
Abstract
In the last decade, the costs of genome sequencing have decreased considerably. The commencement of large-scale cancer sequencing projects has enabled cancer genomics to join the big data revolution. One of the challenges still facing cancer genomics research is determining which are the driver mutations in an individual cancer, as these contribute only a small subset of the overall mutation profile of a tumour. Focusing primarily on somatic single nucleotide mutations in this review, we consider both coding and non-coding driver mutations, and discuss how such mutations might be identified from cancer sequencing datasets. We describe some of the tools and database that are available for the annotation of somatic variants and the identification of cancer driver genes. We also address the use of genome-wide variation in mutation load to establish background mutation rates from which to identify driver mutations under positive selection. Finally, we describe the ways in which mutational signatures can act as clues for the identification of cancer drivers, as these mutations may cause, or arise from, certain mutational processes. By defining the molecular changes responsible for driving cancer development, new cancer treatment strategies may be developed or novel preventative measures proposed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The big data revolution
Sequencing of the first human genome was completed in 2003, at a cost of almost three billion US dollars. In the 15 years that followed, the costs of whole genome sequencing have reduced remarkably, toward the well-known US$1000 target. This has been made possible in the most part through significant technological improvements and the implementation of next-generation sequencing (NGS). NGS platforms allow high-throughput and parallelisable DNA sequencing. These technologies generally utilise short read sequencing, followed by mapping of sequence reads against a reference genome in the analysis stage [reviewed in (Goodwin et al. 2016)]. Reductions in DNA sequencing costs have enabled the commencement of large-scale cancer genome sequencing projects. As cohort sizes have increased, data processing and storage requirements have necessarily become much more demanding. Hosting sequencing data for even a handful of whole human genomes requires hundreds of gigabytes of storage. Further, cancer genomics analyses often incorporate additional datasets from the fields of epigenomics and transcriptomics, thus increasing the complexity of such studies. These factors have enabled cancer genomics to join the big data revolution.
The Cancer Genome Atlas (TCGA) project was launched in 2005 and recently completed, having produced sequencing data from tumour and matched normal tissues from more than 30 cancer types (Tomczak et al. 2015). The International Cancer Genome Consortium (ICGC) commenced in 2008, similarly seeking to whole-genome sequence thousands of cancer samples and provide the data for research access (Zhang et al. 2011). Many processed datasets from these projects are ‘open access’, and raw datasets are generally available after application, for researchers to download and analyse for their own genomics projects. For researchers without the significant computational infrastructure that can be necessary to download and process datasets of these sizes, the National Cancer Institute (NCI) has sponsored the development of three cloud resources, which can enable scientists to analyse and visualise large datasets in a cloud environment (Hinkson et al. 2017).
Driver mutations and cancer development
It has been known for many years that cancer develops as a result of chromosomal abnormalities, and the specific mutation profile of a tumour has important implications for cancer treatment (Nowell 1976). Mutations can develop in cellular DNA through exposure to external DNA-damaging agents or from internal deficiencies in DNA replication or repair (Vogelstein et al. 2013). These processes result in the accumulation of potentially hundreds of thousands of somatic mutations in a single cancer genome, primarily taking the form of single nucleotide mutations, but also including insertions and deletions (indels) or larger structural rearrangements and copy number aberrations (Vogelstein et al. 2013). Of these somatic variants, only a handful will be responsible for malignant transformation, by conferring a selective advantage to the subpopulation of cells that harbour the variant (Tomasetti et al. 2015). Such mutations are termed ‘driver mutations’; they undergo positive selection in a tumour and cause cells to result in the hallmarks that are characteristic of malignancy (Hanahan and Weinberg 2011). Different cancer types harbour different numbers of driver mutations, averaging approximately four per tumour (Martincorena et al. 2017). The remaining variants are termed ‘passenger mutations’, and they confer little functional impact (Stratton et al. 2009). One of the challenges facing cancer genomics research is determining which are the handful of driver mutations from within the vast background of passenger mutations in a cancer genome. The focus of this review will be single nucleotide driver mutations, though we will address indels and larger structural rearrangements and copy number aberrations in some instances.
Types of driver mutations
Cancer develops when cells accumulate somatic mutations, as shown in Fig. 1. It is worth noting that germline variants can also contribute toward how the mutational landscape of a cancer develops [for examples, see (Waszak et al. 2017)], and can contribute to oncogenesis by predisposing cells toward cancer development.

Somatic driver mutations and cancer development. A simplified diagram depicting the process of somatic mutation accumulation and tumour formation from normal tissue
Protein-coding driver mutations
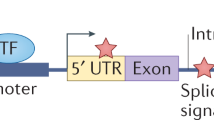
Most cancer driver mutations identified to date lie within gene bodies, and the function of these mutations can generally be ascertained by examining their impact on the encoded protein. Oncogenes are genes that are activated by mutations, allowing cells to acquire a selective advantage (Vogelstein et al. 2013) (Fig. 2a). In contrast, tumour suppressor genes contribute to cancer development through the selective advantage gained by their inactivation, which generally arises through truncating mutations or frameshift indels (Vogelstein et al. 2013) (Fig. 2b). Not all driver mutations have such clear function however. For example, synonymous mutations may also be driver events in cancer if they differentially regulate gene splicing (Supek et al. 2014). Similarly, larger structural variations and copy number aberrations such as genomic deletions may lead to gene fusion events that truncate tumour suppressor genes, or create tumourigenic novel proteins (Mertens et al. 2015). These genetic alterations can subsequently lead to dysregulation of important pathways, resulting in cancer development. When first published in 2004, the Cancer Gene Census [hosted by the Catalogue of Somatic Mutations in Cancer (COSMIC) database (Forbes et al. 2015; Forbes et al. 2011)] had annotated 291 well-characterised ‘cancer genes’ (Futreal et al. 2004). This list now contains more than 500 entries. Some driver genes are commonly mutated across cancer types, including TP53, ARID1A, KRAS and PIK3CA, while other driver genes are more tumour specific (Gonzalez-Perez et al. 2013).

Types of cancer driver mutations. Diagram depicting the formation of driver mutations in a oncogenes, b tumour suppressor genes and c non-coding regions. Bars denote exons, and red triangles depict an example pattern of somatic mutation accumulation across a cancer cohort
Non-coding driver mutations
Many germline variants associated with cancer and other diseases are situated in the non-coding genome (Maurano et al. 2012). In recent years, decreasing genome sequencing costs have enabled the identification of somatic cancer driver mutations in the ~ 98% of the genome that is non-coding. Far fewer non-coding than coding cancer driver mutations have so far been identified, with current examples generally impacting oncogenesis by altering cis-regulation (Fig. 2c).
Non-coding somatic driver mutations may impact transcription factor binding by removing an existing binding motif, or creating a de novo binding site and even an entirely novel regulatory element. For example, the promoter of the TERT gene is mutated in more than 50 cancer types [reviewed in (Bell et al. 2016)]. TERT promoter single nucleotide mutations create a transcription factor binding site that upregulates TERT expression, and were first described in melanoma (Horn et al. 2013; Huang et al. 2013). Other cancer driver mutations in promoter elements have since been discovered, mutating regulatory sites for cancer driver genes such as FOXA1 (Rheinbay et al. 2017). Indels are also able to alter gene cis-regulation by creating or removing transcription factor binding sites [for examples, see (Abraham et al. 2017; Mansour et al. 2014; Rahman et al. 2017)]. On a larger scale, structural variations and copy number aberrations can duplicate, remove or relocate cis-regulatory elements, leading to the dysregulation of enhancer-promoter interactions, and contributing to oncogenesis [for examples, see (Groschel et al. 2014; Zhang et al. 2016)]. In addition to these direct alterations to cis-regulatory elements, the nature of cis-regulation means that these sites are also susceptible to epigenetic dysregulation, through alterations to DNA methylation, nucleosome occupancy or the accessibility of chromatin [reviewed in (Poulos and Wong 2017); please also see this reference for a more comprehensive description of recent efforts undertaken to identify non-coding driver mutations in cancer genomes]. Non-coding driver mutations may also lie outside of cis-regulatory regions, affecting other genomic elements, such as long non-coding RNAs [for example, see (Lanzós et al. 2017)]. Further research efforts will be necessary to fully elucidate the role of non-coding mutations, which may have less clear impacts on cellular function.
Tools for annotating variants to identify driver mutations
A number of computational tools are available for the annotation of putative driver mutations. These tools typically assess a combination of measures in order to determine the likely functional impact of a given variant. Measures of function in the protein-coding genome generally focus on the impact that a somatic variant will have on protein translation, prioritising missense and nonsense mutations over synonymous variants. Measures of function in the non-coding genome generally consider conservation and transcription factor binding motifs, as well as epigenetic features. Table 1 briefly describes a selection of the tools available for the annotation of variants in either the protein-coding or non-coding genome. Many other tools are available for such variant annotation, and this list is not exhaustive. Ultimately, choosing the correct tool for a specific analysis will depend on the downstream applications required.
Positive selection and driver identification
Defining positive selection
Negative selection is common in evolutionary history, but it is rare in cancer development, with only ~ 1% of protein-coding mutations undergoing negative selection in cancer (Martincorena et al. 2017). Instead, positive selection for driver mutations is much more common in oncogenesis. One method commonly used to detect genes undergoing positive selection in coding regions is analysis of the dN/dS ratio, which is a calculation of the ratio of non-synonymous (dN) to synonymous (dS) amino acid substitutions given a certain gene. Researchers can discover cancer driver genes by examining those genes that harbour an excess of non-synonymous mutations. Oncogenes and tumour suppressor genes generally harbour an excess of missense and nonsense mutations, respectively (Martincorena et al. 2017).
Here, we briefly discuss some of the tools that are available for analyses of positive selection in cancer DNA. OncodriveFML (Mularoni et al. 2016) detects positive selection in both coding and non-coding genomic regions by assessing mutation function. e-Driver (Porta-Pardo and Godzik 2014) and OncodriveCLUST (Tamborero et al. 2013a) similarly measure positive selection, specifically examining the internal distribution of variants within a gene to detect domains harbouring an excess of mutations. ActiveDriver (Reimand and Bader 2013; Reimand et al. 2013) is a statistical method that detects positive selection by analysing phosphorylation-associated variants. MuSiC (Dees et al. 2012) relies on measures of mutation recurrence, together with clinical and coverage data in order to statistically evaluate cancer sequencing datasets for potential drivers. Researchers using multiple complementary methods for these types of analyses should detect greater numbers of high-confidence cancer driver events (Tamborero et al. 2013b).
Establishing expected background mutation loads
Mutational processes do not act equally throughout the genome, and certain regions of DNA are more likely to acquire somatic mutations in cancer. For example, lowly expressed genes and regions of heterochromatin are less commonly subjected to transcription-dependent repair mechanisms, and such sites generally accumulate higher mutation loads (Schuster-Bockler and Lehner 2012; Zheng et al. 2014). Similarly, late replicating regions accumulate more mutations, likely due to mismatch repair being less active at such sites (Supek and Lehner 2015), exhaustion of the free nucleotide pool and/or difficulty navigating heterochromatin (Stamatoyannopoulos et al. 2009). Considering mutation rates at smaller scales, exons accumulate fewer mutations than intronic regions due to increased mismatch repair activity at such loci (Frigola et al. 2017). In addition, regions of transcription factor binding, such as at promoter elements or CTCF binding sites, acquire high mutation loads in some cancers because nucleotide excision repair machinery is inhibited from repairing mutagenic DNA lesions (Perera et al. 2016; Poulos et al. 2016; Sabarinathan et al. 2016). At nucleotide resolution, highly methylated cytosines are more often mutated in some cancers, due to the increased tendency for methylated cytosines to deaminate to thymine, and due to particular features of DNA replication and repair at such loci (Poulos et al. 2017).
Driver mutations confer a growth advantage, and they consequently undergo positive selection in a cellular subpopulation. However, accurate inferences of positive selection can be hindered by some of the mutation rate variations described here. It is vital for researchers to understand which combinations of these and other mutational processes may be operative in a given cancer genome. Analyses of this kind are particularly important because researchers typically use the recurrence of a mutation to determine the likelihood of its being a cancer driver, or to select cancer-associated genes. Such analyses can therefore lead to the false-positive identification of cancer driver mutations and genes which simply lie in highly mutated regions of the genome (Lawrence et al. 2013). It should be noted though, that even mutations accumulating due to increased mutability at certain loci may still be driver events. However, by accurately modelling the expected background mutation rates in a cohort under investigation, researchers should be better able to exclude spurious highly mutated regions, instead identifying true driver mutations and genes that will stand out from among the corrected background of passenger mutations.
One commonly used analytical method for calculating mutation rate variation is MutSigCV (Lawrence et al. 2013). This tool combines sample-specific mutation frequency with measures of gene-specific mutation rate, using gene expression and replication timing data (Lawrence et al. 2013). Similar methods have also been developed specifically for analyses of the non-coding genome — such as MutSigNC (Rheinbay et al. 2017) and LARVA (Lochovsky et al. 2015). These tools can assist researchers in the identification of genes that are mutated at low to intermediate frequencies. Though, saturation analyses have demonstrated that even with such models, highly mutated cancer cohorts could require thousands of samples of a single cancer type in order to accurately identify less frequently mutated driver genes (Lawrence et al. 2014).
Tumour heterogeneity and driver identification
Individual cells within a tumour will acquire mutations throughout their lifetime, and the resultant tumour mass will consist of a heterogeneous population of cells (Fig. 1). With the exception of data produced from single-cell sequencing applications, the results of cancer exome or genome sequencing will generally represent the combination of mutation profiles that were present within the subsection of tumour that was sequenced. These mutation profiles can theoretically be separated into distinct clones and subclones, revealing important insights into cancer pathogenesis, and specifically, which coding or non-coding mutations are the drivers that conferred a growth advantage. Research of this kind is particularly important when considering personalised cancer treatments, as mutations that are only present in a small subclone can become key drivers of cancer relapse (Schmitt et al. 2016). Subclones can be identified by analysing copy number-corrected variant allele frequencies for each of the somatic mutations present in a tumour. Mutations in distinct subclones will generally exhibit similar allele frequencies (Yates and Campbell 2012). Some of the tools available for the analysis of cancer clonality include ABSOLUTE (Carter et al. 2012), THetA (Oesper et al. 2013), SubcloneSeeker (Qiao et al. 2014), SciClone (Miller et al. 2014), PyClone (Roth et al. 2014) and SuperFreq (Flensburg et al. 2017). In order to study subclonal heterogeneity in a given cancer sample comprehensively, researchers may require sequencing data from multiple samples from an individual’s tumour [for example, see (Yates et al. 2015)].
Mutational signatures as clues in the cancer genome
One method for understanding and visualising the mutational processes operating in a cancer genome is to generate mutational signatures (Alexandrov et al. 2013a). Mutational signatures represent the frequencies of each type of mutation (C > A, C > G, C > T, T > A, T > C, T > G), together with their flanking nucleotides, and are presented as the counts of the 96 possible trinucleotide mutation combinations. To date, the COSMIC database (Forbes et al. 2015; Forbes et al. 2011) describes 30 distinct mutational signatures that have been identified in cancer samples so far, with each representing the action of a mutational process. For example, signatures have been identified that represent endogenous mutational processes such as defective DNA proofreading following Polymerase Epsilon (POLE) mutation (signature 10), deficient mismatch repair (signature 6) or the action of AID/APOBEC enzymes (signatures 2 and 13) (Alexandrov et al. 2013a). Mutational signatures have also been defined that result from exposure to exogenous mutagens such as cigarette smoke (signature 4) or ultraviolet light (signature 7) (Alexandrov et al. 2013a). A cancer genome will generally harbour mutations arising from a number of different mutational processes, each operating at differing intensities and/or over differing periods of time (Alexandrov et al. 2013b). The final mutational landscape will therefore be combinatorially affected by a number of mutational signatures (Alexandrov et al. 2013b).
By understanding the mutational signatures that are present in a particular cancer, researchers may gain insights into which driver mutation(s) might also be present in that tumour. For example, the presence of signature 10 will not only implicate a likely mutation in the exonuclease domain of POLE, but the modified trinucleotide mutation frequencies that result from POLE mutation may also predispose the cancer to gaining truncating mutations in APC or TP53 (Poulos et al. 2017). In another example, by analysing the DNA of cancers with large numbers of C > T mutations (associated with signature 1, following the deamination of methylated cytosines), researchers uncovered a germline mutation in the DNA glycosylase MBD4 that may predispose cells to subsequently developing certain driver mutations that accelerate oncogenesis (Sanders et al. 2017). Research associating mutational signatures with specific variants may uncover further mutated genes that are responsible for the generation of certain mutational profiles that drive cancer development.
Databases of driver mutations and cancer sequencing data
For researchers seeking robust lists of established cancer driver genes, there are a number of databases available for analysis. Two such databases are the Cancer Gene Census and IntOGen. As previously discussed, the COSMIC database (Forbes et al. 2015; Forbes et al. 2011) hosts the Cancer Gene Census (Futreal et al. 2004), which contains a list of genes, undergoing ongoing curation, that have been well established in cancer development (http://cancer.sanger.ac.uk/census/). Similarly, IntOGen (Gonzalez-Perez et al. 2013) is a web platform that uses annotation tools to provide lists of cancer drivers identified from large cancer sequencing datasets (https://www.intogen.org/). It is worth noting that well-established non-coding driver mutations are still rare in cancer research, and curated databases therefore primarily focus on protein-coding variants. Researchers intending to examine non-coding driver mutations may need to manually examine the literature for such examples [some current examples reviewed in (Cuykendall et al. 2017) and (Poulos and Wong 2017)].
Researchers can also interrogate databases of mutations that have been curated from large-scale cancer sequencing projects. TCGA data is stored at the Genomic Data Commons (GDC), which can be accessed at https://portal.gdc.cancer.gov/ (Grossman et al. 2016). ICGC data is stored at the ICGC Data Portal, which can be accessed at https://dcc.icgc.org/ (Zhang et al. 2011). Both websites provide user-friendly interfaces, allowing searches by gene, cancer type and mutation. Similarly, the COSMIC database (http://cancer.sanger.ac.uk/) contains records of somatic mutations identified in cancer, including manually curated expert data, as well as data from large sequencing projects such as TCGA and ICGC (Forbes et al. 2015; Forbes et al. 2011). cBioPortal (http://www.cbioportal.org/) is another resource that researchers can use to interrogate cancer genomics datasets, via a web interface that allows accessible data visualisation and analysis (Gao et al. 2013).
Future directions in cancer driver discovery
Through the advent of large-scale cancer sequencing projects, many new cancer driver genes and mutations have been identified. This endeavour has been greatly enhanced by the development of new analytical and statistical methods for selecting recurrently mutated loci with an excess of functional variants. However, driver mutations in many cancers have not yet been fully established. Many driver mutations likely lie within cancer driver genes that are yet to be identified (Martincorena et al. 2017), as well as within non-coding regions that have not yet been examined in sufficient detail due to limited sample sizes and availability of epigenomic datasets (Cuykendall et al. 2017; Poulos and Wong 2017). Such mutations may be detected as cancer cohort sizes increase.
The search for driver mutations in cancer genomes is a vital step in the move toward personalised approaches to cancer treatment. By identifying the molecular changes responsible for driving cancer, drugs can be designed that specifically target mutated or dysregulated genes. Further, by defining the mechanisms underlying the formation of such driver events, new strategies may be developed that prevent damage or even enhance repair to commonly mutated regions of DNA.
References
Abraham BJ, Hnisz D, Weintraub AS, Kwiatkowski N, Li CH, Li Z, Weichert-Leahey N, Rahman S, Liu Y, Etchin J et al (2017) Small genomic insertions form enhancers that misregulate oncogenes. Nat Commun 8:14385
Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 7:Unit 7.20
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale A-L et al (2013a) Signatures of mutational processes in human cancer. Nature 500:415–421
Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR (2013b) Deciphering signatures of mutational processes operative in human cancer. Cell Rep 3:246–259
Bell RJA, Rube HT, Xavier-Magalhães A, Costa BM, Mancini A, Song JS, Costello JF (2016) Understanding TERT promoter mutations: a common path to immortality. Mol Cancer Res 14:315–323
Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S et al (2012) Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 22:1790–1797
Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA et al (2012) Absolute quantification of somatic DNA alterations in human cancer. Nat Biotech 30:413–421
Cuykendall TN, Rubin MA, Khurana E (2017) Non-coding genetic variation in cancer. Curr Opin Syst Biol 1:9–15
Dees ND, Zhang Q, Kandoth C, Wendl MC, Schierding W, Koboldt DC (2012) MuSiC: identifying mutational significance in cancer genomes. Genome Res 22:1589–1598
Flensburg C, Sargeant T, Bosma A, Kluin RJC, Kibbelaar RE, Hoogendoorn M, Alexander WS, Roberts AW, Bernards R, de Jong D et al (2017) Dynamic changes in clonal architecture during disease progression in follicular lymphoma. bioRxiv. https://doi.org/10.1101/181792
Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S et al (2015) COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res 43:D805–D811
Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A et al (2011) COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res 39:D945–D950
Frigola J, Sabarinathan R, Mularoni L, Muinos F, Gonzalez-Perez A, Lopez-Bigas N (2017) Reduced mutation rate in exons due to differential mismatch repair. Nat Genet 49:1684–1692
Fu Y, Liu Z, Lou S, Bedford J, Mu XJ, Yip KY, Khurana E, Gerstein M (2014) FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol 15:480
Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR (2004) A census of human cancer genes. Nat Rev Cancer 4:177–183
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:l1
Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Tamborero D, Schroeder MP, Jene-Sanz A, Santos A, Lopez-Bigas N (2013) IntOGen-mutations identifies cancer drivers across tumor types. Nat Meth 10:1081–1082
Goodwin S, McPherson JD, McCombie WR (2016) Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17:333–351
Groschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BA, Erpelinck C, van der Velden VH, Havermans M, Avellino R, van Lom K et al (2014) A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157:369–381
Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, Staudt LM (2016) Toward a shared vision for cancer genomic data. N Engl J Med 375:1109–1112
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Hinkson IV, Davidsen TM, Klemm JD, Kerlavage AR, Kibbe WA (2017) A comprehensive infrastructure for big data in cancer research: accelerating cancer research and precision medicine. Front Cell Dev Biol 5:83
Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K et al (2013) TERT promoter mutations in familial and sporadic melanoma. Science 339:959–961
Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA (2013) Highly recurrent TERT promoter mutations in human melanoma. Science 339:957–959
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protocols 4:1073–1081
Lanzós A, Carlevaro-Fita J, Mularoni L, Reverter F, Palumbo E, Guigó R, Johnson R (2017) Discovery of cancer driver long noncoding RNAs across 1112 tumour genomes: new candidates and distinguishing features. Sci Rep 7:41544
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR (2014) Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505:495–501
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA et al (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499:214–218
Lochovsky L, Zhang J, Fu Y, Khurana E, Gerstein M (2015) LARVA: an integrative framework for large-scale analysis of recurrent variants in noncoding annotations. Nucleic Acids Res 43:8123–8134
Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB et al (2014) Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346:1373–1377
Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, Davies H, Stratton MR, Campbell PJ (2017) Universal patterns of selection in cancer and somatic tissues. Cell 171:1029–1041
Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J et al (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science 337:1190–1195
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F (2016) The Ensembl variant effect predictor. Genome Biol 17:122
Mertens F, Johansson B, Fioretos T, Mitelman F (2015) The emerging complexity of gene fusions in cancer. Nat Rev Cancer 15:371
Miller CA, White BS, Dees ND, Griffith M, Welch JS, Griffith OL, Vij R, Tomasson MH, Graubert TA, Walter MJ et al (2014) SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput Biol 10:e1003665
Mularoni L, Sabarinathan R, Deu-Pons J, Gonzalez-Perez A, López-Bigas N (2016) OncodriveFML: a general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol 17:128
Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194:23–28
Oesper L, Mahmoody A, Raphael BJ (2013) THetA: inferring intra-tumor heterogeneity from high-throughput DNA sequencing data. Genome Biol 14:R80–R80
Perera D, Chacon D, Thoms JA, Poulos RC, Shlien A, Beck D, Campbell PJ, Pimanda JE, Wong JW (2014) OncoCis: annotation of cis-regulatory mutations in cancer. Genome Biol 15:485
Perera D, Poulos RC, Shah A, Beck D, Pimanda JE, Wong JWH (2016) Differential DNA repair underlies mutation hotspots at active promoters in cancer genomes. Nature 532:259–263
Porta-Pardo E, Godzik A (2014) e-Driver: a novel method to identify protein regions driving cancer. Bioinformatics 30:3109–3114
Poulos RC, Olivier J, Wong JWH (2017) The interaction between cytosine methylation and processes of DNA replication and repair shape the mutational landscape of cancer genomes. Nucleic Acids Res 45:7786–7795
Poulos RC, Thoms JAI, Guan YF, Unnikrishnan A, Pimanda JE, Wong JWH (2016) Functional mutations form at CTCF-cohesin binding sites in melanoma due to uneven nucleotide excision repair across the motif. Cell Rep 17:2865–2872
Poulos, R.C., Wong, J.W.H. (2017) cis-regulatory driver mutations in cancer genomes. In eLS (John Wiley & Sons, Ltd), pp. 1–10
Qiao Y, Quinlan AR, Jazaeri AA, Verhaak RGW, Wheeler DA, Marth GT (2014) SubcloneSeeker: a computational framework for reconstructing tumor clone structure for cancer variant interpretation and prioritization. Genome Biol 15:443
Rahman S, Magnussen M, León TE, Farah N, Li Z, Abraham BJ, Alapi KZ, Mitchell RJ, Naughton T, Fielding AK et al (2017) Activation of the LMO2 oncogene through a somatically acquired neomorphic promoter in T-cell acute lymphoblastic leukemia. Blood 129:3221–3226
Reimand J, Bader GD (2013) Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol Syst Biol 9:637
Reimand J, Wagih O, Bader GD (2013) The mutational landscape of phosphorylation signaling in cancer. Sci Rep 3:2651
Rheinbay E, Parasuraman P, Grimsby J, Tiao G, Engreitz JM, Kim J, Lawrence MS, Taylor-Weiner A, Rodriguez-Cuevas S, Rosenberg M et al (2017) Recurrent and functional regulatory mutations in breast cancer. Nature 547:55–60
Ritchie GR, Dunham I, Zeggini E, Flicek P (2014) Functional annotation of noncoding sequence variants. Nat Methods 11:294–296
Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, Ha G, Aparicio S, Bouchard-Côté A, Shah SP (2014) PyClone: statistical inference of clonal population structure in cancer. Nat Methods 11:396–398
Sabarinathan R, Mularoni L, Deu-Pons J, Gonzalez-Perez A, López-Bigas N (2016) Nucleotide excision repair is impaired by binding of transcription factors to DNA. Nature 532:264–267
Sanders MA, Chew E, Flensburg C, Zeilemaker A, Miller SE, al Hinai A, Bajel A, Luiken B, Rijken M, Mclennan T et al (2017) Germline loss of MBD4 predisposes to leukaemia due to a mutagenic cascade driven by 5mC. bioRxiv. https://doi.org/10.1101/180588
Schmitt MW, Loeb LA, Salk JJ (2016) The influence of subclonal resistance mutations on targeted cancer therapy. Nat Rev Clin Oncol 13:335–347
Schuster-Bockler B, Lehner B (2012) Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 488:504–507
Stamatoyannopoulos JA, Adzhubei I, Thurman RE, Kryukov GV, Mirkin SM, Sunyaev SR (2009) Human mutation rate associated with DNA replication timing. Nat Genet 41:393–395
Stratton MR, Campbell PJ, Futreal PA (2009) The cancer genome. Nature 458:719–724
Supek F, Lehner B (2015) Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature 521:81–84
Supek F, Miñana B, Valcárcel J, Gabaldón T, Lehner B (2014) Synonymous mutations frequently act as driver mutations in human cancers. Cell 156:1324–1335
Tamborero D, Gonzalez-Perez A, Lopez-Bigas N (2013a) OncodriveCLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 29:2238–2244
Tamborero D, Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Kandoth C, Reimand J, Lawrence MS, Getz G, Bader GD, Ding L et al (2013b) Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci Rep 3:2650
Tomasetti C, Marchionni L, Nowak MA, Parmigiani G, Vogelstein B (2015) Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc Natl Acad Sci U S A 112:118–123
Tomczak K, Czerwińska P, Wiznerowicz M (2015) The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol 19:A68–A77
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (2013) Cancer genome landscapes. Science 339:1546–1558
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164–e164
Waszak SM, Tiao G, Zhu B, Rausch T, Muyas F, Rodriguez-Martin B, Rabionet R, Yakneen S, Escaramis G, Li Y et al (2017) Germline determinants of the somatic mutation landscape in 2,642 cancer genomes. bioRxiv. https://doi.org/10.1101/208330
Yates LR, Campbell PJ (2012) Evolution of the cancer genome. Nat Rev Genet 13:795–806
Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, Aas T, Alexandrov LB, Larsimont D, Davies H et al (2015) Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med 21:751
Zhang, J., Baran, J., Cros, A., Guberman, J.M., Haider, S., Hsu, J., Liang, Y., Rivkin, E., Wang, J., Whitty, B., et al. (2011) International Cancer Genome Consortium Data Portal—a one-stop shop for cancer genomics data. Database (Oxford) 2011: bar026
Zhang X, Choi PS, Francis JM, Imielinski M, Watanabe H, Cherniack AD, Meyerson M (2016) Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat Genet 48:176–182
Zheng CL, Wang NJ, Chung J, Moslehi H, Sanborn JZ, Hur JS, Collisson EA, Vemula SS, Naujokas A, Chiotti KE et al (2014) Transcription restores DNA repair to heterochromatin, determining regional mutation rates in cancer genomes. Cell Rep 9:1228–1234
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding information
R.C.P is supported by an Australian Government Research Training Program Scholarship. J.W.H.W. is supported by an Australian Research Council Future Fellowship (FT130100096) and a National Health and Medical Research Council Project Grant (APP1119932).
Conflicts of interest
Rebecca C. Poulos declares that she has no conflict of interest. Jason W.H. Wong declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
This article is part of a Special Issue on ‘Big Data’ edited by Joshua WK Ho and Eleni Giannoulatou.
Rights and permissions
About this article

Cite this article
Poulos, R.C., Wong, J.W.H. Finding cancer driver mutations in the era of big data research. Biophys Rev 11, 21–29 (2019). https://doi.org/10.1007/s12551-018-0415-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-018-0415-6