
Abstract
Titin is a filamentous protein spanning the half-sarcomere, with spring-like properties in the I-band region. Various structural, signaling, and mechanical functions have been associated with titin, but not all of these are fully elucidated and accepted in the scientific community. Here, I discuss the primary mechanical functions of titin, including its accepted role in passive force production, stabilization of half-sarcomeres and sarcomeres, and its controversial contribution to residual force enhancement, passive force enhancement, energetics, and work production in shortening muscle. Finally, I provide evidence that titin is a molecular spring whose stiffness changes with muscle activation and actin–myosin-based force production, suggesting a novel model of force production that, aside from actin and myosin, includes titin as a “third contractile” filament. Using this three-filament model of sarcomeres, the stability of (half-) sarcomeres, passive force enhancement, residual force enhancement, and the decrease in metabolic energy during and following eccentric contractions can be explained readily.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
Since the 1950s, muscle contraction and force production have been associated with the relative sliding of the two contractile filaments, actin and myosin (referred to as the sliding filament theory) (Huxley and Hanson 1954; Huxley and Niedergerke 1954), and the cyclic interaction of myosin-based cross-bridges with specialized attachment sites on the actin filaments (referred to as the cross-bridge theory) (Huxley 1957a). However, from the very onset, the cross-bridge theory could not predict well some of the experimentally observed properties in skeletal muscles (Huxley 1957a). For example, the well-recognized and generally accepted residual force enhancement and residual force depression properties of muscles, observed well before the development of the cross-bridge theory (Abbott and Aubert 1952), cannot be predicted without making fundamental changes to the cross-bridge theory (Walcott and Herzog 2008). Furthermore, the stability of myosin filaments in the center of sarcomeres (Iwazumi 1979; Horowits and Podolsky 1987) and that of sarcomeres on the so-called descending limb of the force–length relationship (Zahalak 1997; Novak and Truskinovsky 2014) cannot be predicted with the cross-bridge theory (Iwazumi and Noble 1989; Zahalak 1997), and the forces and metabolic cost predicted by the original cross-bridge theory were much too high for eccentric contractions (Huxley 1957a).
Andrew Huxley, the father of the cross-bridge theory (Huxley 1957a), recognized the shortcomings of his approach. For example, in order to account for the excessive metabolic cost of eccentric contractions, he proposed that there might be multiple cross-bridge cycles for each energy unit hydrolyzed [adenosine triphosphate (ATP)], while for concentric and isometric contractions, the hydrolysis of one ATP molecule was tightly linked to one cross-bridge cycle (Huxley 1957a, 1969; Huxley and Simmons 1971; Rayment et al. 1993). Also, the excessive eccentric forces could be reduced to experimentally observed values by assuming that attached cross-bridges are torn from actin prior to the full completion of the cross-bridge cycle (Huxley 1957a). However, for the residual force enhancement properties of skeletal muscle, Huxley had no solution to offer. In his 1980 book, “Reflections on Muscle,” Huxley acknowledged the insufficiencies of current cross-bridge thinking in eccentric muscle contraction (Huxley 1980). Specifically, he mentions that special features must have evolved that allow the elongation of active muscles to take place without damaging muscles, that these special features allow for explanations of the mechanics and energetics of eccentric contractions, and that force regulation in eccentric contractions bears little relation to what happens in concentric muscle action (Huxley 1980).
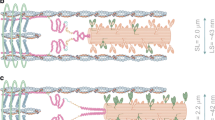
In the mid- and late 1970s, just prior to the publication of Huxley’s book on muscle contraction, titin (initially also referred to as connection) was discovered (Maruyama 1976; Maruyama et al. 1977; Wang et al. 1979). Titin is a filamentous protein spanning the half-sarcomere from the M-band to Z-band (Fig. 1). While thought to be essentially rigidly attached to myosin in the A-band region (except possibly for extreme sarcomere excursions beyond the normal range encountered in typical everyday movements), titin’s I-band structure allows for large elongations and passive force production, and thus has been termed a “spring-like” molecule. Just prior to inserting into the Z-band, titin binds to actin along its most proximal 50 nm, thereby establishing a “permanent” bridge between actin and myosin (Trombitas and Pollack 1993; Linke et al. 1997; Trombitas and Granzier 1997): a bridge that is in parallel with attached cross-bridges and in series with the myosin filament in the passive muscle. With an estimated six titin filaments per half myosin (Granzier and Irving 1995; Cazorla et al. 2000; Liversage et al. 2001; Granzier and Labeit 2007), there is one titin for each actin filament in vertebrate skeletal muscles where actin filaments surround myosin in a hexagonal array (Huxley 1953b, 1957).

Schematic two-dimensional illustration of a sarcomere bordered by Z-bands at either end. Thick, myosin-based filaments are in the center of the sarcomere (green), thin, actin-based filaments insert into the Z-band at either end of the sarcomere (red), and titin filaments (blue) run from the M-line in the middle of the sarcomere to the Z-band. Adapted from Granzier and Labeit (2007) with permission
Ever since its discovery, titin’s functions have been questioned, and titin’s recently proposed roles in active force regulation and mechanical work in muscle shortening are current topics of intense debate in the scientific community. At the recent European Muscle Conference (September 2017), debates on the functional role of titin ended inconclusively. Here, I will attempt to summarize both the acknowledged and the controversial aspects of titin’s mechanical functions, with an emphasis on titin’s proposed role in active force regulation and mechanical work.
Titin’s proposed functions
Titin has been associated with a variety of functions, including mechanical roles in active and passive force regulation in cardiac and skeletal muscles (e.g., Linke et al. 1994, 1996; Granzier and Labeit 2002, 2007; Linke and Fernandez 2002; Herzog et al. 2006; LeWinter and Granzier 2010; Herzog 2014a), structural and developmental roles in sarcomere organization (e.g., Linke and Fernandez 2002; Granzier and Labeit 2007), and functions associated with mechano-sensing and signaling (Schwarz et al. 2008; Kruger and Linke 2009; Linke and Kruger 2010; Granzier et al. 2014). Here, I primarily focus on the proposed mechanical functions of titin in skeletal muscles, although some comparisons with cardiac muscles will be made. However, many extensive reviews on titin’s properties in cardiac muscle have been published recently (Granzier and Labeit 2002, 2007; Granzier et al. 2002), whereas comparatively little has been said on titin’s mechanical role in skeletal muscle.
Passive force contributions of titin
It is generally accepted that titin contributes to the passive forces in skeletal and cardiac muscles. Passive force is defined here as any force that arises from structural elements of muscle, is not associated with metabolic energy consumption, and is not part of the actin–myosin-based cross-bridge forces. The primary passive force contributors in cardiac and skeletal muscles are collagen filaments embedded in the various connective tissue layers of muscles, and the sarcomeric filament titin. In isolated myofibril preparations, titin is the primary passive force contributor (Maruyama 1976; Funatsu et al. 1990; Bartoo et al. 1997; Colomo et al. 1997; Linke and Fernandez 2002; Joumaa et al. 2008b; Leonard and Herzog 2010; Herzog et al. 2012), and the elimination of titin abolishes virtually all passive force (e.g., Leonard and Herzog 2010) .
In skinned single fibers and myofibrils, passive force and titin isoforms are tightly related. Increasing molecular weight, and thus increasing titin subunits and length, are associated with decreasing passive forces. Prado et al. (2005) determined the molecular weights of titin in 37 rabbit skeletal muscles and compared the molecular weights to the passive forces in myofibrils, skinned fibers, and intact and skinned fiber bundles. These authors found a strong inverse relationship between the size of titin and passive force in myofibrils and skinned fibers, i.e., the greater the molecular weight of titin, the smaller the corresponding passive force for a given sarcomere length. However, titin size was not associated in any systematic manner with the passive force in intact fiber bundles (and thus the entire muscle), and the contribution of titin to the total passive force in fiber bundles varied considerably between muscles, ranging from a high of 57% in rabbit psoas (Granzier et al. 2002) to a low of 24% for soleus in the range of 2.0 to 3.2 μm/sarcomere (Prado et al. 2005).
Prado et al. (2005) also found that the contribution of titin to passive muscle force depends on the length of the muscle (i.e., the average sarcomere length). This result agrees with observations in cardiac muscle where titin is thought to contribute more substantially to the passive forces at short (average sarcomere length 2.0–2.2 μm) compared to long (> 2.3 μm/sarcomere) sarcomere length (Cazorla et al. 2000; Freiburg et al. 2000; Granzier et al. 2002). Since the physiologic cardiac cycle occurs between sarcomere lengths that range from approximately 1.9 to 2.3 μm (Ter Keurs et al. 1980), titin plays a significant role in the beating heart.
Whether titin plays an equally important role within the functional range of skeletal muscles has not been determined systematically. It has been observed that in rabbit psoas myofibrils, passive, titin-based forces start to emerge at average sarcomere lengths of approximately 2.6–2.7 μm (Linke et al. 1996; Bartoo et al. 1997; Joumaa et al. 2007; Leonard and Herzog 2010). However, our group measured the shortest (hip fully flexed) and longest (hip fully extended) sarcomere length for rabbit psoas muscles as 1.9 and 2.6 μm, respectively. Knowing that rabbits never fully extend their hip, the maximal sarcomere lengths are probably never reached in the live animal. Furthermore, our measurements were performed on the passive muscle in the anesthetized animal, while in the active muscle, a substantial fiber and sarcomere length shortening would be expected with force production, as elastic elements are stretched and the contractile machinery shortens (Fukunaga et al. 1997; Ichinose et al. 1997; Vaz et al. 2012; de Brito Fontana and Herzog 2016). Therefore, maximal sarcomere lengths in the rabbit psoas likely never exceed about 2.3–2.4 μm, and thus are below the sarcomere lengths where passive titin forces have been first observed. A similar argument could be made for the rabbit soleus and medial gastrocnemius muscles. Thus, it appears that titin passive force does not play a functional role in many skeletal muscles. Whether this statement can be generalized is not yet known, as the functional sarcomere length of most animal muscles are not known. However, should skeletal muscle functional sarcomere length reach values in excess of 2.6 μm, then titin would likely contribute to the passive force of intact muscles (Prado et al. 2005). Also, in the following text I discuss titin’s possible role in shifting its slack length upon muscle activation to shorter sarcomere lengths than those observed in the passive muscle, which could potentially change the argument made here, with titin possibly emerging as a powerful passive force contributor in active skeletal muscles after all (Herzog 2014b; Herzog et al. 2015, 2016).
Titin’s stiffness, and thus passive force at a given sarcomere length, can be modulated in a variety of ways, such as calcium binding to titin upon muscle activation (Labeit et al. 2003; Joumaa et al. 2008b; DuVall et al. 2013), titin phosphorylation (Yamasaki et al. 2002; Borbely et al. 2009; Anderson et al. 2010; Perkin et al. 2015), and titin interactions with actin and other sarcomeric proteins (Li et al. 1995; Linke et al. 1997, 2002; Trombitas and Granzier 1997; Astier et al. 1998; Kulke et al. 2001; Yamasaki et al. 2001; Nagy et al. 2004; Bianco et al. 2007; Leonard and Herzog 2010; Chung et al. 2011), to just name the most common mechanisms. Some of these mechanisms will be discussed below in the section on residual force enhancement properties of skeletal muscles in general (Abbott and Aubert 1952; Edman et al. 1982; Herzog et al. 2006) and the passive residual force enhancement specifically (Herzog and Leonard 2002). Excellent reviews on the regulation of titin’s stiffness in cardiac muscle are abundant (e.g., Granzier and Labeit 2002; Granzier et al. 2002; Linke and Fernandez 2002; LeWinter and Granzier 2010), and these results will not be repeated here, except for comparative purposes.
Titin as a stabilizer of sarcomeres and half-sarcomeres
In the two-filament (actin and myosin) cross-bridge model of muscle contraction, half-sarcomeres and sarcomeres are unstable (Morgan 1990, 1994; Allinger et al. 1996; Zahalak 1997; Epstein and Herzog 1998; Morgan et al. 2000; Novak and Truskinovsky 2014). The half-sarcomere is unstable because small differences in half-sarcomere forces, caused by the stochastic interaction of cross-bridges with actin, will cause an initially centered myosin filament to be displaced from its mid-point position in the sarcomere. This perturbation is unstable as the overlap between actin and myosin will increase in the half-sarcomere of initial myosin drift and thus make this half-sarcomere increasingly stronger, while in the other half-sarcomere, the actin–myosin filament overlap is lost, and force continuously decreases (Iwazumi 1979). An analogous argument can be made for the instability of serially arranged sarcomeres (for example, as they occur in a myofibril; Gordon et al. 1966; Campbell 2009; Stoecker et al. 2009) that are located on the descending limb of the force–length relationship (Hill 1953).
A-band shifts to one end of the sarcomere were observed after prolonged activation in rabbit psoas skinned fibers with an average sarcomere length of 2.6 μm and zero passive force (Horowits and Podolsky 1987, 1988). These shifts, as well as non-uniformities in associated half-sarcomere lengths, were abolished at average sarcomere lengths of about 3.0 μm and a passive stress of approximately 2 N/cm2, corresponding to approximately 20% of the maximum, active, isometric force of these fibers at optimal sarcomere length and 7 °C. The A-band shifts observed in these studies were interpreted as indicating myosin instability in the center of the sarcomere when titin forces were zero or small, while myosin was stabilized in the center of the sarcomere once titin forces had reached a certain passive force level corresponding to approximately 2 N/cm2 (e.g., Horowits 1992). The same observations were made for rabbit soleus fibers, with the notable difference that A-band shifts were greater in soleus than in psoas fibers at corresponding sarcomere lengths and that half-sarcomere uniformity and stability (absence of A-band shifts) were only observed at average sarcomere lengths of about 3.6 μm when passive, titin-based tension had reached 2 N/cm2. The explanation for this result was based on the smaller size of the titin isoforms for rabbit psoas (3.3 and 3.4 MDa in a 70:30% ratio) compared to that of the single titin isoform observed in rabbit soleus (3.6 MDa) (Prado et al. 2005), resulting in reduced titin-based passive forces at a given sarcomere length for rabbit soleus compared to psoas.
Instability of muscles and sarcomeres on the descending limb of the force–length relationship has been used to explain specific muscle properties for more than half a century (Hill 1953; Gordon et al. 1966; Julian et al. 1978; Julian and Morgan 1979). For example, the so-called “creep” property, which is a slow change in isometric force for muscles/fibers on the descending limb of the force–length relationship (Hill 1953; Gordon et al. 1966), and the residual force enhancement and residual force depression properties (Morgan 1990, 1994) have been thought to be caused by instabilities in sarcomere length and the associated development of sarcomere length non-uniformities in active muscles, particularly if the muscles were stretched onto the descending limb of the force–length relationship. Indeed, vast sarcomere length non-uniformities have been observed in entire muscle preparations (Llewellyn et al. 2008; Moo et al. 2016), single fibers (Huxley and Peachey 1961), and isolated myofibrils (Rassier et al. 2003a; Joumaa et al. 2008a; Johnston et al. 2016). However, these sarcomere length non-uniformities occur at all lengths (ascending, plateau, and descending portions of the force–length relationship; Moo et al. 2016) and similarly in isometric, shortening, and stretched muscles (Johnston et al. 2016); thus, they do not seem to be associated with the proposed instability of the negative slope of the descending limb of the force–length relationship.
Surprisingly, Rassier et al. (2003a, b) observed that strictly serially arranged sarcomeres in single, active myofibrils stretched onto the descending limb of the force–length relationship are highly non-uniform, but perfectly stable. Sarcomeres half-way down the descending limb of the force–length relationship, and thus at half of the maximal actin–myosin filament overlap, were perfectly stable and remained at a constant length in the presence of sarcomeres on the plateau of the force–length relationship with maximal overlap between actin and myosin (Fig. 2). It would appear, therefore, that sarcomere length, and thus actin–myosin filament overlap, alone does not predict the isometric force of a sarcomere for a given level of activation within a myofibril. Rather, since the force in serially arranged sarcomeres must be identical (neglecting any inertial effects, which can safely be done), there must be another way other than just actin–myosin filament overlap to determine the isometric, steady-state force of a sarcomere within its natural environment of a myofibril. Active or passive stabilizing mechanisms must ensure that the weakening behavior of sarcomeres on the descending limb of the force–length relationship is compensated for in some (as of yet unknown) manner to ensure stability of the muscle.

Rabbit psoas myofibril comprised of six sarcomeres that is stretched while activated from an average sarcomere length of about 2.4 μm to about 3.0 μm. After active stretching, all individual sarcomeres are on the descending limb of the force–length relationship, but there is no apparent overstretching or popping (quick sarcomere elongations beyond actin–myosin filament overlap: 3.9 μm) as has been proposed by proponents of the sarcomere length non-uniformity theory. Rather, sarcomeres seem to remain at an essentially constant length following the active stretch
Titin has been implicated as a stretch sensor that activates so-called “super-relaxed” cross-bridges, thereby providing a mechanism by which a disadvantage caused by reduced actin–myosin filament overlap (long sarcomere length) can be compensated for by an increased proportion of attached cross-bridges per unit length of myofilament overlap (Fusi et al. 2016). Titin would be an ideal candidate for regulating cross-bridge kinetics in sarcomeres of different lengths, thereby guaranteeing stability. Also, since titin in adjacent half-sarcomeres overlap in the M-band and the Z-band regions (Granzier and Labeit 2007), it is easy to imagine that force transmission across the M-band and Z-band is continuous, thus providing possibilities for force transfer within half-sarcomeres and between sarcomeres. Such a passive, structural force transmission between half-sarcomeres would also provide sarcomere length/force stability and would eliminate the rather odd notion that sarcomeres are the smallest “independent” unit of force production. Muscle mechanics would be simplified greatly if serial sarcomeres in a myofibril were indeed not “independent” but rather mutually dependent force producers, such that forces can be transmitted and “re-distributed” along serially arranged sarcomeres. Aside from structural evidence for such mutual dependence among sarcomeres, there is also functional support for this notion (e.g., Yasuda et al. 1996).
Titin’s role in the residual force enhancement property of skeletal muscle
When an active muscle is stretched, its isometric, steady-state force following the stretch is greater than the corresponding (same length, same activation) purely isometric contraction. This increase in force caused by active stretching has been termed residual force enhancement (RFE) (Edman et al. 1982). RFE can produce forces twice as high as purely isometric contractions (Edman et al. 1982; Leonard and Herzog 2010; Leonard et al. 2010). Force in the enhanced state can easily exceed the isometric force at the plateau of the force–length relationship (Abbott and Aubert 1952; Rassier et al. 2003c; Peterson et al. 2004; Leonard et al. 2010). RFE increases with increasing stretch magnitude (Bullimore et al. 2007; Hisey et al. 2009) but is independent of stretch speed (Edman et al. 1982), is associated with a substantial decrease in the metabolic cost of force production (Joumaa and Herzog 2013), is long-lasting (Abbott and Aubert 1952; Herzog and Leonard 2002; Leonard et al. 2010), and has a passive component that contributes substantially to the enhanced force (Fig. 3a, b) (Herzog and Leonard 2002, 2005; Herzog et al. 2003; Joumaa et al. 2007, 2008b).

Residual force enhancement observed in whole muscle (cat soleus; a), single myofibrils (rabbit psoas; b), and single sarcomeres (rabbit psoas; c). Note the increase in force enhancement (FE; a) with increasing stretch magnitude, and the increased passive force [passive force enhancement (PFE)] following deactivation of the muscles after an active stretch (a, b). Note also the vast FE in a single sarcomere (c) and the substantially greater force after active stretching compared to the isometric, steady-state force prior to stretching which occurred at the plateau of the force–length relationship (2.4 μm)
RFE was first studied and described systematically in 1952 (Abbott and Aubert 1952), and it has subsequently been observed consistently across all structural levels of muscle, from entire muscles (Fig. 3a) (Abbott and Aubert 1952; Lee and Herzog 2002; Oskouei and Herzog 2005; Hahn et al. 2010; Seiberl et al. 2012; Fortuna et al. 2016), to single fibers (Edman et al. 1982; Sugi and Tsuchiya 1988; Peterson et al. 2004; Lee and Herzog 2008) and myofibrils (Fig. 3b) (Rassier et al. 2003a, b; Leonard and Herzog 2010; Leonard et al. 2010; Pun et al. 2010; Rassier and Pavlov 2012), and even in single sarcomeres (Fig. 3c) (Leonard et al. 2010).
For most of the twentieth century, RFE was explained by the instability of sarcomere lengths and the associated development of sarcomere length non-uniformities when muscles are stretched onto the descending limb of the force–length relationship (Morgan 1990, 1994; Edman and Tsuchiya 1996; Morgan et al. 2000; Morgan and Proske 2004, 2006). The concepts of the so-called sarcomere length non-uniformity theory have been well discussed (for review, see Morgan 1994; Herzog et al. 2016) and will not be repeated here, with the exception of experimental results that demonstrate that this theory cannot explain most of the fundamental RFE properties of skeletal muscles.
The most basic predictions of the sarcomere length non-uniformity theory that have been shown to be violated are that:
-
(1)
RFE cannot occur on the ascending limb of the force–length relationship—but it actually does (Abbott and Aubert 1952; Rassier et al. 2003c; Peterson et al. 2004; Lee and Herzog 2008);
-
(2)
Forces in the RFE state cannot exceed the purely isometric forces measured in a muscle at optimal length on the plateau of the force–length relationship—but they do (Fig. 4) (Rassier et al. 2003c; Lee and Herzog 2008; Leonard et al. 2010);
-
(3)
RFE (by definition of the sarcomere length non-uniformity theory) cannot occur in a single sarcomere—but it does (Figs. 3c, 4) (Leonard et al. 2010);
-
(4)
The isometric reference contractions are achieved with essentially uniform sarcomere lengths—but they are not (Fig. 2) (Llewellyn et al. 2008; Johnston et al. 2016; Moo et al. 2016);
-
(5)
There is a distinct increase in sarcomere length non-uniformity, resulting in two distinct groups of sarcomere lengths when a muscle is stretched actively—but that is not the case (Fig. 2) (Joumaa et al. 2008a; Johnston et al. 2016)

Steady-state isometric forces obtained in single, mechanically isolated sarcomeres (rabbit psoas) at sarcomere lengths of 2.4 μm [optimal length = 100% force (filled brown circle)] and 3.4 μm [approximately 50% of maximal isometric force at the plateau length (filled blue diamonds and filled black square = mean force). Also shown are the isometric steady-state forces of these isolated sarcomeres following a stretch from 2.4 to 3.4 μm (filled green triangles and filled black circle). FE Mean force enhancement observed in these sarcomeres, OFE the mean force above the maximal, isometric plateau forces for these sarcomeres. Note the enormous force enhancement and the consistently greater forces in the enhanced state compared to the plateau force. Adapted from Leonard et al. (2010) with permission
For these reasons, it would appear that the sarcomere length non-uniformity theory has little direct support. It will not be discussed further in this review, but interested readers may want to consult the following references for a more in depth treatment of this theory (Morgan 1990, 1994; Herzog 2014a, b; Herzog et al. 2015).
RFE increases with increasing stretch magnitude (Edman et al. 1982; Bullimore et al. 2007; Hisey et al. 2009), is independent of stretch speed (Edman et al. 1982), and is long lasting (Abbott and Aubert 1952; Leonard and Herzog 2010); however, it can be abolished instantaneously if activation of the muscle is interrupted for long enough for the force to drop to zero (Morgan et al. 2000). These properties gave early rise to the notion that RFE might be caused by the engagement of an elastic structural element upon muscle activation (Forcinito et al. 1998). Because of its location, attachment, and mechanical properties, titin was an early favorite for this role (Noble 1992). Simple modeling of passive structural element engagements upon activation allowed for excellent qualitative predictions of the RFE properties (Forcinito et al. 1998). However, direct proof of a passive element playing a role in RFE was missing for a long time.
In 2002, our research group discovered in experiments with cat soleus muscle that RFE was associated with a distinct increase in passive force (Herzog and Leonard 2002). Specifically, we demonstrated that an active stretch resulted in a substantial increase in the passive force (following the active stretch and following deactivation of the muscle) compared to the passive forces measured when the muscle was activated isometrically at the corresponding length or stretched passively to the final length (Fig. 5) (Herzog and Leonard 2002). We termed this increase in passive force following active muscle stretching “passive force enhancement” (PFE) and showed that PFE was long-lasting and increased with stretch magnitude and with final muscle length, but that it could be abolished instantaneously by a quick release of the muscle to a short (prior to stretch) length (Herzog et al. 2003). These studies provided the first direct evidence that a passive component was likely contributing to the RFE property of skeletal muscles.

Force–time histories of cat soleus muscle stretched passively (lowest trace at 6 s), stretched actively (highest trace at 6 s), and activated isometrically at the final stretch length (middle trace at 6 s). Note the increased passive force following muscle deactivation (at 12 s) for the actively stretched muscle, compared to the passively stretched muscle and the muscle activated isometrically at the final stretch length. The increase in passive force following active muscle stretching (here seen at 12 s) was termed passive force enhancement (PFE). Adapted from Herzog and Leonard (2002) with permission
Subsequently, PFE was also observed in isolated myofibrils and sarcomeres (Fig. 3b) (Joumaa et al. 2007, 2008b). Since titin is the primary passive force producing structure in myofibrils (it produces in excess of 95% of the passive force), titin became directly implicated in the mechanisms causing PFE and contributing substantially to the total RFE (Herzog et al. 2006; Leonard and Herzog 2010; Powers et al. 2014).
How might titin contribute to the residual force enhancement?
If titin were to contribute to the PFE and the RFE, its force for a given muscle/sarcomere length would need to be greater when a muscle is stretched actively compared to when it is stretched passively. Such an increase in force could be achieved if titin was stiffer in an active muscle than in a passive muscle. There are two basic mechanisms by which titin (or any molecular spring) can increase its stiffness: (1) it can increase its inherent stiffness (a change in material property) or (2) it can shorten its spring length (resting length) while its material property remains unaltered.
Changes in the inherent stiffness of titin upon activation may occur if calcium binds to titin, thereby increasing its stiffness and force upon stretching. Labeit et al. (2003) showed that there was an approximately 20% increase in non-cross-bridge-based force in skinned mouse soleus fibers activated with calcium ([PCa 4.0]) whose cross-bridge forces were inhibited by 2,3-butanedione monoxime (BDM) compared to fibers stretched passively ([PCa 9.0]). Single molecule experiments with PEVK fragments of titin suggested that specific E-rich motifs in PEVK can bind calcium, thereby becoming stiffer. Our research group showed, using fluorescence spectroscopy, that I27 cardiac immunoglobulin domains also bind calcium in a dose-dependent and reversible manner and demonstrated, with atomic force microscopy, that unfolding of these domains required 20–25% more force in the presence of physiologically relevant concentrations of calcium compared to the passive state with low calcium concentration [PCa 8] (Fig. 6) (DuVall et al. 2013). Further experiments in which single myofibrils were activated with calcium but whose cross-bridge forces were inhibited (with BDM and/or troponin C deletion) also showed an increase in titin-based force (Joumaa et al. 2008b; Leonard and Herzog 2010) compared to low calcium (passive) conditions, as did experiments in which myofibrils were stretched actively and passively beyond the actin–myosin filament overlap (Leonard and Herzog 2010; Powers et al. 2014). These experiments all led to the conclusion that titin is indeed a spring whose stiffnessis changed by calcium, and thus, muscle activation. Aside from calcium activation, titin’s spring stiffness can also be changed with phosphorylation (Yamasaki et al. 2002; Borbely et al. 2009; Anderson et al. 2010; Hudson et al. 2010; Perkin et al. 2015) and disulfide bridging (Scott et al. 2002), among others. In summary, titin is a molecular spring whose inherent stiffness can be modulated by muscle activation. However, changes in the inherent stiffness of titin only seem to account for up to about 20% of the maximal RFE observed experimentally under optimal conditions (Granzier 2010; Leonard and Herzog 2010). Therefore, there must be other mechanisms to explain the remainder of the RFE.

Unfolding force of the first five (out of 8 identical) cardiac I27 immunoglobulin (Ig) domains of titin. Note that unfolding of the I27 Ig domains in the absence of calcium (Control) required about 20% less force than in the presence of calcium (Calcium). Adapted from DuVall et al. (2013) with permission
Another way of increasing titin’s spring stiffness that may potentially account for the full RFE observed experimentally, is a change in titin’s resting or free spring length. A decrease in free spring length could be achieved theoretically if some of titin’s extensible domains were to become inextensible—for example, by binding to a more rigid structure. In vitro, titin fragments have been found to bind to actin (Linke et al. 1997, 2002; Yamasaki et al. 2001; Nagy et al. 2004; Li et al. 1995; Trombitas and Granzier 1997; Astier et al. 1998; Kulke et al. 2001; Bianco et al. 2007; Chung et al. 2011;), thereby slowing the progress of actin–myosin filament sliding in motility assays. However, functionally relevant binding seems to be restricted to cardiac titin’s PEVK domain, which has been found to interact with actin in a calcium-dependent manner (Kulke et al. 2001; Yamasaki et al. 2001). Specifically, calcium seems to release actin from titin, thereby reducing titin-based passive force and stiffness and facilitating the cardiac cycle (Yamasaki et al. 2001). Permanent titin–actin binding has been shown to occur in the most proximal titin segments near the Z-band (Trombitas and Pollack 1993; Trombitas and Granzier 1997). Together with the relatively rigid association of titin with myosin, this titin link to actin results in a passive molecular spring that is arranged in parallel with attached cross-bridges, while it is in series with the myosin filament and the actin filament near its insertion into the Z-band in the relaxed muscle. However, in vitro assays have largely excluded strong binding of skeletal titin sub-fragments to actin (Kulke et al. 2001; Yamasaki et al. 2001).
Antibody labeling of titin demonstrates that titin is extensible along its entire I-band length (except for titin’s most proximal 50–100 nm) in passive muscle (Horowits et al. 1989; Trombitas et al. 2000; Minajeva et al. 2001; Linke and Fernandez 2002). Horowits et al. (1989) used monoclonal antibodies that bind in the I-band region of titin to observe distal and proximal (relative to the antibody) titin segment elongation in passive and active rabbit psoas fibers. Passive fibers were analyzed for sarcomere lengths ranging from 2.1 to 3.8 μm, while activated fibers were kept at a constant sarcomere length of 2.6 μm (fibers were activated for 5 min to produce A-band shifts to one side of the sarcomere, thus compressing one-half of the sacromere and extending the other half). These authors found that in both passive and active fibers, the lengths of the proximal and distal titin segments depended only on the length of the half-sarcomere, leading them to conclude that activation did not change the mechanical properties of titin (Horowits et al. 1989).
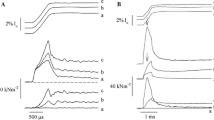
In contrast, DuVall et al. (2017), using a variety of I-band-specific titin antibodies, found that stretching of rabbit psoas myofibrils resulted in different segmental elongations in the active and passive conditions. While these authors replicated previous results for segmental titin elongations for passive myofibril stretching (Horowits et al. 1989), they observed that proximal segments of titin stopped elongating after a short stretch amplitude in active conditions (Fig. 7). They interpreted their results with an activation- and stretch-induced entanglement of titin with myosin or cross-bridges that rendered some of the extensible distal regions of titin inextensible. However, an equally valid explanation would be that titin’s proximal segments bind to actin after a short stretch, thereby rendering the proximal segments inextensible. Since elongations of proximal titin segments stop occurring at sarcomere lengths as short as 2.3 μm, which is a sarcomere length at which titin is known to be unstrained in rabbit psoas myofibrils, this latter explanation seems the more feasible of the two. If correct, such titin–actin interactions in actively stretched muscles change titin’s free spring length, thereby increasing titin’s stiffness and consequently its force upon sarcomere elongation (Herzog 2014a). Theoretical modeling of titin–actin interactions, as observed experimentally (DuVall et al. 2017), demonstrate that even the largest RFE observed experimentally (e.g., Leonard et al. 2010) can be explained using titin binding to actin in actively stretched muscle (Schappacher-Tilp et al. 2015).

Passive (a) and active (b) stretching of proximal titin segments labeled using an antibody [F146 that binds to the PEVK region (diamond symbols)] region that allows for measurements of proximal and distal titin segment elongations during passive and active stretching of single rabbit psoas myofibrils. Figures on the left show elongation of the half-sarcomere [top traces (circular symbols) using an M-line label) and elongations of the proximal titin segment (bottom traces: from X-band to F146 label) for two representative sarcomeres from two different myofibrils. Note in a (passive stretching) that the two proximal titin segments elongate continuously with half-sarcomere elongations, reaching final lengths of approximately 0.95 μm (at a sarcomere length of 4.0 μm) and about 0.6 μm (at a sarcomere length of about 3.5 μm). In contrast, when the myofibrils are stretched while activated, the proximal segments elongate similarly to the elongations observed in the passive condition, but then stop elongating and remain substantially shorter than in the passive case (i.e., with a length of about 0.6 and 0.35 μm, respectively). The panels on the right illustrate schematically what we believe might be happening. In the passive stretch (a), the proximal and distal titin segments elongate in accordance with their stiffness properties. In the active stretch (b), titin is thought to attach to actin at some point, thereby shortening titin’s free spring length, increasing its stiffness, eliminating elongation of proximal titin, and increasing titin-based force
The results by DuVall et al. (2017) do not agree with those observed by Horowits et al. (1989). However, in the DuVall study (DuVall et al. 2017), some sarcomere stretching was required prior to the loss of proximal titin segment elongation, while in the Horowits study (Horowits et al. 1989), sarcomeres were kept at a constant isometric length. Also, in the DuVall study, segmental elongations were measured continuously during the active and passive stretching, while in the Horowits study, the active and passive fibers were fixed at specific lengths, and thus measurements of titin segment lengths were not continuous, but were made at a single (average) sarcomere length following fixation. It is not known how fixation might affect possible titin-to-actin binding in active fibers, and if indeed some sarcomere stretching is required to initiate titin–actin interaction; both these aspects might have been missed in the Horowits experiments.
Linke et al. (1996) used epitope tracking to determine the elongations of specific titin segments in passive rabbit cardiac myofibrils and in rabbit soleus and psoas myofibrils. Their results using an N2A-based epitope do not mimic those by DuVall et al. (2017) for passive stretching of titin segments in rabbit psoas. While they found that proximal titin segment elongation stopped at average sarcomere lengths of about 2.5 μm with little if any further elongation, DuVall et al. (2017) found continuous elongations of proximal titin segments up to a sarcomere length of 4.0 μm (Fig. 7). Leonard and Herzog (2010) showed that titin-based forces were much greater in psoas myofibrils stretched actively than in those stretched passively from optimal lengths to lengths beyond the actin–myosin filament overlap (Fig. 8). When cross-bridge interaction in the active [pCa 3.5] myofibrils was inhibited by BDM or troponin C deletion (Joumaa et al. 2008b), titin-based forces were still greater than in myofibrils under passive [pCa 8.5] conditions but much smaller than those when cross-bridge interactions with actin were allowed to occur normally (Leonard and Herzog 2010; Leonard et al. 2010). Starting active stretching from a longer length (3.4 μm) than optimal (2.3–2.5 μm) also decreased the titin-based forces compared to when stretches were initiated at optimal length (Leonard and Herzog 2010). Although the molecular mechanisms associated with segment-specific changes in titin’s mechanical properties need further elucidation, it appears that changes in the titin’s mechanical properties that occur with muscle activation and stretching contribute substantially to the experimentally observed RFE and PFE in skeletal muscles. Furthermore, the results by Leonard and Herzog (2010) suggest that it is not calcium activation that produces these substantial changes in passive/titin-based forces; rather, active cross-bridge binding is required to observe substantial force enhancement. A possible explanation could be that strong cross-bridge binding causes a movement of the regulatory proteins (troponin and tropomyosin) that may expose titin binding sites on actin that cannot be accessed by titin in the passive muscle or in calcium-activated muscle in which strong cross-bridge binding to actin is inhibited.

Stress (force/cross-sectional area) versus sarcomere length relationship for single rabbit psoas myofibrils stretched from an average sarcomere length of 2.0 μm to 6.0 μm. Myofibrils were stretched passively (Passive), actively (Active), actively from an average sarcomere length of 3.4 μm (Half-Force), and after deletion of titin (No Titin). Actin myosin filament overlap is lost at an average sarcomere length of about 4.0 μm (indicated by the shaded area). According to the cross-bridge theory, one would expect the forces beyond actin myosin filament overlap (non-shaded area) to be purely passive and the same for all conditions with intact titin filaments. However, the forces in the actively stretched myofibrils were substantially greater than those for the passively stretched myofibrils in the area where myofilament overlap was lost. Deactivation of selected myofibrils at an average sarcomere length of 5.0 μm did not result in a loss of force (results not shown), indicating that there was no remnant cross-bridge-based force at these lengths. Elimination of titin from the myofibrils abolished all passive and all active force in myofibrils, indicating that titin is not only an essential protein for passive force production but is absolutely essential for active force transmission from the cross-bridges to the Z-bands and for centering the myosin filaments in the sarcomere. Adapted from Leonard and Herzog (2010) with permission
Stretching psoas sarcomeres to very long lengths, i.e., in excess of 5.0 μm, has been criticized for its potential to damage sarcomeres permanently by dislodging titin from its attachment to the Z-band or the myosin filament. However, Herzog et al. (2012) found that stretching of myofibrils to an average sarcomere length of > 5 μm (and individual sarcomeres within these myofibrils up to 6 μm) was not associated with permanent damage (i.e., loss of force). Rather, given sufficient time (about 10 min) and recovery at a very short average sarcomere length (1.8–2.0 μm), full recovery of titin-based passive force was observed, indicating that for stretches of sarcomeres up to 5.0 μm, there does not appear to be permanent damage to the structural network of (rabbit psoas) sarcomeres that would prevent full force recovery (Fig. 9). The fact that force recovery required several minutes and needed to be done at short sarcomere lengths (recovery for < 10 min or at sarcomere lengths averaging ≥ 2.6 μm was always incomplete) was interpreted as indicating that titin immunoglobulin unfolding, which would have taken place at these long sarcomere lengths (Kellermayer et al. 1997; Herzog et al. 2012), is slow and only occurs when forces acting on titin are essentially zero (Kellermayer et al. 1997).

Examples of two separate rabbit psoas myofibrils that were repeatedly stretched to sarcomere lengths beyond actin myosin filament overlap. Note that in both cases repeat stretches did not result in a decrease in peak force or loading energy, thus indicating that even stretching to lengths up to 5.0 μm did not result in permanent damage and loss of force. a Myofibril stretched to an average sarcomere length of approximately 5.2 μm (stretch 1), then shortened and rested for 10 min at an average sarcomere length of 2.6 μm and re-stretched (stretch 2). Two sets of three stretch-shortening cycles were performed and the third stretch of the first set (stretch 1), and the first stretch of the second set (stretch 2) are shown b Myofibril stretched to an average sarcomere length of approximately 4.2 μm, then shortened and rested for 10 min at an average sarcomere length of approximately 1.8 μm. Two sets of three stretch-shortening cycles were performed (with a 10-min rest in between) and the first cycles (stretch 1) of the first and second set (stretch 2) are shown. Adapted from Herzog et al. (2014) with permission
This interpretation has been challenged recently where immunoglobulin refolding has been thought to occur at force levels acting on titin of about 8 pN, a relatively large force corresponding to the force exerted by about two to four attached cross-bridges (Rivas-Pardo et al. 2016). If these latest results receive further support, then the notion of unfolding and subsequent refolding of titin’s immunoglobulin domain having physiological relevance may have to be revisited.
Conclusion
Titin is a multi-purpose spring with many acknowledged mechanical functions, including the provision of passive force, stability of the myosin filaments, and stability of sarcomeres on the descending limb of the force–length relationship (Granzier et al. 1996, 2002; Linke et al. 1998; Granzier and Labeit 2002, 2007; Anderson et al. 2010; Linke and Kruger 2010). Less clear are its possible functions in explaining the RFE property of skeletal and cardiac muscle and the molecular details of its function as an adaptable molecular spring (Herzog et al. 2006, 2016; Herzog 2014a). Specifically, many unexplained phenomena in eccentric muscle action (increased force, RFE, decreased energetic requirements) can easily be accounted for within the framework of the cross-bridge theory, assuming that titin is an adaptable spring (Schappacher-Tilp et al. 2015). It will be an exciting challenge in this upcoming decade to elucidate the detailed molecular properties of titin in actively elongating muscle.
References
Abbott BC, Aubert XM (1952) The force exerted by active striated muscle during and after change of length. J Physiol 117:77–86
Allinger TL, Epstein M, Herzog W (1996) Stability of muscle fibers on the descending limb of the force- length relation. A theoretical consideration. J Biomech 29:627–633
Anderson BR, Bogomolovas J, Labeit S, Granzier HLM (2010) The effects of PKCalpha phosphorylation on the extensibility of titin’s PEVK element. J Struct Biol 170:270–277
Astier C, Raynaud F, Lebart MC, Roustan C, Benyamin Y (1998) Binding of a native titin fragment to actin is regulated by PIP2. FEBS Lett 429:95–98
Bartoo ML, Linke WA, Pollack GH (1997) Basis of passive tension and stiffness in isolated rabbit myofibrils. Am J Phys 273:C266–C276
Bianco P, Nagy A, Kengyel A et al (2007) Interaction forces between F-actin and titin PEVK domain measured with optical tweezers. Biophys J 93:2102–2109
Borbely A, Falcao-Pires I, van Heerebeek L et al (2009) Hypophosphorylation of the stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res 104(6):780–786
Bullimore SR, Leonard TR, Rassier DE, Herzog W (2007) History-dependence of isometric muscle force: effect of prior stretch or shortening amplitude. J Biomech 40:1518–1524
Campbell KS (2009) Interactions between connected half-sarcomeres produce emergent mechanical behavior in a mathematical model of muscle. PLoS Comput Biol 5:e1000560
Cazorla O, Freiburg A, Helmes M et al (2000) Differential expression of cardiac tintin isoforms and modulation of cellular stiffness. Circ Res 86:59–67
Chung CS, Bogomolovas J, Gasch A et al (2011) Titin-actin interaction: PEVK-actin-based viscosity in a large animal. J Biomed Biotechnol 2011:310791
Colomo F, Piroddi N, Poggesi C, Te KG, Tesi C (1997) Active and passive forces of isolated myofibrils from cardiac and fast skeletal muscle of the frog. J Physiol 500(Pt 2):535–548
de Brito Fontana H, Herzog W (2016) Vastus lateralis maximum force-generating potential occurs at optimal fascicle length regardless of activation level. Eur J Appl Physiol 116:1267–1277
DuVall MM, Gifford JL, Amrein M, Herzog W (2013) Altered mechanical properties of titin immunoglobulin domain 27 in the presence of calcium. Eur Biophys J 42:301–307
DuVall M, Jinha A, Schappacher-Tilp G, Leonard T, Herzog W (2017) Differences in Titin segmental elongation between passive and active stretch in skeletal muscle. J Exp Biol 220(Pt 23):4418–4425
Edman KAP, Tsuchiya T (1996) Strain of passive elements during force enhancement by stretch in frog muscle fibres. J Physiol 490(1):191–205
Edman KAP, Elzinga G, Noble MIM (1982) Residual force enhancement after stretch of contracting frog single muscle fibers. J Gen Physiol 80:769–784
Epstein M, Herzog W (1998) Theoretical models of skeletal muscle:biological and mathematical considerations. John Wiley & Sons Ltd., New York
Forcinito M, Epstein M, Herzog W (1998) Can a rheological muscle model predict force depression/enhancement? J Biomech 31:1093–1099
Fortuna R, Power GA, Mende E, Seiberl W, Herzog W (2016) Residual force enhancement following shortening is speed-dependent. Sci Rep 5:21513
Freiburg A, Trombitas K, Hell W et al (2000) Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circ Res 86:1114–1121
Fukunaga T, Ichinose Y, Ito M, Kawakami Y, Fukashiro S (1997) Determination of fascicle length and pennation in a contracting human muscle in vivo. J Appl Physiol 82:354–358
Funatsu T, Higuchi H, Ishiwata S (1990) Elastic filaments in skeletal muscle revealed by selective removal of thin filaments with plasma gelsolin. J Cell Biol 110:53–62
Fusi L, Brunello E, Yan Z, Irving M (2016) Thick filament mechano-sensing is a calcium-independent regulatory mechanism in skeletal muscle. Nat Commun 7:13281
Gordon AM, Huxley AF, Julian FJ (1966) The variation in isometric tension with sarcomere length in vertebrate muscle fibres. J Physiol 184:170–192
Granzier HLM (2010) Activation and stretch-induced passive force enhancement—are you pulling my chain? Focus on “regulation of muscle force in the absence of actin-myosin-based cross-bridge interaction”. Am J Physiol Cell Physiol 299:C11–C13
Granzier HLM, Irving TC (1995) Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J 68:1027–1044
Granzier HLM, Labeit S (2002) Cardiac titin: an adjustable multi-functional spring. J Physiol 541(2):335–342
Granzier HLM, Labeit S (2007) Structure-function relations of the giant elastic protein titin in striated and smooth muscle cells. Muscle Nerve 36:740–755
Granzier HLM, Trombitas K, Kellermayer MSZ, Helmes M, Stockman B (1996) Titin: a bi-directional spring and modulator of filament sliding. Proc Can Soc Biomech 9:10–11
Granzier HLM, Labeit D, Wu Y, Labeit S (2002) Titin as a modular spring: emerging mechanisms for elasticity control by titin in cardiac physiology and pathophysiology. J Muscle Res Cell Motil 23:457–471
Granzier HLM, Hutchinson KR, Tonino P et al (2014) Deleting titin’s I-band/A-band junction reveals critical roles for titin in biomechanical sensing and cardiac function. Proc Natl Acad Sci USA 111:14589–14594
Hahn D, Seiberl W, Schmidt S, Schweizer K, Schwirtz A (2010) Evidence of residual force enhancement for multi-joint leg extension. J Biomech 43:1503–1508
Herzog W (2014a) Mechanisms of enhanced force production in lengthening (eccentric) muscle contractions. J Appl Physiol 116:1407–1417
Herzog W (2014b) The role of titin in eccentric muscle contraction. J Exp Biol 217:2825–2833
Herzog W, Leonard TR (2002) Force enhancement following stretching of skeletal muscle: a new mechanism. J Exp Biol 205:1275–1283
Herzog W, Leonard TR (2005) The role of passive structures in force enhancement of skeletal muscles following active stretch. J Biomech 38:409–415
Herzog W, Schachar R, Leonard TR (2003) Characterization of the passive component of force enhancement following active stretching of skeletal muscle. J Exp Biol 206:3634–3643
Herzog W, Lee EJ, Rassier DE (2006) Residual force enhancement in skeletal muscle. J Physiol Lond 574:635–642
Herzog JA, Leonard TR, Jinha A, Herzog W (2012) Are titin properties reflected in single myofibrils? J Biomech 45:1893–1899
Herzog JA, Leonard TR, Jinha A, Herzog W (2014) Titin (visco-) elasticity in skeletal muscle myofibrils. MCB 11:1–17
Herzog W, Powers K, Johnston K, DuVall M (2015) A new paradigm for muscle contraction: review. Front Physiol 6:174–185
Herzog W, Schappacher G, DuVall M, Leonard TR, Herzog JA (2016) Residual force enhancement following eccentric contractions: a new mechanism involving Titin. Physiology 31:300–312
Hill AV (1953) The mechanics of active muscle. Proc R Soc Lond 141:104–117
Hisey B, Leonard TR, Herzog W (2009) Does residual force enhancement increase with increasing stretch magnitudes? J Biomech 42:1488–1492
Horowits R (1992) Passive force generation and titin isoforms in mammalian skeletal muscle. Biophys J 61:392–398
Horowits R, Podolsky RJ (1987) The positional stability of thick filaments in activated skeletal muscle depends on sarcomere length: evidence for the role of titin filaments. J Cell Biol 105:2217–2223
Horowits R, Podolsky RJ (1988) Thick filament movement and isometric tension in activated skeletal muscle. Biophys J 54:165–171
Horowits R, Maruyama K, Podolsky RJ (1989) Elastic behaviour of connecting filaments during thick filament movement in activated skeletal muscle. J Cell Biol 109:2169–2176
Hudson BD, Hidalgo CG, Gotthardt M, Granzier HLM (2010) Excision of titin’s cardiac PEVK spring element abolishes PKCalpha-induced increases in myocardial stiffness. J Mol Cell Cardiol 48:972–978
Huxley HE (1953) Electron microscope studies of the organization of the filaments in striated muscle. Biochim Biophys Acta 12:387–394
Huxley AF (1957a) Muscle structure and theories of contraction. Prog Biophys Biophys Chem 7:255–318
Huxley HE (1957b) The double array of filaments in cross-striated muscle. Biochem Biophys Acta 12:387–394
Huxley HE (1969) The mechanism of muscular contraction. Science 164:1356–1366
Huxley AF (1980) Reflections on muscle. Liverpool University Press, Liverpool
Huxley HE, Hanson J (1954) Changes in cross-striations of muscle during contraction and stretch and their structural implications. Nature 173:973–976
Huxley AF, Niedergerke R (1954) Structural changes in muscle during contraction. Interference microscopy of living muscle fibres. Nature 173:971–973
Huxley AF, Peachey LD (1961) The maximum length for contraction in vertebrate striated muscle. J Physiol Lond 156:150–165
Huxley AF, Simmons RM (1971) Proposed mechanism of force generation in striated muscle. Nature 233:533–538
Ichinose Y, Kawakami Y, Ito M, Fukunaga T (1997) Estimation of active force-length characteristics of human vastus lateralis muscle. Acta Anat (Basel) 159:78–83
Iwazumi T (1979) In: Sugi H, Pollack GH (eds) Crossbridge mechanism in muscle contraction. University of Tokyo Press, Tokyo, pp 611–632
Iwazumi T, Noble M (1989) An electrostatic mechanism of muscular contraction. Int J Cardiol 24:267–275
Johnston K, Jinha A, Herzog W (2016) The role of sarcomere length non-uniformities in residual force enhancement of skeletal muscle myofibrils. Royal Soc Open Sci 3:150657
Joumaa V, Herzog W (2013) Energy cost of force production is reduced after active stretch in skinned muscle fibres. J Biomech 46:1135–1139
Joumaa V, Rassier DE, Leonard TR, Herzog W (2007) Passive force enhancement in single myofibrils. Pflügers Arch 455:367–371
Joumaa V, Leonard TR, Herzog W (2008a) Residual force enhancement in myofibrils and sarcomeres. Proc R Soc B 275:1411–1419
Joumaa V, Rassier DE, Leonard TR, Herzog W (2008b) The origin of passive force enhancement in skeletal muscle. Am J Physiol Cell Physiol 294:C74–C78
Julian FJ, Morgan DL (1979) The effects of tension on non-uniform distribution of length changes applied to frog muscle fibres. J Physiol 293:379–392
Julian FJ, Sollins MR, Moss RL (1978) Sarcomere length non-uniformity in relation to tetanic response of stretched skeletal muscle fibres. Proc R Soc Lond B 200:109–116
Kellermayer MSZ, Smith SB, Granzier HLM, Bustamante C (1997) Folding-unfolding transitions in single titin molecules characterized with laser tweezers. Science 276:1112–1116
Kruger M, Linke WA (2009) Titin-based mechanical signaling in normal and failing myocardium. J Mol Cell Cardiol 46(4):490–498
Kulke M, Fuijita-Becker S, Rostkova E et al (2001) Interaction between PEVK-titin and actin filaments: origin of a viscous force component in cardiac myofibrils. Circ Res 89:874–881
Labeit D, Watanabe K, Witt C et al (2003) Calcium-dependent molecular spring elements in the giant protein titin. Proc Natl Acad Sci USA 100:13716–13721
Lee HD, Herzog W (2002) Force enhancement following muscle stretch of electrically and voluntarily activated human adductor pollicis. J Physiol 545:321–330
Lee EJ, Herzog W (2008) Residual force enhancement exceeds the isometric force at optimal sarcomere length for optimized stretch conditions. J Appl Physiol 105:457–462
Leonard TR, Herzog W (2010) Regulation of muscle force in the absence of actin-myosin based cross-bridge interaction. Am J Physiol Cell Physiol 299:C14–C20
Leonard TR, Duvall M, Herzog W (2010) Force enhancement following stretch in a single sarcomere. Am J Physiol Cell Physiol 299(6):C1398–C1401
LeWinter MM, Granzier HLM (2010) Cardiac titin: a multifunctional giant. Circulation 121:2137–2145
Li Q, Jin J-P, Granzier HLM (1995) The effect of genetically expressed cardiac titin fragments on in vitro actin motility. Biophys J 69:1508–1518
Linke WA, Fernandez JM (2002) Cardiac titin: molecular basis of elasticity and cellular contribution to elastic and viscous stiffness components in myocardium. J Muscle Res Cell Motil 23:483–497
Linke WA, Kruger M (2010) The giant protein titin as an integrator of myocyte signaling pathways. Physiology (Bethesda) 25:186–198
Linke WA, Popov VI, Pollack GH (1994) Passive and active tension in single cardiac myofibrils. Biophys J 67:782–792
Linke WA, Ivemeyer M, Olivieri N et al (1996) Towards a molecular understanding of the elasticity of titin. J Mol Biol 261:62–71
Linke WA, Ivemeyer M, Labeit S et al (1997) Actin-titin interaction in cardiac myofibrils: probing a physiological role. Biophys J 73:905–919
Linke WA, Ivemeyer M, Mundel P, Stockmeier MR, Kolmerer B (1998) Nature of PEVK-titin elasticity in skeletal muscle. Proc Natl Acad Sci U S A 95:8052–8057
Linke WA, Kulke M, Li H et al (2002) PEVK domain of titin: an entropic spring with actin-binding properties. J Struct Biol 137:194–205
Liversage AD, Holmes D, Knight PJ, Tskhovrebova L, Trinick J (2001) Titin and the sarcomere symmetry paradox1. J Mol Biol 305:401–409
Llewellyn ME, Barretto RPJ, Delp SL, Schnitzer MJ (2008) Minimally invasive high-speed imaging of sarcomere contractile dynamics in mice and humans. Nature 454:784–788
Maruyama K (1976) Connectin, an elastic protein from myofibrils. J Biochem 80:405–407
Maruyama K, Kimura S, Kuroda M, Handa S (1977) Connectin, an elastic protein of muscle. J Biochem 82:347–350
Minajeva A, Kulke M, Fernandez JM, Linke WA (2001) Unfolding of Titin domains explains the Viscoelastic behavior of skeletal myofibrils. Biophys J 80(3):1442–1451
Moo EK, Fortuna R, Sibole SC, Abusara Z, Herzog W (2016) In vivo sarcomere lengths and sarcomere elongations are not uniform across an intact muscle. Front Physiol 7:1–9
Morgan DL (1990) New insights into the behavior of muscle during active lengthening. Biophys J 57:209–221
Morgan DL (1994) An explanation for residual increased tension in striated muscle after stretch during contraction. Exp Physiol 79:831–838
Morgan DL, Proske U (2004) Popping sarcomere hypothesis explains stretch-induced muscle damage. Clin Exp Pharmacol Physiol 31:541–545
Morgan DL, Proske U (2006) Can all residual force enhancement be explained by sarcomere non-uniformities? J Physiol 578(2):613–615
Morgan DL, Whitehead NP, Wise AK, Gregory JE, Proske U (2000) Tension changes in the cat soleus muscle following slow stretch or shortening of the contracting muscle. J Physiol 522(3):503–513
Nagy A, Cacciafesta P, Grama L et al (2004) Differential actin binding along the PEVK domain of skeletal muscle titin. J Cell Sci 117:5781–5789
Noble MIM (1992) Enhancement of mechanical performance of striated muscle by stretch during contraction. Exp Physiol 77:539–552
Novak I, Truskinovsky L (2014) Nonaffine response of skeletal muscles on the ‘descending limb’. Math Mech Solids 20:1–24
Oskouei AE, Herzog W (2005) Observations on force enhancement in sub-maximal voluntary contractions of human adductor pollicis muscle. J Appl Physiol 98:2087–2095
Perkin J, Slater R, Del Favero G et al (2015) Phosphorylating Titin’s cardiac N2B element by ERK2 or CaMKII delta lowers the single molecule and cardiac muscle force. Biophys J 109:2592–2601
Peterson D, Rassier DE, Herzog W (2004) Force enhancement in single skeletal muscle fibres on the ascending limb of the force-length relationship. J Exp Biol 207:2787–2791
Powers K, Schappacher-Tilp G, Jinha A et al (2014) Titin force is enhanced in actively stretched skeletal muscle. J Exp Biol 217:3629–3636
Prado LG, Makarenko I, Andresen C et al (2005) Isoform diversity of giant proteins in relation to passive and active contractile properties of rabbit skeletal muscles. J Gen Physiol 126:461–480
Pun C, Syed A, Rassier DE (2010) History-dependent properties of skeletal muscle myofibrils contracting along the ascending limb of the force–length relationship. Proc Biol Sci 277:475–484
Rassier DE, Pavlov I (2012) Force produced by isolated sarcomeres and half-sarcomeres after an imposed stretch. Am J Physiol Cell Physiol 302:C240–C248
Rassier DE, Herzog W, Pollack GH (2003a) Stretch-induced force enhancement and stability of skeletal muscle myofibrils. Adv Exp Med Biol 538:501–515
Rassier DE, Herzog W, Pollack GH (2003b) Dynamics of individual sarcomeres during and after stretch in activated single myofibrils. Proc R Soc Lond B 270:1735–1740
Rassier DE, Herzog W, Wakeling JM, Syme D (2003c) Stretch-induced, steady-state force enhancement in single skeletal muscle fibers exceeds the isometric force at optimal fibre length. J Biomech 36:1309–1316
Rayment I, Holden HM, Whittaker M et al (1993) Structure of the actin-myosin complex and its implications for muscle contraction. Science 261:58–65
Rivas-Pardo JA, Eckels EC, Popa I et al (2016) Work done by Titin protein folding assists muscle contraction. Cell Rep 14:1–9
Schappacher-Tilp G, Leonard T, Desch G, Herzog W (2015) A novel three-filament model of force generation in eccentric contraction of skeletal muscles. PLoS One 10:e0117634
Schwarz ML, Witt SH, Schneider-Wald B et al (2008) Titin expression in human articular cartilage and cultured chondrocytes: a novel component in articular cartilage biomechanical sensing? Biomed Pharmacother 62:339–347
Scott KA, Steward A, Fowler SB, Clarke J (2002) Titin: a multidomain protein that behaves as the sum of its parts. J Mol Biol 315:819–829
Seiberl W, Hahn D, Herzog W, Schwirtz A (2012) Feedback controlled force enhancement and activation reduction of voluntarily activated quadriceps femoris during sub-maximal muscle action. J Electromyogr Kinesiol 22:117–123
Stoecker U, Telley IA, Stüssi E, Denoth J (2009) A multisegmental cross-bridge kinetics model of the myofibril. J Theor Biol 259:714–726
Sugi H, Tsuchiya T (1988) Stiffness changes during enhancement and deficit of isometric force by slow length changes in frog skeletal muscle fibres. J Physiol Lond 407:215–229
Ter Keurs HE, Rijnsburger WH, van Heuningen R, Nagelsmit MJ (1980) Tension development and sarcomere length in rat cardiac trabeculae: evidence of length-dependent activation. Circ Res 46:703–714
Trombitas K, Granzier HLM (1997) Actin removal from cardiac myocytes shows that near Z line titin attaches to actin while under tension. Am J Phys 273:C662–C670
Trombitas K, Pollack GH (1993) Elastic properties of the titin filament in the z-line region of vertebrate striated-muscle. J Muscle Res Cell Motil 14:416–422
Trombitas K, Redkar A, Centner T et al (2000) Extensibility of isoforms of cardiac titin: variation in contour length of molecular subsegments provides a basis for cellular passive stiffness diversity. Biophys J 79:3226–3234
Vaz MA, de la Rocha FC, Leonard T, Herzog W (2012) The force-length relationship of the cat soleus muscle. Muscles Ligaments Tendons J 2:79–84
Walcott S, Herzog W (2008) Modeling residual force enhancement with generic cross-bridge models. Math Biosci 216:172–186
Wang K, Mcclure J, Tu A (1979) Titin: major myofibrillar components of striated muscle. Proc Natl Acad Sci USA 76:3698–3702
Yamasaki R, Berri M, Wu Y et al (2001) Titin-Actin interaction in mouse myocardium: passive tension modulation and its regulation by calcium/S100A1. Biophys J 81:2297–2313
Yamasaki R, Wu Y, McNabb M et al (2002) Protein kinase a phosphorylates titin’s cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ Res 90:1181–1188
Yasuda K, Shindo Y, Ishwata S (1996) Synchronous behavior of spontaneous oscillations of sarcomeres in skeletal myofibrils under isotonic conditions. Biophys J 70:1823–1829
Zahalak GI (1997) Can muscle fibers be stable on the descending limbs of their sarcomere length–tension relations? J Biomech 30:1179–1182
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Walter Herzog declares that he has no conflict of interest.
Ethical approval
Ethics approvals for all experiments described in this study were obtained by the Life Sciences and Animal Research Ethics Commitee of the University of Calgary.
Rights and permissions
About this article

Cite this article
Herzog, W. The multiple roles of titin in muscle contraction and force production. Biophys Rev 10, 1187–1199 (2018). https://doi.org/10.1007/s12551-017-0395-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-017-0395-y