE, Santiago AG, Fu B, Srivastava A, Jung W, Wang LM, Krzeminsk L, Mao X, Sun X, Hu C-Y, Chen P, Hernandez CJ (2019) Mechanical stress compromises multicomponent efflux complexes in bacteria. Proc Natl Acad Sci USA 116(51):25462–25467. https://doi.org/10.1073/pnas.1909562116
Papadimitriou K, Alegría Á, Bron PA, de Angelis M, Gobbetti M, Kleerebezem M, Lemos JA, Linares DM, Ross P, Stanton C, Turroni F, van Sinderen D, Varmanen P, Ventura M, Zúñiga M, Tsakalidou E, Kok J (2016) Stress physiology of lactic acid bacteria. Microbiol Mol Biol Rev 80(3):837–890. https://doi.org/10.1128/mmbr.00076-15
Brandstaeter S, Fuchs SL, Aydin RC, Cyron CJ (2019) Mechanics of the stomach: a review of an emerging field of biomechanics. GAMM-Mitteilungen 42(3):e201900001. https://doi.org/10.1002/gamm.201900001
Martinsen TM, Fossmark R, Waldum HL (2019) The phylogeny and biological function of gastric juice – microbiological consequences of removing gastric acid. Int J Mol Sci 20(23):6031. https://doi.org/10.3390/ijms20236031
Shani-Levi C, Alvito P, Andrés A, Assunção R, Barberá R, Blanquet-Diot S, Bourlieu C, Brodkorb A, Cilla A, Deglaire A, Denis S, Dupont D, Heredia A, Karakaya S, Giosafatto CVL, Mariniello L, Martins C, Ménard O, El SN, Vegarud GE, Ulleberg E, Lesmes U (2017) Extending in vitro digestion models to specific human populations: perspectives, practical tools and bio-relevant information. Trends Food Sci Technol 60:52–63. https://doi.org/10.1016/j.tifs.2016.10.017
Guan N, Liu L (2020) Microbial response to acid stress: mechanisms and applications. Appl Microbiol Biotechnol 104:51–65. https://doi.org/10.1007/s00253-019-10226-1
Sanhueza E, Paredes-Osses E, González CL, García A (2015) Effect of pH in the survival of Lactobacillus salivarius strain UCO_979C wild type and the pH acid acclimated variant. Electron J Biotechnol 18(5):343–346. https://doi.org/10.1016/j.ejbt.2015.06.005
Mackie A, Mulet-Cabero A-I, Torcello-Gómez A (2020) Simulating human digestion: developing our knowledge to create healthier and more sustainable foods. Food Funct 11:9397–9431. https://doi.org/10.1039/d0fo01981j
Lund P, Tramonti A, De Biase D (2014) Coping with low pH: molecular strategies in neutralophilic bacteria. FEMS Microbiol Rev 38(6):1091–1125. https://doi.org/10.1111/1574-6976.12076
Kim HJ, Li H, Collins JJ, Ingber DE (2015) Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc Natl Acad Sci USA 113(1):E7–E15. https://doi.org/10.1073/pnas.1522193112
Lee MG, Kim Y, Jun I, Aoun J, Muallem S (2020) Molecular mechanisms of pancreatic bicarbonate secretion. Pancreapedia: Exocrine Pancreas Knowledge Base. Version 2.0, 1–24. https://pancreapedia.org/sites/default/files/Molecular%20Mechanisms%20of%20Pancreatic%20Bicarbonate%20Secretion_Version_2-0-.pdf. Accessed 25 May 2021
Boyer JL, Soroka CJ (2021) Bile formation and secretion: an update. J Hepatol 75(1):190–201. https://doi.org/10.1016/j.jhep.2021.02.011
Brüggenwirth IMA, Porte RJ, Martins PN (2020) Bile composition as a diagnostic and prognostic tool in liver transplantation. Liver Transpl 26(9):1177–1187. https://doi.org/10.1002/lt.25771
Tian Y, Gui W, Koo I, Smith PB, Allman EL, Nichols RG, Rimal B, Cai J, Liu Q, Patterson AD (2020) The microbiome modulating activity of bile acids. Gut Microbes 11(4):979–996. https://doi.org/10.1080/19490976.2020.1732268
Urdaneta V, Casadesús J (2017) Interactions between bacteria and bile salts in the gastrointestinal and hepatobiliary tracts. Front Med 4:163. https://doi.org/10.3389/fmed.2017.00163
Chairatana P, Nolan EM (2017) Defensins, lectins, mucins, and secretory immunoglobulin A: microbe-binding biomolecules that contribute to mucosal immunity in the human gut. Crit Rev Biochem Mol Biol 52(1):45–56. https://doi.org/10.1080/10409238.2016.1243654
Neffe-Skocińska K, Rzepkowska A, Szydłowska A, Kołożyn-Krajewska D (2018) Trends and possibilities of the use of probiotics in food production. In: Holban AM, Mihai A (eds) Alternative and Replacement Foods-Handbook of Food Bioengineering. Academic Press & Elsevier B.V, Amsterdam, Netherlands, pp 65–94
Sauer J-M, Merchant H (2018) Physiology of the gastrointestinal system. In: McQueen C (ed) Comprehensive Toxicology. Elsevier B.V, Amsterdam, Netherlands, pp 16–44
Oliphant K, Allen-Vercoe E (2019) Macronutrient metabolism by the human gut microbiome: major fermentation by-products and their impact on host health. Microbiome 7(1):91. https://doi.org/10.1186/s40168-019-0704-8
Vernocchi P, Del Chierico F, Putignani L (2020) Gut microbiota metabolism and interaction with food components. Int J Mol Sci 21(10):3688. https://doi.org/10.3390/ijms21103688
Mörkl S, Butler MI, Holl A, Cryan JF, Dinan TG (2020) Probiotics and the microbiota-gut-brain axis: focus on psychiatry. Curr Nutr Rep 9(3):171–182. https://doi.org/10.1007/s13668-020-00313-5
Wen C, Wei S, Zong X, Wang Y, Jin M (2021) Microbiota-gut-brain axis and nutritional strategy under heat stress. Animal Nutrition 7(4):1329–1336. https://doi.org/10.1016/j.aninu.2021.09.008.e
Carabotti M, Scirocco A, Maselli MA, Severi C (2016) The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol 29(2):240. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4367209/#ref70. Accessed 25 Jul 2022
Appleton J (2018) The gut-brain axis: influence of microbiota on mood and mental health. Integr Med Encinitas 17(4):28–32. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6469458/. Accessed 25 Jul 2022
Flint A, Butcher J, Stintzi A (2016) Stress responses, adaptation, and virulence of bacterial pathogens during host gastrointestinal colonization. Microbiol. Spectr 4(2):VMBF-0007–2015. https://doi.org/10.1128/microbiolspec.VMBF-0007-2015
Wendel U (2022) Assessing viability and stress tolerance of probiotics—a review. Front Microbiol 12:818468. https://doi.org/10.3389/fmicb.2021.818468
Rocca JD, Simonin M, Blaszczak JR, Ernakovich JG, Gibbons SM, Midani FS, Washburne AD (2018) The microbiome stress project: towards a global meta-analysis of environmental stressors and their effects on microbial communities. Front Microbiol 9:3272. https://doi.org/10.3389/fmicb.2018.03272
Guillén S, Nadal L, Álvarez I, Mañas P, Cebrián G (2021) Impact of the resistance responses to stress conditions encountered in food and food processing environments on the virulence and growth fitness of non-typhoidal Salmonellae. Foods 10:617. https://doi.org/10.3390/foods10030617
Schroeder M, Brooks B, Brooks A (2017) The complex relationship between virulence and antibiotic resistance. Genes 8(1):39. https://doi.org/10.3390/genes8010039
Mills S, Stanton C, Fitzgerald GF, Ross RP (2011) Enhancing the stress responses of probiotics for a lifestyle from gut to product and back again. Microb Cell Factories 10(Suppl 1):S19. https://doi.org/10.1186/1475-2859-10-s1-s19
van Zyl WF, Deane SM, Dicks LMT (2020) Molecular insights into probiotic mechanisms of action employed against intestinal pathogenic bacteria. Gut Microbes 12(1):1831339. https://doi.org/10.1080/19490976.2020.1831339
Jabbar Z, Mukhtar H, Tayyeb A, Manzoor A (2020) Next-generation sequencing to elucidate adaptive stress response and plantaricin genes among Lactobacillus plantarum strains. Future Microbiol 15(5):333–348. https://doi.org/10.2217/fmb-2019-0158
Jung S, Lee J-H (2020) Characterization of transcriptional response of Lactobacillus plantarum under acidic conditions provides insight into bacterial adaptation in fermentative environments. Sci Rep 10(1):19203. https://doi.org/10.1038/s41598-020-76171-6
Wei Y, Gao J, Liu D, Li Y, Liu W (2019) Adaptational changes in physiological and transcriptional responses of Bifidobacterium longum involved in acid stress resistance after successive batch cultures. Microb Cell Factories 18(1):156. https://doi.org/10.1186/s12934-019-1206-x
Wang W, He J, Pan D, Wu Z, Guo Y, Zeng X, Lian L (2018) Metabolomics analysis of Lactobacillus plantarum ATCC 14917 adhesion activity under initial acid and alkali stress. PLoS ONE 13(5):e0196231. https://doi.org/10.1371/journal.pone.0196231
Jin J, Qin Q, Guo H, Liu S, Ge S, Zhang H, Cui J, Ren F (2015) Effect of pre-stressing on the acid-stress response in Bifidobacterium revealed using proteomic and physiological approaches. PLoS ONE 10(2):e0117702. https://doi.org/10.1371/journal.pone.0117702
Bagon BB, Valeriano VDV, Oh JK, Pajarillo EAB, Lee JY, Kang D-K (2021) Exoproteome perspective on the bile stress response of Lactobacillus johnsonii. Proteomes 9:10. https://doi.org/10.3390/proteomes9010010
Wang G, Zhai Z, Ren F, Li Z, Zhang B, Hao Y (2020) Combined transcriptomic and proteomic analysis of the response to bile stress in a centenarian-originated probiotic Lactobacillus salivarius Ren. Food Res Int 137:109331. https://doi.org/10.1016/j.foodres.2020.109331
Ma X, Wang G, Zhai Z, Zhou P, Hao Y (2018) Global transcriptomic analysis and function identification of malolactic enzyme pathway of Lactobacillus paracasei L9 in response to bile stress. Front Microbiol 9:1978. https://doi.org/10.3389/fmicb.2018.01978
Bustos AY, de Valdez GF, Raya R, de Almeida AM, Fadda S, Taranto MP (2015) Proteomic analysis of the probiotic Lactobacillus reuteri CRL1098 reveals novel tolerance biomarkers to bile acid-induced stress. Food Res Int 77:599–607. https://doi.org/10.1016/j.foodres.2015.10.001
Yao W, Yang L, Shao Z, Xie L, Chen L (2020) Identification of salt tolerance-related genes of Lactobacillus plantarum D31 and T9 strains by genomic analysis. Ann Microbiol 70(1):10. https://doi.org/10.1186/s13213-020-01551-2
Luo X, Li M, Zhang H, Li R, Zou Y, Wu R, Chen Y (2020) Comparative proteomic analysis of three Lactobacillus plantarum under salt stress by iTRAQ. J Sci Food Agric 101(8):3457–3471. https://doi.org/10.1002/jsfa.10976
Qi NL, Gong X, Yang CL, Cheng ZH, Zhou W, Li JH (2018) 1H NMR-based metabolic profile of Lactobacillus salivarius FDB89 under osmotic stress. Appl Ecol Environ Res 16:3489–3500. https://doi.org/10.15666/aeer/1603_34893500
Palomino MM, Waehner PM, Fina Martin J, Ojeda P, Malone L, Sánchez Rivas C, Prado Acosta M, Allievi MC, Ruzal SM (2016) Influence of osmotic stress on the profile and gene expression of surface layer proteins in Lactobacillus acidophilus ATCC 4356. Appl Microbiol Biotechnol 100(19):8475–8484. https://doi.org/10.1007/s00253-016-7698-y
Zhang H, Liu J, Wen R, Chen Q, Kong B (2020) Metabolomics profiling reveals defense strategies of Pediococcus pentosaceus R1 isolated from Harbin dry sausages under oxidative stress. LWT - Food Sci Technol 135:110041. https://doi.org/10.1016/j.lwt.2020.110041
Zuo F, Yu R, Xiao M, Khaskheli GB, Sun X, Ma H, Ren F, Zhang B, Chen S (2018) Transcriptomic analysis of Bifidobacterium longum subsp longum BBMN68 in response to oxidative shock. Sci Rep 8:17085. https://doi.org/10.1038/s41598-018-35286-7
Calderini E, Celebioglu HU, Villarroel J, Jacobsen S, Svensson B, Pessione E (2017) Comparative proteomics of oxidative stress response of Lactobacillus acidophilus NCFM reveals effects on DNA repair and cysteine de novo synthesis. Proteomics 17(5):1600178. https://doi.org/10.1002/pmic.201600178
Oberg TS, Ward RE, Steele JL, Broadbent JR (2015) Transcriptome analysis of Bifidobacterium longum strains that show a differential response to hydrogen peroxide stress. J Biotechnol 212:58–64. https://doi.org/10.1016/j.jbiotec.2015.06.405
Siroli L, Braschi G, Rossi S, Gottardi D, Patrignani F, Lanciotti R (2020) Lactobacillus paracasei A13 and high-pressure homogenization stress response. Microorganisms 8(3):439. https://doi.org/10.3390/microorganisms8030439
Guo J, Li X, Li B, Yang J, Jin D, Li K (2020) Transcriptome analysis of Lactobacillus paracasei SMN-LBK under ethanol stress. J Dairy Sci 103(9):7813–7825. https://doi.org/10.3168/jds.2019-16955
Parlindungan E, May B, Jones OA (2019) Metabolic insights into the effects of nutrient stress on Lactobacillus plantarum B21. Front Mol Biosci 6:75. https://doi.org/10.3389/fmolb.2019.00075
Pan L, Yu J, Ren D, Yao C, Chen Y, Menghe B (2019) Metabolomic analysis of significant changes in Lactobacillus casei Zhang during culturing to generation 4,000 under conditions of glucose restriction. J Dairy Sci 102:1–17. https://doi.org/10.3168/jds.2018-15702
Kajfasz JK, Quivey RG (2011) Responses of lactic acid bacteria to acid stress. In: Tsakalidou E, Papadimitriou K (eds) Stress responses of lactic acid bacteria. Springer, Boston, Massachusetts, EE.UU., pp 23–53
Zhang J, Wu C, Du G, Chen J (2012) Enhanced acid tolerance in Lactobacillus casei by adaptive evolution and compared stress response during acid stress. Biotechnol Bioprocess Eng 17(2):283–289. https://doi.org/10.1007/s12257-011-0346-6
Whitehead K, Versalovic J, Roos S, Britton RA (2008) Genomic and genetic characterization of the bile stress response of probiotic Lactobacillus reuteri ATCC 55730. Appl Environ Microbiol 74(6):1812–1819. https://doi.org/10.1128/AEM.02259-07
De Mey E, De Maere H, Paelinck H, Fraeye I (2017) Volatile N-nitrosamines in meat products: potential precursors, influence of processing, and mitigation strategies. Crit Rev Food Sci Nutr 57(13):2909–2923. https://doi.org/10.1080/10408398.2015.1078769
Mann S, Park MS, Johnston TV, Ji GE, Hwang KT, Ku S (2021) Isolation, characterization and biosafety evaluation of Lactobacillus fermentum OK with potential oral probiotic properties. Probiotics Antimicrob Proteins. https://doi.org/10.1007/s12602-021-09761-z
Spano G, Russo P, Lonvaud-Funel A, Lucas P, Alexandre H, Grandvalet C, Lolkema JS (2010) Biogenic amines in fermented foods. Eur J Clin Nutr 64(3):S95–S100. https://doi.org/10.1038/ejcn.2010.218
Vrancken G, Rimaux T, Wouters D, Leroy F, De Vuyst L (2009) The arginine deiminase pathway of Lactobacillus fermentum IMDO 130101 responds to growth under stress conditions of both temperature and salt. Food Microbiol 26(7):720–727. https://doi.org/10.1016/j.fm.2009.07.006
Jin YY, Singh P, Chung HJ, Hong ST (2018) Blood ammonia as a possible etiological agent for Alzheimer’s disease. Nutrients 10(5):564. https://doi.org/10.3390/nu10050564
Arcari T, Feger ML, Guerreiro DN, Wu J, O’Byrne CP (2020) Comparative review of the responses of Listeria monocytogenes and Escherichia coli to low pH stress. Genes 11(11):1330. https://doi.org/10.3390/genes11111330
Lucas PM, Blancato VS, Claisse O, Magni C, Lolkema JS, Lonvaud-Funel A (2007) (Agmatine deiminase pathway genes in Lactobacillus brevis are linked to the tyrosine decarboxylation operon in a putative acid resistance locus. Microbiology 153(7):2221–2230. https://doi.org/10.1099/mic.0.2007/006320-0
del Rio B, Linares DM, Ladero V, Redruello B, Fernández M, Martin MC, Alvarez MA (2015) Putrescine production via the agmatine deiminase pathway increases the growth of Lactococcus lactis and causes the alkalinization of the culture medium. Appl Microbiol Biotechnol 99(2):897–905. https://doi.org/10.1007/s00253-014-6130-8
Widodo W, Yana N, Surya SA, Dwi WT (2020) Detection and expression analysis of the bile salt hydrolase gene in Pediococcus and Lactobacillus. Biodiversitas 21(12):5901. https://doi.org/10.13057/biodiv/d211255
Yuan J, Wang B, Sun Z, Bo X, Yuan X, Zhao H, Du X, Wang F, Jiang Z, Zhang L, Jia L, Wang Y, Wei K, Wang J, Zhang X, Sun Y, Huan L, Zeng M (2008) Analysis of host-inducing proteome changes in Bifidobacterium longum NCC2705 grown in vivo. J Proteome Res 7:375–385. https://doi.org/10.1021/pr0704940
Vankerckhoven V, Huys G, Vancanneyt M, Vael C, Klare I, Romond MB, Goossens H (2008) Biosafety assessment of probiotics used for human consumption: recommendations from the EU-PROSAFE project. Trends Food Sci Technol 19(2):102–114. https://doi.org/10.1016/j.tifs.2007.07.013
Hernández-Gómez JG, López-Bonilla A, Trejo-Tapia G, Ávila-Reyes SV, Jiménez-Aparicio AR, Hernández-Sánchez H (2021) In vitro bile salt hydrolase (BSH) activity screening of different probiotic microorganisms. Foods 10(3):674. https://doi.org/10.3390/foods10030674
Kundu S, Kumar S, Bajaj A (2015) Cross-talk between bile acids and gastrointestinal tract for progression and development of cancer and its therapeutic implications. IUBMB Life 67(7):514–523. https://doi.org/10.1002/iub.1399
Sánchez B, Ruiz L, Gueimonde M, Ruas-Madiedo P, Margolles A (2013) Adaptation of bifidobacteria to the gastrointestinal tract and functional consequences. Pharmacol Res 69(1):127–136. https://doi.org/10.1016/j.phrs.2012.11.004
Derrien M, van Passel MW, van de Bovenkamp JH, Schipper RG, de Vos WM, Dekker J (2010) Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes 1(4):254–268. https://doi.org/10.4161/gmic.1.4.12778
Noriega L, Gueimonde M, Sánchez B, Margolles A, de los Reyes-Gavilán CG (2004) Effect of the adaptation to high bile salts concentrations on glycosidic activity, survival at low pH and cross-resistance to bile salts in Bifidobacterium. Int J Food Microbiol 94(1):79–86. https://doi.org/10.1016/j.ijfoodmicro.2004.01.003
Chen W, Yu L, Shi Y (2019) Safety evaluation of lactic acid bacteria. In: Chen W (ed) Lactic Acid Bacteria. Springer Nature Singapore Pte Ltd. and Science Press, Singapore, pp 371–409
Chen MJ, Tang HY, Chiang ML (2017) Effects of heat, cold, acid and bile salt adaptations on the stress tolerance and protein expression of kefir-isolated probiotic Lactobacillus kefiranofaciens M1. Food Microbiol 66:20–27. https://doi.org/10.1016/j.fm.2017.03.020
Liu D-M, Huang Y-Y, Liang M-H (2022) Analysis of the probiotic characteristics and adaptability of Lactiplantibacillus plantarum DMDL 9010 to gastrointestinal environment by complete genome sequencing and corresponding phenotypes. LWT - Food Sci Technol 158:113129. https://doi.org/10.1016/j.lwt.2022.113129
Castro-López C, García HS, Martínez-Ávila GCG, González-Córdova AF, Vallejo-Cordoba B, Hernandez-Mendoza A (2021) Genomics-based approaches to identify and predict the health-promoting and safety activities of promising probiotic strains – a probiogenomics review. Trends Food Sci Technol 108:148–163. https://doi.org/10.1016/j.tifs.2020.12.017
Chen L, Gu Q, Li P, Chen S, Li Y (2019) Genomic analysis of Lactobacillus reuteri WHH1689 reveals its probiotic properties and stress resistance. Food Sci Nutr 7(2):844–857. https://doi.org/10.1002/fsn3.934
Bang M, Yong C-C, Ko H-J, Cho I-G, Oh S (2018) Transcriptional response and enhanced intestinal adhesion ability of Lactobacillus rhamnosus GG after acid stress. J Microbiol Biotechnol 28(10):1604–1613. https://doi.org/10.4014/jmb.1807.07033
Pérez-Montoro B, Benomar N, Caballero Gómez N, Ennahar S, Horvatovich P, Knapp CW, Gálvez A, Abriouel H (2018) Proteomic analysis of Lactobacillus pentosus for the identification of potential markers involved in acid resistance and their influence on other probiotic features. Food Microbiol 72:31–38. https://doi.org/10.1016/j.fm.2017.11.006
Heunis T, Deane S, Smit S, Dicks LMT (2014) Proteomic profiling of the acid stress response in Lactobacillus plantarum 423. J Proteome Res 13(9):4028–4039. https://doi.org/10.1021/pr500353x
Ma J, Xu C, Liu F, Hou J, Shao H, Yu W (2021) Stress adaptation and cross-protection of Lactobacillus plantarum KLDS 1.0628. CYTA J Food 19(1):72–80. https://doi.org/10.1080/19476337.2020.1859619
Kelly SM, Lanigan N, O’Neill IJ, Bottacini F, Lugli GA, Viappiani A, Turron F, Ventura M, van Sinderen D (2020) Bifidobacterial biofilm formation is a multifactorial adaptive phenomenon in response to bile exposure. Sci Rep 10(1):11598. https://doi.org/10.1038/s41598-020-68179-9
Lv L-X, Yan R, Shi H-Y, Shi D, Fang D-Q, Jiang H-Y, Wu W-R, Guo F-F, Jiang X-W, Gu S-L, Chen Y-B, Yao J, Li L-J (2017) Integrated transcriptomic and proteomic analysis of the bile stress response in probiotic Lactobacillus salivarius LI01. J Proteom 150:216–229. https://doi.org/10.1016/j.jprot.2016.08.021
An H, Douillard FP, Wang G, Zhai Z, Yang J, Song S, Cui J, Ren F, Luo Y, Zhang B, Hao Y (2014) Integrated transcriptomic and proteomic analysis of the bile stress response in a centenarian-originated probiotic Bifidobacterium longum BBMN68. Mol Cell Proteom 13(10):2558–2572. https://doi.org/10.1074/mcp.m114.039156
Costantini PE, Firrincieli A, Fedi S, Parolin C, Viti C, Cappelletti M, Vitali B (2021) Insight into phenotypic and genotypic differences between vaginal Lactobacillus crispatus BC5 and Lactobacillus gasseri BC12 to unravel nutritional and stress factors influencing their metabolic activity. Microb Genom 7(6):000575. https://doi.org/10.1099/mgen.0.000575
van der Meulen SB, de Jong A, Kok J (2017) Early transcriptome response of Lactococcus lactis to environmental stresses reveals differentially expressed small regulatory RNAs and tRNAs. Front Microbiol 8:1704. https://doi.org/10.3389/fmicb.2017.01704
Gandhi A, Shah NP (2016) Effect of salt stress on morphology and membrane composition of Lactobacillus acidophilus, Lactobacillus casei, and Bifidobacterium bifidum, and their adhesion to human intestinal epithelial-like Caco-2 cells. J Dairy Sci 99(4):2594–2605. https://doi.org/10.3168/jds.2015-10718
Li C, Li P-Z, Sun J-W, Huo G-C, Liu L-B (2014) Proteomic analysis of the response to NaCl stress of Lactobacillus bulgaricus. Biotechnol Lett 36(11):2263–2269. https://doi.org/10.1007/s10529-014-1601-7
Gao Y, Liu Y, Ma F, Sun M, Mu G, Tuo Y (2020) Global transcriptomic and proteomics analysis of Lactobacillus plantarum Y44 response to 2,2-azobis(2-methylpropionamidine) dihydrochloride (AAPH) stress. J Proteom 226:103903. https://doi.org/10.1016/j.jprot.2020.103903
Wu P, An J, Chen L, Zhu Q, Li Y, Mei Y, Chen Z, Liang Y (2020) Differential analysis of stress tolerance and transcriptome of probiotic Lacticaseibacillus casei Zhang produced from solid-state (SSF-SW) and liquid-state (LSF-MRS) fermentations. Microorganisms 8(11):1656. https://doi.org/10.3390/microorganisms8111656
Schott A-S, Behr J, Quinn J, Vogel RF (2016) MALDI-TOF mass spectrometry enables a comprehensive and fast analysis of dynamics and qualities of stress responses of Lactobacillus paracasei subsp. paracasei F19. PLoS ONE 11(10):e0165504. https://doi.org/10.1371/journal.pone.0165504
Ricciardi A, Castiglione Morelli MA, Ianniello RG, Parente E, Zotta T (2015) Metabolic profiling and stress response of anaerobic and respiratory cultures of Lactobacillus plantarum C17 grown in a chemically defined medium. Ann Microbiol 65(3):1639–1648. https://doi.org/10.1007/s13213-014-1003-z
Butorac A, Dodig I, Bačun-Družina V, Tishbee A, Mrvčić J, Hock K, Diminić J, Cindrić M (2013) The effect of starvation stress on Lactobacillus brevis L62 protein profile determined by de novo sequencing in positive and negative mass spectrometry ion mode. Rapid Commun Mass Spectrom 27(9):1045–1054. https://doi.org/10.1002/rcm.6528
Wu L, Wang W, Wu Z, Pan D, Zeng X, Guo Y, Lian L (2020) Effect of acid and alkali stress on extracellular metabolite profile of Lactobacillus plantarum ATCC 14917. J Basic Microbiol 60(8):722–729. https://doi.org/10.1002/jobm.202000203
Acknowledgements
Cecilia Castro-López wants to thank the National Council for Science and Technology (CONACyT) of Mexico for the graduate fellowship.
Author information
Authors and Affiliations
Contributions
CCL: data collection, content design, figure preparation, and writing‒original draft; HERL: data collection and writing‒original draft; HSG: writing‒review and editing; BVC: visualization and writing‒review; AFGC: visualization and writing‒review; AHM: conceptualization, supervision and writing‒review, editing, and responsible for the final content of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
• Gastrointestinal tract (GIT) conditions affect probiotic viability, functionality, and efficacy.
• Probiotic cells display intrinsic phenotypes to overcome or resist the GIT stress.
• Stress factors alter the gene expression and protein profile of probiotics.
• Changes in specific metabolic traits of probiotics may compromise their biosafety.
• There is a lack of trials studying actual in vivo probiotic cellular alterations.
Revised manuscript submitted to Probiotics and Antimicrobial Proteins for possible publication August 2022.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Castro-López, C., Romero-Luna, H.E., García, H.S. et al. Key Stress Response Mechanisms of Probiotics During Their Journey Through the Digestive System: A Review. Probiotics & Antimicro. Prot. 15, 1250–1270 (2023). https://doi.org/10.1007/s12602-022-09981-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12602-022-09981-x