
Abstract
The structural engineering of hydrated ammonium vanadate as a cathode for aqueous Zn-ion batteries has attracted significant research interest because of its ability to suppress vanadium dissolution and accelerate the electrochemical dynamics. Herein, a feasible fabrication strategy for oxygen-deficient (NH4)2V10O25·xH2O/GO (NVOH@GO) composites was proposed, and the charge storage mechanism was discussed. The results of characterization analysis showed that the introduction of graphene oxide (GO) not only enlarged the layer spacing and improved electrical conductivity, providing spacious channels for Zn2+ (de)intercalation and accelerating the ion diffusion dynamics, but also induced more oxygen vacancies, inhibited the dissolution of vanadium, and reduced self-discharging, offering additional and stable active sites for ion storage. The optimized NVOH@GO electrode delivered extraordinarily stable capacities of 334 mAh·g−1 after 2000 cycles at 5 A·g−1 and 238 mAh·g−1 after 10,000 cycles at 20 A·g−1. Furthermore, ex-situ X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and Raman results systematically revealed the electrochemical mechanism, including a phase transition reaction and subsequent Zn2+/H2O co-(de)intercalation process. This study provides an effective strategy for expanding the interlayer spacing, inducing defect engineering, and enhancing the structural stability of vanadium-based cathodes for Zn-ion batteries and other multivalent aqueous ion batteries.
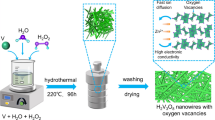
Graphical abstract

摘要
作为水系锌离子电池的正极材料, 水合钒酸铵能够抑制钒溶解、加速电化学动力学, 因此, 其结构工程研究引起了广泛的研究兴趣。本文, 我们提出了一种缺氧(NH4)2V10O25∙xH2O/GO (NVOH@GO)复合材料的制备策略, 并探讨了其电荷存储机制。表征结果表明, 氧化石墨烯(GO)的引入, 不仅扩大了层间距、提高了导电性, 为Zn2+(脱)插层提供了更宽敞的通道、加快了离子扩散动力学; 而且诱导产生了更多氧空位、抑制了钒的溶解、降低了自放电, 为离子存储提供了稳定的活性位点。优化的NVOH@GO电极在5 A·g−1循环2000次、20 A·g−1循环10,000次后, 分别具有334和238 mAh·g−1的稳定容量。此外, 非原位X射线衍射、X射线光电子能谱和拉曼结果, 系统地揭示了其电化学机制, 包括相变反应和Zn2+/H2O共(脱)插层过程。本文提供了一种具有较大层间距、丰富缺陷和较强结构稳定性的锌离子电池和其他多价水离子电池用钒基正极的制备策略。
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Toxic and flammable organic electrolytes, fire or explosion safety hazards, and the scarcity and high price of lithium and cobalt make lithium-ion batteries uncompetitive in sustainable and scalable energy devices, encouraging the development of aqueous multivalent metal ion (Mg2+, Zn2+, Al3+, Ca2+, etc.) batteries with the characteristics of non-toxicity, low price, high theoretical capacity, and environmental friendliness [1,2,3,4,5,6,7]. Aqueous rechargeable zinc-ion batteries (ARZIBs) have been considered one of the most promising and competitive battery candidates because of the low redox potential (Zn2+/Zn ≈ − 0.76 V vs. standard hydrogen electrode (SHE)), high specific capacity (820 mAh·g−1, 5851 mAh·cm−3), non-toxicity, considerable chemical stability, and the natural abundance of Zn metal [3, 8,9,10,11,12]. However, the development of ARZIBs is plagued by unfavorable specific capacity, poor cyclic stability, and sluggish electrochemical kinetics of the cathode hosts because of the high polarization properties of Zn2+ and strong electrostatic interaction between the cathode and Zn2+, which induces undesirable cathode dissolution into the electrolyte and insufficient insertion/extraction/diffusion of Zn2+ in cathode hosts [9, 13,14,15,16]. Therefore, exploiting suitable high-performance cathode materials is imperative for the commercial application of ARZIBs.
Two types of cathodes have been commonly reported for ARZIBs: intercalation-type (Prussian blue analogs, manganese-based oxides, vanadium-based oxides/sulfides, polyanionic compounds and MoS2, etc.) and conversion-type (Co3O4, NiO, organic compounds, etc.) [1, 2, 9, 16]. Among these, vanadium oxides with open frameworks and multiple oxidation states have become the most competitive cathode candidates owing to their high theoretical specific capacity [17,18,19]. However, narrow interlayer spacing, low conductivity, and the strong electrostatic interaction between the V–O skeleton and Zn2+ restrict the diffusion of Zn2+ and induce vanadium dissolution into the electrolyte, resulting in sluggish kinetics and severe capacity decay of vanadium oxides [16, 17, 20]. The pre-intercalation of cations and the introduction of structural water could effectively expand the interlayer spacing of vanadium oxides, and structural water could also play an electrostatic shielding role for Zn2+ and weaken the electrostatic interactions between the V–O framework and Zn2+ [10, 19, 21,22,23,24,25,26]. Nevertheless, the high atomic weight and inert electrochemical activity of the pre-intercalated metal cations hinder the advantages of the high theoretical capacity of vanadium oxides [17, 18, 27,28,29].
Recently, hydrated ammonium vanadate has attracted considerable research interest owing to the low atomic weight and large size of the NH4+ ion, and the cohesion of the layered structure can also be efficiently enhanced by hydrogen bonding between NH4+ and the V–O framework [12, 17, 20, 30,31,32]. From these perspectives, (NH4)2V10O25·8H2O (NVOH) with a 1 nm interlayer distance and NH4+/H2O pillars between the VO layers has obvious structural advantages for reversible Zn2+ (de)intercalation without volume expansion [17, 31, 33]. Furthermore, defect engineering is considered to be an effective strategy to regulate the electronic structure of electrode materials, and could be combined with other strategies to achieve multiple structural optimization, thereby improving the electrical conductivity and facilitating the ion diffusion dynamics [3, 17, 34, 35]. Therefore, exploring (NH4)2V10O25·8H2O with extended layer spacing, plenty of oxygen defects, and high electrical conductivity should be an effective strategy to improve its Zn2+ storage performance [3, 31].
Herein, oxygen-deficient (NH4)2V10O25·xH2O/GO (NVOH@GO) composites with expanded layer spacing were fabricated via a facile solution synthesis strategy. The encapsulation of (NH4)2V10O25·xH2O into graphene oxide (GO) increased the layer spacing and improved the electrical conductivity of NVOH, accelerating ion diffusion. The electron transfer and strong interaction between NVOH and GO induced more oxygen vacancies in the NVOH@GO composites, offering additional active sites for ion storage and providing additional electron transfer paths. Furthermore, the as-prepared NVOH@GO suppressed the dissolution of vanadium and effectively reduced self-discharging, indicating stable structure of the electrode and favoring superior cyclic stability. Consequently, the optimized NVOH@GO delivered a high specific capacity of 418 mAh·g−1 at 0.5 A·g−1 and a stable capacity of 238 mAh·g−1 after 10,000 cycles at 20 A·g−1. Finally, the electrochemical mechanism, including a phase transition reaction and subsequent Zn2+/H2O co-(de)intercalation processes, was elaborated using ex-situ technologies.
2 Experimental
2.1 Preparation of NVOH and NVOH@GO-x
Preparation of NVOH@GO-x: 0.4 g VCl3 and 0.4 g NH4HCO3 was dissolved in 10 and 40 mL of deionized water, respectively. After adding the VCl3 solution dropwise into the NH4HCO3 solution while stirring, the mixture was stirred at 50 °C for 4 h. A graphene oxide solution (0.0059 g·mL−1) was added to the above mixture under stirring. After ultrasonic treatment for 30 min, the uniform dispersion was heated at 80 °C for 2 h. Subsequently, 6 mL of H2O2 (30 wt%) was added dropwise to the above uniform dispersion at 80 °C, and the resulting dispersion was stirred at 80 °C for another 2 h. Finally, NVOH@GO-x was obtained by centrifugation, washed with deionized water, and dried at 100 °C for 12 h in a vacuum oven. The samples with the addition of 0.5, 1.0, and 1.5 g graphene oxide solutions were labeled as NVOH@GO-1, NVOH@GO-2, and NVOH@GO-3, respectively.
Preparation of NVOH: The NVOH preparation process was similar to that of NVOH@GO-x, except that graphene oxide was not added.
2.2 Material characterization
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images were captured using a Zeiss Sigma 500 field-emission scanning electron microscope and JEOL JEM-2100F field-emission transmission electron microscope, respectively. X-ray diffraction (XRD) patterns were obtained on a Bruker D8-Focus X-ray diffractometer using Cu-Kα radiation (λ = 0.15406 nm). Fourier transform infrared spectroscopy (FTIR), Raman spectroscopy, and X-ray photoelectron spectroscopy (XPS) were conducted using a Bruker Smart Lab infrared spectrometer, HR-800 high-resolution confocal micro-Raman system, and PHI-1600 photoelectron spectrometer, respectively. Thermogravimetric analysis (TGA) was performed using a TG/DTA 6300 thermogravimetric analyzer in air. Inductively coupled plasma (ICP) analysis was performed on a Thermo Scientific iCAP RQ mass spectrometer. Electron paramagnetic resonance (EPR) spectra were acquired using a Bruker EMX Micro spectrometer.
2.3 Electrochemical characterization
CR2032 coin cells with metallic Zn as the counter electrode were assembled in air to investigate the electrochemical performance of the fabricated materials. To prepare the working electrode, a slurry of 70 wt% active materials, 20 wt% acetylene black and 10 wt% polyvinylidene fluoride (PVDF) with N-methyl-pyrrolidone as a solvent was coated on carbon paper and dried in a vacuum oven at 80 °C for 12 h; and the areal mass loading of the active materials was ~ 1 mg·cm−2. The electrolyte used was 2 mol·L−1 Zn(CF3SO3)2, and glass fiber was used as the separator. Cyclic voltammetry (CV) at various scan rates and electrochemical impedance spectroscopy (EIS) at frequencies of 10 mHz–100 kHz were measured on a Parstat 4000 + electrochemical workstation. Galvanostatic intermittent titration technique (GITT) and galvanostatic charge/discharge (GCD) curves were obtained using a Neware (CT-4008 T) instrument, and the GITT tests were performed using a pulse of 20 min at 50 m A·g−1 followed by a 120-min interruption.
3 Results and discussion
3.1 Structural and morphological characterization
NVOH@GO-x composites were fabricated via a facile solution synthesis method using VCl3 as the vanadium source and H2O2 as the oxidant (Fig. 1a). First, VCl3 dissolved in deionized water was decomposed into VCl2, HCl, and H2V4O9 (Reaction (1)), which gradually reacted with NH4HCO3 to form ammonium vanadate. Then, GO was anchored on the ammonium vanadate, and low-valence vanadium was gradually oxidized to high-valence vanadium by H2O2, which was confirmed by a change in the color of the solution after adding H2O2 (Fig. 1b). Finally, ammonium vanadate with high-valence vanadium covered with GO was fabricated.

a Schematic illustration of fabrication process and b color change during H2O2 oxidation of NVOH@GO-2; structural characterization of NVOH and NVOH@GO-x: c XRD patterns; d FTIR spectra; e Raman spectra; f XPS survey spectra; high-resolution XPS spectra of g C 1s, h N 1s, i V 2p and j O 1s; k EPR spectra
XRD patterns of NVOH and NVOH@GO-x in Fig. 1c exhibit similar diffraction peaks around 8.2°, 25.6°, 34.8°, 47.3°, 50.5°, and 60.7°, which match well with the diffraction peaks of (NH4)2V10O25·8H2O (JCPDS No. 26-0097). The main diffraction peak at 8.2° corresponding to the (001) plane of NVOH shifts to 7.9° progressively with the addition of GO, indicating enlarged layer spacing in NVOH@GO-x. No additional peaks are detected for NVOH@GO-x, indicating that the addition of GO does not affect the phase structure of NVOH. The structures of NVOH and NVOH@GO were further characterized using FTIR spectroscopy. As shown in Fig. 1d, the two bands at 523.8 and 767.9 cm−1 are assigned to the symmetric and asymmetric stretching vibration of V–O–V bonds, respectively [20, 21, 36]. The absorption peak located at approximately 1004.6 cm−1 is attributed to the V=O stretching vibration of V5+ [20, 37, 38]. The peaks at approximately 1402 and 3139.8 cm−1 are the symmetric bending and asymmetric stretching vibration of the N–H bond of NH4+, confirming the presence of NH4+ in NVOH and NVOH@GO-x [20, 36, 38]. The other two bands at 3597.8 and 1651.7 cm−1 correspond to H–O–H stretching vibration and O–H bending vibration, implying the presence of coordinated water in NVOH and NVOH@GO-x [38, 39]. The typical Raman signals at 162, 273, and 409 cm−1 are attributed to the (V2O2)n, O3–V=O, and V–O3–V bending vibrations, respectively, while the Raman peaks at 521, 711, and 1020 cm−1 are assigned to the stretching vibrations of V3–O, V2–O, and V=O, respectively (Fig. 1e) [17, 19, 20, 40, 41]. The Raman peaks at 1341.5 and 1608.3 cm−1 in NVOH@GO-x can be indexed to the D and G bands of graphitized carbon, indicating the integration of GO into NVOH [35, 42,43,44,45]. The red shifting of the O3–V=O bending vibrations suggests the elongation of V=O bonds along the c-direction, resulting in an enlarged interlayer distance in NVOH@GO-x, whereas the blue shifting of other V–O–V bonds originates from the strong interaction between V and O in NVOH@GO-x [20, 41]. XRD, FTIR, and Raman results confirm the successful fabrication of NVOH and NVOH@GO composites, and the addition of GO expands the interlayer spacing of NVOH. The enlarged layer spacing provides capacious channels for the insertion and extraction of Zn2+ from NVOH and weakens the interaction between the adjacent V2O5 layers, which favors the fast electrochemical kinetics of NVOH@GO-x [20, 27, 46].
TGA curves for NVOH in Fig. S1 show three weight loss steps: below 150 °C, 150–250 °C and 250–350 °C, which can be ascribed to the release of physically absorbed water, structural water, and NH3, respectively [17, 20, 47]. The weight loss above 350 °C in NVOH@GO-x is due to the decomposition of GO [48, 49]. The GO content in NVOH@GO-1, NVOH@GO-2, and NVOH@GO-3 is calculated to be 0.6 wt%, 1.7 wt%, and 3.5 wt%, respectively. For NVOH@GO-x, the non-coincident TGA curves for NH3 release show that a strong interaction between GO and NVOH exists in NVOH@GO-x, which provides additional electron transfer paths and improves electronic conductivity [50, 51].
XPS spectra in Fig. 1f reveal the co-existence of V, O, C, and N in NVOH@GO-x. The high-resolution C 1s peaks at 284.6 and 285.0 eV in Fig. 1g can be attributed to C–C/C=C and C=O bonds, respectively [42, 52]. For N 1s (Fig. 1h), the two peaks at 401.9 and 399.9 eV correspond to protonated –NH4+– and –NH– segments, respectively [19, 37, 53]. The characteristic peaks of V 2p at 517.7/525.4 eV and 516.5/524.2 eV can be assigned to V5+ and V4+, respectively (Fig. 1i) [17, 20, 47, 53,54,55]. The V4+/V5+ ratio increases from 0.14 for NVOH to 0.26 for NVOH@GO-3, implying that more V4+ and oxygen defects are present in NVOH@GO composites. The N 1s and V 2p spectra of NVOH@GO-x shift to lower binding energies in contrast to those of NVOH, which can be ascribed to electron transfer and strong interaction between NVOH and GO [51]. The O 1s spectra can be deconvoluted into three peaks at 530.3, 530.8, and 532.2 eV (Fig. 1j), which can be ascribed to the V–O bond, O–H bond, and H2O molecules, respectively [17, 56]. The obvious XPS signals for H2O molecules indicate that more adsorbed H2O is present in the NVOH@GO composites, which is conducive to the infiltration of the aqueous electrolyte. V4+ has one electron in its 3d orbital, and the EPR intensity represents the concentration of V4+ and oxygen defects [17, 18]. The stronger EPR signals with a spectral splitting factor (g) value of 1.969 in NVOH@GO-x reveal that GO addition induces more oxygen vacancies in NVOH (Fig. 1k) [18, 57, 58]. The XPS and EPR results confirm that the electron transfer and strong interaction between NVOH and GO induce more oxygen vacancies in the NVOH@GO composites, which offers additional active sites for ion storage and jump sites for charge transfer [17, 18].
The morphologies of NVOH and NVOH@GO-x are shown in Figs. 2, S2. NVOH exhibits a honeycomb structure with wrinkled nanosheets (Fig. 2a, b). In NVOH@GO-x (Figs. 2c, d, S2), the honeycomb structure can hardly be distinguished because of coverage by GO on the NVOH, and wrinkled sheets are observed in the enlarged images, indicating the GO coating on the NVOH. The TEM images in Fig. 2e, g confirm the sheet morphology of NVOH and NVOH@GO-2, respectively. High-resolution TEM (HRTEM) images exhibit the clear lattice dislocations (marked by yellow ovals) with lattice spacings of 0.99 and 1.10 nm in NVOH and NVOH@GO-2 (Fig. 2f, h), respectively, matching the (001) crystal lattice plane of (NH4)2V10O25·8H2O, which is consistent with the XRD and XPS results. Figure 2i, j displays the uniform distribution of all elements in NVOH and NVOH@GO-2, proving the successful intercalation of NH4+ in NVOH and the encapsulation of NVOH into GO. Morphological characterization verifies the formation of NVOH and NVOH@GO composites, and the introduction of GO expands the layer spacing.

Morphological characterization of NVOH and NVOH@GO-2: SEM images of a, b NVOH and c, d NVOH@GO-2; TEM images of e, f NVOH and g, h NVOH@GO-2; TEM images and corresponding energy-dispersive spectroscopy elemental mapping for i NVOH and j NVOH@GO-2
3.2 Electrochemical performance and reaction kinetic
CV curves at 0.1 mV·s−1 in Figs. 3a, S3a–c both display two pairs of broad peaks at 0.97/0.95 V and 0.67/0.62 V, which are related to the redox couples of V5+/V4+ and V4+/V3+ during the Zn2+ insertion/extraction process, respectively [18, 20, 27]. After the first activation cycle, the NVOH, NVOH@GO-1, NVOH@GO-2, and NVOH@GO-3 electrodes deliver average capacities of 303, 332, 349, and 342 mAh·g−1 at 0.5 A·g−1 respectively; and the GCD curves exhibit similar voltage plateaus to the CV results (Figs. 3b, S3d–f). The overlapping CV and GCD curves suggest high reversibility of the fabricated electrodes [18, 38]. Impressively, all NVOH@GO-x electrodes deliver better rate capability than NVOH through three consecutive loops at current densities of 0.5 to 20.0 A·g–1 (Figs. 3c, d, S3g–i). In particular, at 0.5, 1.0, 2.0, 5.0, 10.0, and 20.0 A·g–1, the average discharge capacities of the optimized NVOH@GO-2 electrode are 418, 396, 373, 339, 303, and 260 mAh·g–1, respectively; and the capacities at 1.0, 2.0, 5.0, 10.0, and 20.0 A·g−1 are equivalent to 95%, 89%, 81%, 72%, and 62% of that at 0.5 A·g−1, while the capacities of NVOH at the above current densities are only 336, 318, 292, 246, 205, and 159 mAh·g−1. In addition, the GCD curves of NVOH@GO-x exhibit virtually unchanged shapes at various current densities, whereas the voltage plateaus for NVOH almost vanish at 20.0 A·g−1 (Figs. 3c, S3g–i). After 2000 cycles at 5.0 A·g−1 (Figs. 3e, S4), NVOH, NVOH@GO-1, NVOH@GO-2, and NVOH@GO-3 electrodes deliver reversible capacities of 226, 299, 334, and 282 mAh·g−1, respectively, with 100% Coulombic efficiency and well-retained GCD curves. After 10,000 cycles at 20.0 A·g−1 (Fig. 3f), NVOH@GO-2 also exhibits remarkable cyclic reversibility with a high specific capacity of 238 mAh·g−1 and ~ 100% Coulombic efficiency. In addition, the NVOH@GO-2 electrode exhibits superior cyclic stability and cyclic capability with high mass loadings of 4 and 11 mg·cm−2 (Fig. S5a, b). At 50 and 100 mA·g−1, under deep charging/discharging conditions, the NVOH@GO-2 electrode delivers higher capacities of 469 and 430 mAh·g−1, respectively, with overlapping GCD curves (Fig. S5c–f). Furthermore, the 120 h standing result indicates that the NVOH@GO-2 electrode displays much lower self-discharge with 91% capacity retention after resting for 5 days (Fig. S5g, h), showing stable charge storage, which is a prerequisite for practical application of the NVOH@GO-2 electrode [59, 60]. The above results validate the high electrochemical reversibility, rapid reaction kinetics, and superior electrochemical stability of the optimized NVOH@GO-2 electrode, which can also be verified by comparing the electrochemical performance of the NVOH@GO-2 electrode in this work with other vanadium-based cathodes reported in the literatures (Table S1).

Electrochemical performance: a CV curves at 0.1 mV·s−1, b GCD curves at 0.5 A·g−1, and c GCD curves at different current densities of NVOH@GO-2; d rate performance and e cyclic performance at 5.0 A·g−1 of NVOH and NVOH@GO-x; f cyclic performance at 20 A·g−1 of NVOH@GO-2
CV curves were measured at various scan rates to investigate the Zn2+ intercalation kinetics of the as-prepared electrodes. The redox peaks in the CV curves at 0.1 and 0.8 mV·s−1 of the NVOH@GO-2 electrode display narrower voltage gaps and higher peak current densities than those of the NVOH electrode (Figs. 4a, S6a), suggesting lower polarization and faster redox reaction kinetics in the NVOH@GO-2 electrode [20, 27, 51]. As the scan rates increase, the peak centers of the redox couples change significantly and the V4+/V3+ redox pairs are suppressed in NVOH (Fig. S7a), while the CV curves of the NVOH@GO-x electrodes remain similar, with slightly shifted peak positions (Figs. 4b, S7b, c), further validating the low polarization and fast electrochemical kinetics of the NVOH@GO-x electrodes [34, 61]. It is generally accepted that the relationship between current (i) and scan rates (ν) can be expressed by Eq. (2), where a and b are adjustable parameters. Values for b of 0.5 and 1 illustrate diffusion-controlled and surface-controlled pseudocapacitive processes for charge storage, respectively [34, 62]. The calculated b values of peaks 1–4 for NVOH and NVOH@GO-x range from 0.57 to 0.92 (Figs. 4c and S7d–f), which suggests that the redox reactions are controlled by a combination of capacitive-controlled and diffusion-controlled processes [35, 62]. The proportion of capacitive-controlled and diffusion-controlled capacities can be quantitatively determined by Eq. (3), where k1 and k2 are the percentage coefficients of capacitive and diffusion contribution in the total capacity, respectively. The fitted CV curves (shadow area) at 1 mV·s−1 in Figs. 4d, S7g–i demonstrate that the percentages of capacitive contributions are 73%, 69%, 56%, and 40% in NVOH, NVOH@GO-1, NVOH@GO-2, and NVOH@GO-3, respectively. Figure 4e shows the contribution ratios of diffusion-controlled and capacitive-controlled capacities in the total capacity at different scan rates. Although the capacitive contribution increases with increasing scan rate, the NVOH@GO-x electrodes display lower capacitive contribution ratios, which indicates that the NVOH-GO interface modulates the electrostatic interaction and accelerates ion diffusion into the NVOH framework [16, 35, 61].

Electrochemical kinetics: a the fifth CV curves of NVOH and NVOH@GO-2 at 0.1 mV·s−1; b CV curves at various scan rates, c lgi versus lgv plots and d capacitive contribution (shadow region) to total current at 1.0 mV·s−1 of NVOH@GO-2; e individual contribution ratios of capacitive and diffusion-controlled behavior at various scan rates; f GITT profiles and calculated Zn2+ diffusion coefficient (D) during g discharging process and h charging process of NVOH and NVOH@GO-2
EIS and GITT were conducted to further examine the reaction kinetics and solid-state diffusion dynamics, respectively. A semicircle in the high-frequency region and a slanted line in the low-frequency region are observed in all the Nyquist plots (Fig. S6b), which are related to the ohmic resistance (Re), charge-transfer resistance (Rct), and Warburg impedance (Zw). The Re and Rct values of the NVOH@GO-x electrodes are much lower than those of NVOH (Table S2), demonstrating the improved electrical conductivity and accelerated reaction kinetics of NVOH@GO-x electrodes [21, 41, 57]. The GITT profiles in Fig. 4f indicate that the NVOH@GO-2 electrode delivers less overpotential during charging/discharging, further indicating its fast kinetics. The ion diffusion coefficients can be calculated according to Eq. (4), where τ, mm, Vm, S, ΔEs, and ΔEτ are the current pulse time, moles, molar volume, electrode area, steady-state voltage change, and voltage change during the pulse, respectively [19, 35, 57]. The calculated diffusion coefficients of NVOH@GO-2 during charging and discharging are higher than those of NVOH (Fig. 4g, h), indicating the efficient diffusion dynamics of the NVOH@GO-2 electrode. The CV, EIS, and GITT results reveal that the electron transfer and the strong interaction between NVOH and GO, together with oxygen vacancies, improve the electronic conductivity and enhance the ion diffusion kinetics of the NVOH@GO-x electrodes, contributing to their superior electrochemical performance.
3.3 Electrochemical reaction mechanism
The electrochemical reaction mechanism of the NVOH@GO electrode was characterized using a series of ex-situ experiments. Ex-situ XRD patterns and the corresponding GCD curves are shown in Fig. 5a–c. At all investigated charging/discharging states, the diffraction peaks corresponding to (NH4)2V10O25 vanish, and new diffraction peaks emerging at 12.3°, 20.9°, 30.1°, 32.0°, 34.2°, and 36.5° can be indexed to Zn3V2O7(OH)2·2H2O (JCPDS No. 50-0570), which is generated by the coordination of Zn2+-intercalated V–O with H2O molecules, suggesting that Zn2+ and H2O are co-intercalated into NVOH@GO [20, 26, 63]. In addition, as can be observed in Fig. 5b, the (001) plane of Zn3V2O7(OH)2·2H2O shifts to lower 2θ values during the discharging process and moves back during the charging process, indicating the reversible Zn2+ (de)intercalation process of Zn3V2O7(OH)2·2H2O, which is consistent with the literatures [14, 64].

Electrochemical reaction mechanism of NVOH@GO-2 electrode: a, b ex-situ XRD patterns at selected states and c corresponding GCD curves of initial two cycles at 0.05 A·g−1; d Raman spectra at different states; XPS spectra of e Zn 2p, f V 2p, g O 1s and h N 1s at different states; i schematic diagram of reaction mechanism for NVOH@GO electrode; j V concentration in electrolyte after electrodes were immersed in 2 mol·L−1 Zn(CF3SO3)2 for 50 days
To further confirm the reversible Zn2+ (de)intercalation of Zn3V2O7(OH)2·2H2O, ex-situ Raman spectroscopy and XPS were performed. Three Raman peaks at 248, 369, and 498 cm−1 are assigned to Zn2+–O2− symmetrical vibrations, two Raman peaks at 141 and 315 cm−1 are ascribed to V–O bending vibrations, and two Raman peaks at 806 and 866 cm−1 are assigned to V–O stretching vibration (Fig. 5d), suggesting the presence of Zn3V2O7(OH)2·2H2O [14, 21, 64, 65]. The Raman peak at 498 cm−1 for the fully charged electrode and the Raman peak at 315 cm−1 for the fully discharged electrode disappear because the extraction of Zn2+ weakens the Zn–O vibration and enhances the V–O vibration [14]. In both the fully discharged and charged states, obvious Zn peaks located at 1021.9 and 1045.0 eV ascribed to intercalated and surface-adsorbed Zn2+ are observed in Fig. 5e, indicating the presence of Zn-containing compounds and the reversible Zn2+ (de)intercalation of Zn3V2O7(OH)2·2H2O [14, 40, 64, 66, 67]. V5+ at 517.7 and 525.4 eV accompanied by V4+ at 517.2 and 524.4 eV are de-convoluted in the V 2p XPS spectra of the charged and discharged electrodes (Fig. 5f), and the charged electrode displays a higher V5+/V4+ ratio due to Zn2+ de-intercalation [17, 28, 29, 38]. The O 1s spectra consist of three parts (Fig. 5g), corresponding to V–O bonds (530.6 eV), H2O molecules (532.1 eV), and Zn–O bonds (533.0 eV), respectively [38, 64, 65]. In the discharged state, the O 1s XPS peak for H2O molecules increases owing to the co-intercalation of Zn2+ and H2O, while the weakened peak for V–O bonds can be ascribed to the insertion of Zn2+, in agreement with the ex-situ Raman results [23, 38]. The N 1s spectra exhibits two peaks at 402.1 and 400.5 eV (Fig. 5h), ascribed to protonated –NH4+– and –NH– segments, respectively, which implies that the intercalated NH4+ may be present as NH3 [19, 53]. Moreover, the intensity of the –NH4+– peak increases, and the N 1s XPS spectrum shifts to higher binding energies in the discharged state, indicating that NH4+ is involved in the redox reaction [16, 53]. The two charged electrodes display similar ex-situ Raman peaks and XPS spectra (Fig. 5d–h), indicating the high reversibility of the electrode in subsequent cycles. The ex-situ XRD, Raman, and XPS results reveal the charge storage mechanism of the NVOH@GO electrode, including a phase transition from (NH4)2V10O25 to Zn3V2O7(OH)2·2H2O during the first discharge process and a reversible Zn2+/H2O co-(de)intercalation of Zn3V2O7(OH)2·2H2O during the subsequent cycle, as described in Fig. 5i.
The structural stability of the electrode materials was evaluated by a static soaking experiment in 2 mol·L−1 Zn(CF3SO3)2 for 50 days and 2 mol·L−1 ZnSO4 for 10 days. Although the electrolyte soaked with NVOH and the NVOH@GO-2 electrode remains transparent and colorless during the static soaking experiment (Figs. 5j, S8), the V concentration in the electrolyte immersed with NVOH is much higher than that immersed with the NVOH@GO-2 electrode, suggesting that increased interaction between V and O in NVOH@GO-x induced by GO addition suppresses the dissolution of vanadium into the electrolyte, indicating the stability of the electrode and favoring its superior cyclic stability [38, 65].
4 Conclusion
In summary, a solution synthesis strategy was successfully employed to engineer oxygen-deficient (NH4)2V10O25·xH2O/GO composites, which exhibited satisfactory electrochemical performance as cathodes for ARZIBs. In particular, the optimized NVOH@GO delivered an extraordinary rate capability of 303 mAh·g−1 at 10 A·g−1 and an ultra-stable capacity of 238 mAh·g−1 after 10,000 cycles at 20 A·g−1. The superior electrochemical performance of the NVOH@GO composites can be attributed to the following favorable features: the encapsulation of (NH4)2V10O25·xH2O into GO enlarged the layer spacing and improved the electrical conductivity of NVOH, which provided spacious channels for Zn2+ (de)intercalation and accelerated ion diffusion, favoring fast electrochemical dynamics; the electron transfer and strong interaction between NVOH and GO provided additional electron transfer paths; abundant oxygen vacancies offered additional active sites for ion storage and jump sites for charge transfer, leading to a high-specific capacity; and inhibition of vanadium dissolution and limited self-discharge contributed to the superior cyclic stability. In addition, the electrochemical mechanism, including the phase transition reaction and subsequent Zn2+/H2O co-(de)intercalation process, was elucidated using ex-situ XRD, XPS, and Raman techniques. This work not only provides a strategy for the construction of V-based cathodes with dissolution inhibition, but also proposes a charge storage mechanism.
References
Zhang N, Chen XY, Yu M, Niu ZQ, Cheng FY, Chen J. Materials chemistry for rechargeable zinc-ion batteries. Chem Soc Rev. 2020;49(13):4203. https://doi.org/10.1039/c9cs00349e.
Wang X, Zhang ZCY, Xi BJ, Chen WH, Jia YX, Feng JK, Xiong SL. Advances and perspectives of cathode storage chemistry in aqueous zinc-ion batteries. ACS Nano. 2021;15(6):9244. https://doi.org/10.1021/acsnano.1c01389.
Guo C, Yi S, Si R, Xi B, An X, Liu J, Li J, Xiong S. Advances on defect engineering of vanadium-based compounds for high-energy aqueous zinc-ion batteries. Adv Energy Mater. 2022;12(40):2202039. https://doi.org/10.1002/aenm.202202039.
Wu B, Luo W, Li M, Zeng L, Mai L. Achieving better aqueous rechargeable zinc ion batteries with heterostructure electrodes. Nano Res. 2021;14(9):3174. https://doi.org/10.1007/s12274-021-3392-1.
Wu HH, Zhuo F, Qiao H, Kodumudi Venkataraman L, Zheng M, Wang S, Huang H, Li B, Mao X, Zhang Q. Polymer-/ceramic-based dielectric composites for energy storage and conversion. Energ Environ Mater. 2022;5(2):486. https://doi.org/10.1002/eem2.12237.
Wang F, Liu Y, Wei HJ, Li TF, Xiong XH, Wei SZ, Ren FZ, Volinsky AA. Recent advances and perspective in metal coordination materials-based electrode materials for potassium-ion batteries. Rare Met. 2021;40(2):448. https://doi.org/10.1007/s12598-020-01649-1.
Song M, Zhong CL. Achieving both high reversible and stable Zn anode by a practical glucose electrolyte additive toward high-performance Zn-ion batteries. Rare Met. 2021;41(2):356. https://doi.org/10.1007/s12598-021-01858-2.
Tao F, Liu Y, Ren X, Wang J, Zhou Y, Miao Y, Ren F, Wei S, Ma J. Different surface modification methods and coating materials of zinc metal anode. J Energy Chem. 2022;66:397. https://doi.org/10.1016/j.jechem.2021.08.022.
Chen X, Zhang H, Liu JH, Gao Y, Cao X, Zhan C, Wang Y, Wang S, Chou SL, Dou SX, Cao D. Vanadium-based cathodes for aqueous zinc-ion batteries: mechanism, design strategies and challenges. Energy Storage Mater. 2022;50:21. https://doi.org/10.1016/j.ensm.2022.04.040.
Li Y, Zhang D, Huang S, Yang HY. Guest-species-incorporation in manganese/vanadium-based oxides: towards high performance aqueous zinc-ion batteries. Nano Energy. 2021;85:105969. https://doi.org/10.1016/j.nanoen.2021.105969.
Zhu QN, Wang ZY, Wang JW, Liu XY, Yang D, Cheng LW, Tang MY, Qin Y, Wang H. Challenges and strategies for ultrafast aqueous zinc-ion batteries. Rare Met. 2021;40(2):309. https://doi.org/10.1007/s12598-020-01588-x.
Geng L, Meng J, Wang X, Han C, Han K, Xiao Z, Huang M, Xu P, Zhang L, Zhou L, Mai L. Eutectic electrolyte with unique solvation structure for high-performance zinc-ion batteries. Angew Chem Int Ed. 2022;61(31):202206717. https://doi.org/10.1002/anie.202206717.
Sui D, Wu M, Shi K, Li C, Lang J, Yang Y, Zhang X, Yan X, Chen Y. Recent progress of cathode materials for aqueous zinc-ion capacitors: carbon-based materials and beyond. Carbon. 2021;185:126. https://doi.org/10.1016/j.carbon.2021.08.084.
Chen X, Wang L, Li H, Cheng F, Chen J. Porous V2O5 nanofibers as cathode materials for rechargeable aqueous zinc-ion batteries. J Energy Chem. 2019;38:20. https://doi.org/10.1016/j.jechem.2018.12.023.
Pam ME, Yan D, Yu J, Fang D, Guo L, Li XL, Li TC, Lu X, Ang LK, Amal R, Han Z, Yang HY. Microstructural engineering of cathode materials for advanced zinc-ion aqueous batteries. Adv Sci. 2020;8(1):2002722. https://doi.org/10.1002/advs.202002722.
Li W, Han C, Gu Q, Chou SL, Wang JZ, Liu HK, Dou SX. Electron delocalization and dissolution-restraint in vanadium oxide superlattices to boost electrochemical performance of aqueous zinc-ion batteries. Adv Energy Mater. 2020;10(48):2001852. https://doi.org/10.1002/aenm.202001852.
Cao J, Zhang D, Yue Y, Wang X, Pakornchote T, Bovornratanaraks T, Zhang X, Wu ZS, Qin J. Oxygen defect enriched (NH4)2V10O25·8H2O nanosheets for superior aqueous zinc-ion batteries. Nano Energy. 2021;84:105876. https://doi.org/10.1016/j.nanoen.2021.105876.
He T, Ye Y, Li H, Weng S, Zhang Q, Li M, Liu T, Cheng J, Wang X, Lu J, Wang B. Oxygen-deficient ammonium vanadate for flexible aqueous zinc batteries with high energy density and rate capability at −30 °C. Mater Today. 2021;43:53. https://doi.org/10.1016/j.mattod.2020.11.019.
Sun J, Zhao Y, Liu Y, Jiang H, Huang C, Cui M, Hu T, Meng C, Zhang Y. “Three-in-One” strategy that ensures V2O5·nH2O with superior Zn2+ storage by simultaneous protonated polyaniline intercalation and encapsulation. Small Struct. 2022;3(4):2100212. https://doi.org/10.1002/sstr.202100212.
Zong Q, Du W, Liu C, Yang H, Zhang Q, Zhou Z, Atif M, Alsalhi M, Cao G. Enhanced reversible zinc ion intercalation in deficient ammonium vanadate for high-performance aqueous zinc-ion battery. Nanomicro Lett. 2021;13(1):116. https://doi.org/10.1007/s40820-021-00641-3.
Dong W, Du M, Zhang F, Zhang X, Miao Z, Li H, Sang Y, Wang JJ, Liu H, Wang S. In situ electrochemical transformation reaction of ammonium-anchored heptavanadate cathode for long-life aqueous zinc-ion batteries. ACS Appl Mater Interfaces. 2021;13(4):5034. https://doi.org/10.1021/acsami.0c19309.
Li Y, Wang Z, Cai Y, Pam ME, Yang Y, Zhang D, Wang Y, Huang S. Designing advanced aqueous zinc-ion batteries: principles, strategies, and perspectives. Energ Environ Mater. 2022;5(3):823. https://doi.org/10.1002/eem2.12265.
He T, Weng S, Ye Y, Cheng J, Wang X, Wang X, Wang B. Cation- deficient Zn0.3(NH4)0.3V4O10·0.91H2O for rechargeable aqueous zinc battery with superior low-temperature performance. Energy Storage Mater. 2021;38:389. https://doi.org/10.1016/j.ensm.2021.03.025.
Zhang Y, Wan F, Huang S, Wang S, Niu Z, Chen J. A chemically self-charging aqueous zinc-ion battery. Nat Commun. 2020;11(1):2199. https://doi.org/10.1038/s41467-020-16039-5.
Deng W, Zhou Z, Li Y, Zhang M, Yuan X, Hu J, Li Z, Li C, Li R. High-capacity layered magnesium vanadate with concentrated gel electrolyte toward high-performance and wide-temperature zinc-ion battery. ACS Nano. 2020;14(11):15776. https://doi.org/10.1021/acsnano.0c06834.
Yang Y, Tang Y, Liang S, Wu Z, Fang G, Cao X, Wang C, Lin T, Pan A, Zhou J. Transition metal ion-preintercalated V2O5 as high-performance aqueous zinc-ion battery cathode with broad temperature adaptability. Nano Energy. 2019;61:617. https://doi.org/10.1016/j.nanoen.2019.05.005.
Kim J, Lee SH, Park C, Kim HS, Park JH, Chung KY, Ahn H. Controlling vanadate nanofiber interlayer via intercalation with conducting polymers: cathode material design for rechargeable aqueous zinc ion batteries. Adv Funct Mater. 2021;31(26):2100005. https://doi.org/10.1002/adfm.202100005.
Su ZH, Wang RH, Huang JH, Sun R, Qin ZX, Zhang YF, Fan HS. Silver vanadate (Ag0.33V2O5) nanorods from Ag intercalated vanadium pentoxide for superior cathode of aqueous zinc-ion batteries. Rare Met. 2022;41(8):2844. https://doi.org/10.1007/s12598-022-02026-w.
Yu X, Hu F, Guo ZQ, Liu L, Song GH, Zhu K. High-performance Cu0.95V2O5 nanoflowers as cathode materials for aqueous zinc-ion batteries. Rare Met. 2022;41(1):29. https://doi.org/10.1007/s12598-021-01771-8.
Lai J, Tang H, Zhu X, Wang Y. A hydrated NH4V3O8 nanobelt electrode for superior aqueous and quasi-solid-state zinc ion batteries. J Mater Chem A. 2019;7(40):23140. https://doi.org/10.1039/C9TA07822C.
Wang X, Wang Y, Jiang Y, Li X, Liu Y, Xiao H, Ma Y, Huang YY, Yuan G. Tailoring ultrahigh energy density and stable dendrite-free flexible anode with Ti3C2Tx MXene nanosheets and hydrated ammonium vanadate nanobelts for aqueous rocking-chair zinc ion batteries. Adv Funct Mater. 2021;31(35):2103210. https://doi.org/10.1002/adfm.202103210.
Gan Y, Wang C, Li JY, Zheng JJ, Wan HZ, Wang H. Stability optimization strategy of aqueous zinc ion batteries. Chin J Rare Met. 2022;46(6):753. https://doi.org/10.13373/j.cnki.cjrm.XY21100036.
Jiang H, Zhang Y, Pan Z, Xu L, Zheng J, Gao Z, Hu T, Meng C. Facile hydrothermal synthesis and electrochemical properties of (NH4)2V10O25·8H2O nanobelts for high-performance aqueous zinc ion batteries. Electrochim Acta. 2020;332:135506.
Zhang Z, Xi B, Wang X, Ma X, Chen W, Feng J, Xiong S. Oxygen defects engineering of VO2-x·H2O nanosheets via in situ polypyrrole polymerization for efficient aqueous zinc ion storage. Adv Funct Mater. 2021. https://doi.org/10.1002/adfm.202103070.
Liu G, Xiao F, Zhang T, Gu Y, Li J, Guo D, Xu M, Wu N, Cao A, Liu X. In-situ growth of MoO2@N doped carbon on Mo2C-MXene for superior lithium storage. Appl Surf Sci. 2022;597:153688. https://doi.org/10.1016/j.apsusc.2022.153688.
Xu L, Zhang Y, Jiang H, Zheng J, Dong X, Hu T, Meng C. Facile hydrothermal synthesis and electrochemical properties of (NH4)2V6O16 nanobelts for aqueous rechargeable zinc ion batteries. Colloids Surf Physicochem Eng Aspects. 2020;593:124621. https://doi.org/10.1016/j.colsurfa.2020.124621.
Chen K, Zhang G, Xiao L, Li P, Li W, Xu Q, Xu J. Polyaniline encapsulated amorphous V2O5 nanowire-modified multi-functional separators for lithium-sulfur batteries. Small Methods. 2021;5(3):2001056. https://doi.org/10.1002/smtd.202001056.
Zhang L, Hu J, Zhang B, Liu J, Wan H, Miao L, Jiang J. Suppressing cathode dissolution via guest engineering for durable aqueous zinc-ion batteries. J Mater Chem A. 2021;9(12):7631. https://doi.org/10.1039/d1ta00263e.
Zhang Y, Wang Y, Lu L, Sun C, Yu DYW. Vanadium hexacyanoferrate with two redox active sites as cathode material for aqueous Zn-ion batteries. J Power Sources. 2021;484:229263. https://doi.org/10.1016/j.jpowsour.2020.229263.
Liu Y, Pan Z, Tian D, Hu T, Jiang H, Yang J, Sun J, Zheng J, Meng C, Zhang Y. Employing, “one for two” strategy to design polyaniline-intercalated hydrated vanadium oxide with expanded interlayer spacing for high-performance aqueous zinc-ion batteries. Chem Eng J. 2020;399:125842. https://doi.org/10.1016/j.cej.2020.125842.
Liu C, Neale Z, Zheng J, Jia X, Huang J, Yan M, Tian M, Wang M, Yang J, Cao G. Expanded hydrated vanadate for high-performance aqueous zinc-ion batteries. Energ Environ Sci. 2019;12(7):2273. https://doi.org/10.1039/c9ee00956f.
Liu G, Xu L, Li Y, Guo D, Wu N, Yuan C, Qin A, Cao A, Liu X. Metal-organic frameworks derived anatase/rutile heterostructures with enhanced reaction kinetics for lithium and sodium storage. Chem Eng J. 2022;430:132689. https://doi.org/10.1016/j.cej.2021.132689.
Tao F, Liu Y, Ren X, Jiang A, Wei H, Zhai X, Wang F, Stock HR, Wen S, Ren F. Carbon nanotube-based nanomaterials for high-performance sodium-ion batteries: recent advances and perspectives. J Alloys Compd. 2021;873:159742. https://doi.org/10.1016/j.jallcom.2021.159742.
Sui D, Xu L, Zhang H, Sun Z, Kan B, Ma Y, Chen Y. A 3D cross-linked graphene-based honeycomb carbon composite with excellent confinement effect of organic cathode material for lithium-ion batteries. Carbon. 2020;157:656. https://doi.org/10.1016/j.carbon.2019.10.106.
Li J, Huang H, Cao X, Wu HH, Pan K, Zhang Q, Wu N, Liu X. Template-free fabrication of MoP nanoparticles encapsulated in N-doped hollow carbon spheres for efficient alkaline hydrogen evolution. Chem Eng J. 2021;416:127677. https://doi.org/10.1016/j.cej.2020.127677.
He D, Peng Y, Ding Y, Xu X, Huang Y, Li Z, Zhang X, Hu L. Suppressing the skeleton decomposition in Ti-doped NH4V4O10 for durable aqueous zinc ion battery. J Power Sources. 2021;484:229284. https://doi.org/10.1016/j.jpowsour.2020.229284.
Huang C, Liu S, Feng J, Wang Y, Fan Q, Kuang Q, Dong Y, Zhao Y. Optimizing engineering of rechargeable aqueous zinc ion batteries to enhance the zinc ions storage properties of cathode material. J Power Sources. 2021;490:229528. https://doi.org/10.1016/j.jpowsour.2021.229528.
Luo H, Wang B, Wu F, Jian J, Yang K, Jin F, Cong B, Ning Y, Zhou Y, Wang D, Liu H, Dou S. Synergistic nanostructure and heterointerface design propelled ultra-efficient in-situ self-transformation of zinc-ion battery cathodes with favorable kinetics. Nano Energy. 2021;81:105601. https://doi.org/10.1016/j.nanoen.2020.105601.
Cao H, Zheng Z, Norby P, Xiao X, Mossin S. Electrochemically induced phase transition in V3O7·H2O nanobelts/reduced graphene oxide composites for aqueous zinc-ion batteries. Small. 2021;17(24):2100558. https://doi.org/10.1002/smll.202100558.
Jiang H, Zhang Y, Liu Y, Yang J, Xu L, Wang P, Gao Z, Zheng J, Meng C, Pan Z. In situ grown 2D hydrated ammonium vanadate nanosheets on carbon cloth as a free-standing cathode for high-performance rechargeable Zn-ion batteries. J Mater Chem A. 2020;8(30):15130. https://doi.org/10.1039/d0ta05065b.
Bin D, Huo W, Yuan Y, Huang J, Liu Y, Zhang Y, Dong F, Wang Y, Xia Y. Organic-inorganic-induced polymer intercalation into layered composites for aqueous zinc-ion battery. Chem. 2020;6(4):968. https://doi.org/10.1016/j.chempr.2020.02.001.
Guo D, Yang M, Yang M, Yang T, Hu G, Liu H, Liu G, Wu N, Qin A, Liu X. Stabilized covalent interfacial coupling design of Li3V2(PO4)3 with carbon framework for boosting lithium storage kinetics. CrystEngComm. 2021;23:8506. https://doi.org/10.1039/D1CE01254A.
Yu D, Wei Z, Zhang X, Zeng Y, Wang C, Chen G, Shen ZX, Du F. Boosting Zn2+ and NH4+ storage in aqueous media via in-situ electrochemical induced VS2/VOx heterostructures. Adv Funct Mater. 2020;31(11):2008743. https://doi.org/10.1002/adfm.202008743.
Guo D, Wang F, Yang M, Hu G, Liu G, Wu N, Qin A, Liu X. Constructing abundant oxygen vacancies in NaVPO4F@C for boosting sodium storage kinetics. Electrochim Acta. 2022;424:140695. https://doi.org/10.1016/j.electacta.2022.140695.
Zhu T, Mai B, Hu P, Liu Z, Cai C, Wang X, Zhou L. Ammonium ion and structural water Co-assisted Zn2+ intercalation/de-intercalation in NH4V4O10·0.28H2O. Chin J Chem. 2021;39(7):1885.
Yang W, Dong L, Yang W, Xu C, Shao G, Wang G. 3D oxygen-defective potassium vanadate/carbon nanoribbon networks as high-performance cathodes for aqueous zinc-ion batteries. Small Methods. 2019;4(1):1900670. https://doi.org/10.1002/smtd.201900670.
Du Y, Wang X, Sun J. Tunable oxygen vacancy concentration in vanadium oxide as mass-produced cathode for aqueous zinc-ion batteries. Nano Res. 2020;14(3):754. https://doi.org/10.1007/s12274-020-3109-x.
Luo H, Wang B, Wang C, Wu F, Jin F, Cong B, Ning Y, Zhou Y, Wang D, Liu H, Dou S. Synergistic deficiency and heterojunction engineering boosted VO2 redox kinetics for aqueous zinc-ion batteries with superior comprehensive performance. Energy Storage Mater. 2020;33:390. https://doi.org/10.1016/j.ensm.2020.08.011.
Yang Z, Wang B, Chen Y, Zhou W, Li H, Zhao R, Li X, Zhang T, Bu F, Zhao Z, Li W, Chao D, Zhao D. Activating sulfur oxidation reaction via six-electron-redox mesocrystal NiS2 for sulfur-based aqueous battery. Natl Sci Rev. 2022. https://doi.org/10.1093/nsr/nwac268.
Hou Z, Zhang T, Liu X, Xu Z, Liu J, Zhou W, Qian Y, Fan HJ, Chao D, Zhao D. A solid-to-solid metallic conversion electrochemistry toward 91% zinc utilization for sustainable aqueous batteries. Sci Adv. 2022;8(41):eapb8960. https://doi.org/10.1126/sciadv.abp8960.
Zheng J, Liu C, Tian M, Jia X, Jahrman EP, Seidler GT, Zhang S, Liu Y, Zhang Y, Meng C, Cao G. Fast and reversible zinc ion intercalation in Al-ion modified hydrated vanadate. Nano Energy. 2020;70:104519. https://doi.org/10.1016/j.nanoen.2020.104519.
Zhao J, Xu Z, Zhou Z, Xi S, Xia Y, Zhang Q, Huang L, Mei L, Jiang Y, Gao J, Zeng Z, Tan C. A safe flexible self-powered wristband system by integrating defective MnO2-x nanosheet-based zinc-ion batteries with perovskite solar cells. ACS Nano. 2021;15(6):10597. https://doi.org/10.1021/acsnano.1c03341.
Zhu K, Wu T, Huang K. Understanding the dissolution and phase transformation mechanisms in aqueous Zn/α-V2O5 batteries. Chem Mater. 2021;33(11):4089. https://doi.org/10.1021/acs.chemmater.1c00715.
Ding J, Gao H, Zhao K, Zheng H, Zhang H, Han L, Wang S, Wu S, Fang S, Cheng F. In-situ electrochemical conversion of vanadium dioxide for enhanced zinc-ion storage with large voltage range. J Power Sources. 2021;487:229369. https://doi.org/10.1016/j.jpowsour.2020.229369.
Zhu M, Wang H, Lin W, Chan D, Li H, Wang K, Tang Y, Hao T, Chen S, Malyi OI, Tang Y, Zhang Y. Amphipathic molecules endowing highly structure robust and fast kinetic vanadium-based cathode for high-performance zinc-ion batteries. Small Struct. 2022. https://doi.org/10.1002/sstr.202200016.
Chen H, Chen L, Meng J, Yang Z, Wu J, Rong Y, Deng L, Shi Y. Synergistic effects in V3O7/V2O5 composite material for high capacity and long cycling life aqueous rechargeable zinc ion batteries. J Power Sources. 2020;474:228569. https://doi.org/10.1016/j.jpowsour.2020.228569.
Chen Q, Jin J, Kou Z, Liao C, Liu Z, Zhou L, Wang J, Mai L. Zn2+ pre-intercalation stabilizes the tunnel structure of MnO2 nanowires and enables zinc-ion hybrid supercapacitor of battery-level energy density. Small. 2020;16(14):2000091. https://doi.org/10.1002/smll.202000091.
Acknowledgements
This study was financially supported by the Natural Science Foundations of China (Nos. 51904152 and 42002040), Natural Science Foundations of Henan Province (No. 222300420502), Key Science and Technology Program of Henan Province (No. 222102240044), and Key Scientific Research Projects in Colleges and Universities of Henan Province (No. 21B610010).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, GL., Zhang, T., Li, XJ. et al. Oxygen-deficient ammonium vanadate/GO composites with suppressed vanadium dissolution for ultra-stable high-rate aqueous zinc-ion batteries. Rare Met. 42, 3729–3740 (2023). https://doi.org/10.1007/s12598-023-02364-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12598-023-02364-3