
Abstract
The northern Arabian Sea is highly productive in terms of abundant and diverse fish fauna (≈1,000 species) due to nutrient-rich waters. A total of 303 specimens were collected from the coastal waters of Pakistan, representing 100 genera, 61 families, and 15 orders identifiable to 120 morphological species and 18 others that could not be morphologically assigned to any known taxa. However, the COI analysis via neighbor-joining (NJ) tree, Bayesian inference (BI), and Maximum-likelihood (ML) trees and Automatic Barcode Gap Discovery (ABGD) produced nearly similar topologies and revealed 138 single-species-representing phylogenetic clusters including 18 highly supported previously unknown species clusters showing deep intraspecific divergences from the closely related morphospecies. The calculated average Kimura 2-parameter (K2P) genetic distances were 0.25%, 6.34%, 17.12%, and 26.8% within the species, genera, families, and orders, respectively. We analyzed species from eight groups (clupeiforms, sciaenids, flatfishes, spiny-flathead, lizardfish, sillaginids, grunts, stargazers) through BI and ML and found cryptic genetic diversity in these groups. Barcoding gap analysis (BGA) demonstrated that the barcode gaps were present for 39 of 70 species having multiple specimens (50.7%). Our study provides the first-ever addition of COI sequences for five species (Cociella punctata Cuvier, 1829; Epinephelus stoliczkae Day, 1875; Pomadasys aheneus McKay & Randall, 1995; Scorpaenopsis possi Randall & Eschmeyer, 2002; and Sillago panhwari Panhwar et al., 2017 in Panhwar et al. 2018) into the GenBank database. The study concluded that DNA barcoding is an efficient approach in species identification, supplementing the morphological identification of the known forms and highlighting the cryptic diversity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fishes are the largest vertebrate groups on the earth with more than 34,000 known species (Froese and Pauly 2020). They range in size from 60 feet long (Rhincodon typus Smith, 1828) to less than one-third of an inch (Paedocypris progenetica Kottelat, Britz, Tan & Witte, 2006) and with varying geographical distributions to several endemics to their local habitats. Traditionally, the information allied to the identification of a fish is based on morphological characteristics, involving the preparation of identification keys. The species description is often used for comparison of different species but they sometimes fail to treat all life stages and genders (Ribeiro et al. 2012). The advancement in the field of systematic and taxonomy through involving DNA barcodes has now become a popular tool for reliable identification at every stage of life (egg or larvae, damaged or semi-cooked specimen). As such, DNA barcoding has resulted in the discovery of new species and helped to resolve the issues of cryptic diversity (Thongtam na Ayudhaya et al. 2017; Lim et al. 2016; Locke et al. 2010). The concept of DNA barcode premise that every species has specific mitochondrial cytochrome c oxidase gene I (COI) sequences or barcodes, like an internal species tags that can be derived by sequencing of at least 650 bp region COI (Hebert et al. 2003). The COI sequences remain nearly constant within the species but vary among different species and can be identified through matching against expert-identified voucher specimen’s sequences (Ribeiro et al. 2012). With the help of this technique, more than 14,400 fish species have been successfully barcoded, in a global initiative to barcode all fish species (Steinke et al. 2017). Several studies have been done by the experts to establish a barcode reference library for their fish fauna: 117 species from Arabian Gulf (Rabaoui et al. 2019); 272 Perciform fishes from the South China Sea (Hou et al. 2018); 194 species from Congo and Lower Guinean (Sonet et al. 2019); 41 species from Rongcheng Bay, China (Wang et al. 2018); 135 species from Sao Paulo State, Brazil (Ribeiro et al. 2012); 23 species from Taal Lake, Philippines (Aquilino et al. 2011); 89 species from Turkey (Keskin and Atar 2013); or 79 freshwater fish species from the Lower Paraná River in South America (Díaz et al. 2016). The most comprehensive work from the Indian Ocean was carried out by Lakra et al. (2011) who generated COI barcodes for 115 species. According to Basheer et al. (2016), the database on Indian fishes comprises a total sequence of 23,429 fish specimens including 1,083 marine, 349 freshwater, and 49 brackish-water species.
The level of endemism in the well-characterized shore fishes of the Red Sea (12.9%) exceeds those from Mascarene Island (3.4%), the Arabian Gulf (4.0%), and southern Oman (2.8%) (DiBattista et al. 2016). The coastal areas of Pakistan, as a part of the northern Arabian Sea, is highly productive in terms of fish diversity, abundance, and endemism as it receives nutrient-rich waters from up-sloping in the Gulf of Oman and surrounding areas. These areas are bestowed with a variety of marine habitats including large Indus Delta (600,000 ha), numerous major and minor creeks, mudflats, and mangrove forests. Commercially important finfish species reported in a recently published FAO fish guide is over 600 based on identification through morphological/traditional taxonomic examination (Psomadakis et al. 2015). On the other hand, there is enormous pressure on the marine biodiversity of the Arabian Sea because of the increasing human population, coastal development, and overexploitation of fisheries resources. A number of fisheries stocks have already been overexploited, e.g., shellfishes (Mohsin et al. 2017). Some finfish species have now completely disappeared from Pakistani waters, e.g., four species of sawfishes (Harrison and Dulvy 2014).
The DNA barcoding of coastal fishes has not been carried out for Pakistan. Thus, this is the first report that aims to identify the marine ichthyofauna and explore the cryptic diversity based on genetic variations in COI sequences. Furthermore, the information would be made readily available to non-taxonomists, researchers, and policymakers to aid them in their efforts in the effective management of the marine environment and help create conservation strategies.
Materials and methods
Sampling and identification
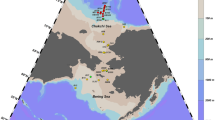
The fish sampling was carried out from commercial landings sites of Sindh (West Wharf Fish Harbor and Ibrahim Hyedri Fish Harbor) and Makran coast (Gawader and Jewani fish landing areas) from 2016 to 2018 (Fig. 1).

A physiographic map of the coastal areas of Pakistan. Sampling was carried out from Sindh and Makran coast during 2016 to 2018. (Source: National Geographic, Esri, Garmin, HERE, UNEP-WCMC, USGS, NASA, ESA, METI, NRCAN, GEBCO, NOAA, increment P Corp)
A total of 303 specimens were collected from the study areas. Specimens were kept on ice; selected specimens were tagged and subsequently, images were taken of the fresh specimens. Specimens were identified using relevant literature and identification keys for various fish groups (Psomadakis et al. 2015; Panhwar et al. 2018; Liu et al. 2017; Xiao et al. 2016; Pathak et al. 2016; Russell et al. 2015; Khamees et al. 2018; Qamar et al. 2016). We used “sp.” when the examined specimens deviated in important morphological characters from the known taxa and most likely represented an unknown species. The morphological data is available for all the species represented as “sp.” and will be later on published separately.
For DNA analysis, lateral muscle (≈100 mg) from the right side of the fresh fish specimens was removed and preserved in 96% ethanol in small cryo-vials. All specimens were cataloged and deposited at the Ichthyological section of the Pakistan Museum of Natural History, Islamabad, Pakistan (PMNH). The catalog numbers of the specimens and their sequences are provided without mentioning PMNH.
DNA extraction, amplification, and sequencing
DNA was successfully extracted for 303 fishes from muscle tissue of each specimen using a standard phenol-chloroform method that is a relatively fast and easy technique for recovering high-quality DNA. Two sets of forward and reverse primers were used for the amplification of the COI gene following Ward et al. (2005):
-
FishF1-5TCAACCAACCACAAAGACATT GGCAC3,
-
FishR1-5TAGACTTCTGGGTGGCCAAAGAATCA3;
-
FishF2-5TCGACTAATCATAAAGATATCGGCAC3,
-
FishR2-5′ACTTCAGGGTGACCGAAGAATCAGAA3.
Polymerase chain reaction (PCR) was conducted in 25-μl volumes, which contained PCR mix 12.5 μl, forward primer 0.5 μl, reverse primer 0.5 μl, ultrapure water 10.5 μl, and DNA template 1 μl. The barcode region of COI was subsequently amplified under the following thermal conditions: 4 min at 95°C; 35 cycles of 0.5 min at 95°C, 0.5 min at 52°C, and 1 min at 72°C; 5 min at 72°C; then held at 4°C. PCR products were visualized on a 1.2% agarose gel. All 303 successful PCR products were sent to commercial laboratories for bidirectional sequencing. All here obtained sequences were deposited in the GenBank database under the numbers MN511846–MN512146, MN530052, and MN530053 (for details see Table 1).
Data analysis
The DNA sequences were assembled and aligned from forward and reverse reads and edited manually by using DNASTAR multiple packages (Hou et al. 2018). The generated sequences were ranging from 652 to 655bp and were compared with available sequences of fishes in the GenBank. The pairwise genetic distance was calculated to analyze sequence divergences using the Kimura two-parameter (K2P) distance model (Kimura 1980) and uncorrected p distances. To create a neighbor-joining (NJ) tree for all 303 sequences, only K2P distances were considered, because both distance models were producing nearly the same results. Node support with 1,000 bootstrap replications was created to provide a graphic representation of the patterning of divergence between species (Saitou and Nei 1987). The K2P distance, p distance, and the NJ tree were calculated and generated using Molecular Evolutionary Genetics Analysis software “MEGA X.”
For the haplotype data set of 303 sequences with additional sequences obtained from GenBank, evolutionary relationships were inferred using maximum likelihood (ML) and Bayesian inference (BI) approaches. PARTITIONFINDER 2 (Lanfear et al. 2017) and the implemented Bayesian Information Criterion were used to determine the best partitioning scheme and the best model of sequence evolution. ML analyses were conducted with raxml 7.2.8. Five independent searches were then performed using different starting conditions and the fast bootstrap algorithm to explore the robustness of the results by comparing the best trees. Then, 1,000 non-parametric thorough bootstrap replicates were calculated and plotted against the best tree. BI was performed using MRBAYES 3.2.1 (Ronquist et al. 2012) with two parallel runs (each with four chains) and default parameters using the codon-partitioned data set. The chains ran for 20 million generations with every 1,000th generation sampled. The values less than 60 and 50 in BI and ML, respectively, are not provided in figures as well as text due to their low significance.
Automatic Barcode Gap Discovery (ABGD) was performed online using a web interface (www.abi.snv.jussieu.fr/public/abgd/). The default value for the relative gap width was set as X = 1.5, which did not produce a result; two lower values (X = 0.8, 1.0) were used on the genetic distance metrics of K2P and p distance to delimit species (Kekkonen et al. 2015). All results using prior limits for intraspecific divergence ranging from P = 0.001–0.1 were recorded.
Barcoding gap analysis (BGA) was performed for species-level comparisons of the maximum intraspecific genetic distance to the minimum interspecific distance. BGA was carried out using the “Barcoding Gap Analysis” tool in the BOLD system (Ratnasingham and Hebert 2007). Singleton species were excluded in BGA.
Barcode Index Numbers (BIN) analysis was applied to assess the agreement of the taxonomic species-COI sequence data sets obtained in our present study to the available data in the BOLD system (Ratnasingham and Hebert 2013). The sequences were automatically assigned to BINs using the BOLD Workbench application (version 3.6; http://www.boldsystems.org; analyses performed on May 2020).
Additional GenBank sequences
COI sequences of species related to our eight main problematic taxonomic groups (see the “Results” section) were obtained from the GenBank database for use in the construction of BI and ML trees. Details of the additional GenBank accession numbers of the specimens and their locality are given in the “Results” section for each group.
Results
Based on the external morphological features, all the 303 specimens were identified into 100 genera, 61 families, and 15 orders in 120 morphospecies and 18 unidentified taxa that did not entirely correspond to any known species. A maximum number of 13 sequences were analyzed for Nemipterus japonicus Bloch, 1791, followed by nine for Upeneus vittatus Forsskål, 1775, and eight for Planiliza klunzingeri Day, 1888, whereas 68 sequences (22.44%) were represented by a single species each (Table 1). A sequence of 655 bp (for 265 specimens) was generated using universal primers FishF1 and FishR1, whereas 652 bp (for 38 specimens) using primers FishF2 and FishR2. No stop codons, insertions, or deletions were detected in any of the amplified sequences. The average nucleotide composition in a complete data set was 29.4% for T, 28.4% C, 23.6% A, and 18.6% for G. The average GC content was 47.03%; the highest was 52.8% for Chirocentrus dorab Forsskål, 1775, and the lowest of 37.3% was for Chiloscyllium burmensis Dingerkus & DeFino, 1983. Nucleotide pair frequency analysis of the complete data set resulted in conserved sites of 319 of 655 bp (48.7%), variables sites 336 of 655 bp (51.14%), parsimony informative sites of 317 (48.39%), and singleton sites of 17 (2.59%). The average number of identical pairs (ii) was 516 of which 198, 208, and 110 were found for the first, second, and third codon, respectively. Transitional pairs (s) were found to be higher (77) than transversional pairs (sv=59). Both transitional and transversional pairs were highest at the third codon position (57 and 48 for si and sv, respectively). The average ratio (R) of si/sv was 1.31 for the data set.
The average Kimura 2-parameter/uncorrected p distances within species, genus, families, and orders were 0.25/0.22 %, 7.4/6.6 %, 17.1/15.7 %, and 26.8/24.7%, respectively. Among the sequences, the highest genetic distance (K2P = 0.365, p distance = 0.359) was calculated between Narcine timlei Bloch & Schneider, 1801 (55296) and Otolithes sp.2 (55080).
A neighbor-joining tree (Fig. S1) and BI and ML tree (Fig. 2) analyses showed mostly high nodal support values (NJ ≥ 95%, BI/ML≥0.90/95) with distinct clusters in correspondence with the taxonomic status of the fish species. A total of 138 discernible lineages were generated. At the known species level, no taxonomic deviation was noticed and the specimens of the same species were clustered together whereas 18 single-species-representing clusters showing deep interspecific divergence from their closely related morphospecies represent unidentified taxa. Nodal support values from their close congeners for NJ and BI/ML are as follows for five taxa: Scorpaenopsis sp. (99%, 1.0/98), Scolopsis sp. (99%, 0.80/71), Siganus sp. (99% 1.0/91), Himantura sp. (99%, 1.0/83), and Narke sp. (95% 0.99/81), while thirteen others (Saurida sp., Thryssa sp., Johnius sp., Otolithes sp.1, Otolithes sp.2, Sillaginodes sp., Sillago sp., Brachirus sp., Phyllichthys sp., Cynoglossus sp.1, Cynoglossus sp.2, Pseudorhombus sp., Uranoscopus sp.) are separately highlighted under eight groups (clupeiforms, sciaenids, flatfishes, spiny flathead, lizardfish, sillaginids, grunts, stargazers) with analysis of additional sequences obtained from GenBank. Our study provides the first-ever addition of COI sequences for five species (Cociella punctata; Epinephelus stoliczkae; Pomadasys aheneus; Scorpaenopsis possi; Sillago panhwari) in the GenBank database.

RaxML tree for 303 fish sequences reflecting 138 single-species clusters. Bootstrap and posterior probability values from a Bayesian tree of identical topology are indicated at nodes. Nodal support values less than 0.70% for BI and 50% for ML are denoted by ---
The ABGD performed on both distance metrics (K2P, p) generated nearly similar counts of single-species representing clusters ranging from 138 to 159 (K2P, X = 0.8) and from 138 to 156 (K2P, X = 1.0) on initial and recursive partitions. However, ABGD on p matrix generated 138 single-species representing clusters on initial and recursive partition (X=1) in concordance with the results of our NJ, BI, and ML analysis while it generated 139 on recursive partition when X=0.8 (Table 2).
A total of 68 sequences were represented by a single specimen each. BGA demonstrated that the barcode gaps were present for 39 of 70 species with multiple specimens and no gap was found in 31 sequences. The maximum intraspecific value of BGA, i.e., 2.51, was recorded for Terapon jarbua Forsskål, 1775, followed by Lethrinus nebulosus Forsskål, 1775 (2.5), Pomadasys aheneus (1.39), Otolithes sp.1 (1.23), Nemipterus japonicus (1.23), Pseudorhombus sp. (1.2), Sardinella gibbosa Bleeker 1849 (1.09), and Lagocephalus guentheri Miranda Ribeiro, 1915 (0.92) (Fig. 3).

Barcode Gap Analysis report: Species with the maximum intraspecific K2P divergence ranged from 0.92 to 2.51 among 138 single-species representing clusters
Higher-level taxonomic mismatch
At higher taxonomic levels (genus, family, and order), the phylogenetic placement of some species showed taxonomic deviations and clustered at different positions instead with their congeners, confamilials, and conorders with mostly low or insignificant BI and ML support (Fig. 2). A puffer fish Takifugu oblongus Bloch, 1786 (Tetraodontiformes) nested close to Moray eels Strophidon sathete Hamilton, 1822, and Gymnothorax favagineus Day 1875 (both Anguilliformes), far from other Tetraodontiformes (Lagocephalus lunaris Bloch & Schneider, 1801, L. guentheri). A grunt species Plectorhinchus sordidus Klunzinger, 1870, a member of family Haemulidae (Perciformes), nested with members of order Clupeiformes instead with other grunts (Pomadasys stridens Forsskål, 1775, P. aheneus, P. olivaceus Day, 1875). A wolf-herring species Chirocentrus dorab (Clupeiformes) nested with Megalops cyprinoides Broussonet, 1782 (Elopiformes) with a moderate BI support (0.78/--). Cheilopogon furcatus Mitchill, 1815 (Beloniformes) clustered as a sister clade to all Sillaginids (Perciformes) with a low BI support (0.63/--). All members of order Mugiliformes lumped together on the BI/ML tree except Mugil cephalus Linnaeus, 1758, which formed a separate clade far from other mugils. Members of the flatfishes (Pleuronectiformes) were placed in several unrelated taxonomic hierarchies in the BI/ML tree. For example, Pseudorhombus sp. clustered as a sister clade to Lactarius lactarius Bloch & Schneider, 1801 (Perciformes) with a moderate BI support (BI/ML, 0.73/--). Two species of the genus Sphyraena (S. putnamae Jordan & Seale, 1905, and S. pinguis Günther, 1874), members of the order Perciformes, nested within the members of order Pleuronectiformes without any nodal support.
The genera Trachinocephalus and Saurida of the family Synodontidae (lizardfish) nested in different families: Trachinocephalus myops Forster, 1801 nested with the species of family Leiognathidae (Leiognathus robustus Sparks & Dunlap, 2004, Karalla daura Cuvier, 1829) while Saurida species (S. tumbil Bloch, 1795, S. lesepsianus Russell, Golani & Tikochinski, 2015, Saurida sp.) nested as a sister clade to Pempheridae (Pempheris rhomboidea Kossmann & Räuber 1877, Ostorhinchus fasciatus Shaw, 1790) with no any nodal support. Interestingly, two species of the genus Gerres (G. filamentosus Cuvier, 1829, G. oyena Forsskål, 1775), members of family Gerreiadae, were placed far from each other. G. oyena was placed as a sister clade with the members of family Mullidae (Upeneus vittatus, U. moluccensis Bleeker, 1855) while G. filamentosus clustered with the members of order Beloniformes (Ablennes hians, Strongylura strongylura, Hyporhamphus sindensis). In the family Scombridae, two species (Euthynnus affinis, Rastrelliger kanagurta) splitted in separate clusters; E. affinis nested as a sister clade to Pampus argenteus (Stromateidae) (BI/ML, 0.89/50) and Ariomma indicum (Ariommatidae) and R. kanagurta nested with Trichiurus lepturus (Trichiuridae) with the high nodal support only in BI (BI/ML, 0.99/--). Five species in the family Clupeidae (Sardinella albella, S. gibbosa, S. longiceps, Nematalosa nasus, Escualosa thoracata) clustered close together but S. abella positioned itself as a sister clade to N. nasus (BI/ML, 0.88/--) whereas E. thoracata clustered as a sister clade to S. gibbosa and S. longiceps with no nodal support (BI/ML, --/--). In the family Platycephalidae, Cociella heemstrai nested with Grammoplites suppositus rather than Cociella punctata with high nodal support (BI/ML, 1.0/98).
Candidate species and conflicting taxonomic designations
The following groups, based on DNA barcoding and preliminary morphological examination (not presented here), reveal cases where new information confirming species delimitations or requiring further taxonomic reexamination is warranted. The information on potential candidate species is given in the following eight broader categories.
Clupeiforms
A total of nineteen haplotypes of clupeiform fishes were successfully generated belonging to five families including Chirocentridae, Clupeidae, Dussumieriidae, Engraulidae, and Pristigasteridae (Table 1). An additional 16 sequences were obtained from GenBank from the nearby regions for comparison and verification. The analysis of our Clupeiformes through BI and Ml tree shows higher nodal support values (Fig. 4). The sequences of Pakistani Clueipormes (Sardinella albella, S. gibbosa, S. longiceps, Esculosa thoracata, Ilisha elongate, Thryssa dussumieri, T. hamiltonii, T. vitrirostris) clustered/nested with specimens of these species from India and Bangladesh. The analyzed sequences of Thryssa sp. in the present study grouped into a strongly supported clade (BI/ML = 1.0/99). A single sequence of Thryssa sp. (55314) formed a separate lineage with an average uncorrected p distance of 15% from the closely nested four sequences of T. vitrirostris (55309-12; n=4), which in turn formed a well-supported cluster with sequences from Iran (KU937393) and South Africa (JF494689). A sequence of T. hamiltonii (MF594610) from Bangladesh was nested separately from the known sequences of T. hamiltonii and could be a misidentification. Our sequences of Nematalosa nasus (55185, 55297) formed a cluster (BI/ML = 1.0/100) with a specimen from Saudi Arabia (KU499652). A sequence of Dussumieria elopsoides (55260) clustered (BI/ML = 1/100) with an average uncorrected p distance of 3% with three sequences, each from China (EF607363), Taiwan (KU942886), and Vietnam (MK777250).

RaxML tree of our sequences of Clupeiformes and those of related species from GenBank database. Bootstrap and posterior probability values from a Bayesian tree of identical topology are indicated at nodes
Sciaenids
Sciaenids were represented in our study by five genera including Daysciaena, Pennahia, Otolithoides, Otolithes, and Johnius, the former three by single species each. Our sequences of Otolithes (O. ruber, 55060-55061; Otolithes sp.1, 55217, 55057-55059; Otolithes sp.2, 55077, 55080-55081, 55359) appeared in three distinct clusters. All the sequences of O. ruber clustered together (100% matched; BI/ML 0.89/82) including sequences obtained from India (FJ237585) and Bangladesh (KY024208). Two clusters of Otolithes sp.1 and Otolithes sp.2 appeared a sister clades to each other with basal support of BI (73) and an interspecific p distance of 14%. Our ten sequences of Johnius species distinctly appeared in the BI/ML tree in two clusters representing J. amblycephalus and Johnius sp., with a high nodal support value of 1.0/92 (Fig. 5). The intraspecific p distance in both Johnius sp. (55088, 55089, 55101, 55102, 55169) and J. amblycephalus (54715, 54819, 55096, 55175, 55275) was nil where as a K2P divergence of 2% appeared between a sequence of J. amblycephalus from UAE (KX777910) and our sequences. In the present study, the interspecific divergence (K2P/p) of Johnius sp. from all other sciaenids ranged from 17–32/15–25% and 15–28/13–23% for J. amblycephalus. The strong nodal support in BI/ML tree (1.0/100) and intraspecific divergence showed that our Otolithoides biauritus (55367) and Daysciaena albida (55093) matched 100% with sequences from India (MF383188) and Indo-west Pacific (KP722719), respectively. Our single sequence of Pennahia anea (55302) also showed 100% similarity with sequence of this species from Myanmar (MH235684) whereas deviating in K2P/p genetic distances by 4% from a sequence of P. anea from China (MG574454).

RaxML tree of our sequences of sciaenids and those of related species from GenBank database. Bootstrap and posterior probability values from a Bayesian tree of identical topology are indicated at nodes
Flatfishes
Twenty haplotypes of flatfishes were successfully generated falling in three families including Paralichthyidae, Soleidae, and Cynoglossidae. An additional 10 sequences were obtained from GenBank for comparison and verification of our species (Fig. 6). In the family Paralichthyidae, all the Pseudorhombus species (P. elevates, P. arsius, and Pseudorhombus sp.) grouped by high nodal support of BI/ML values (1.0/96). Our five sequences for Pseudorhombus sp. (55304-07, 55139) formed a single cluster differing from the known species of Pseudorhombus arsius from South Africa (JF494303, JF494302) and South China Sea (FJ237895, FJ237894) by the genetic divergence (p distance) of 7% and 12%, respectively. We are, therefore, reluctant to suggest this single-species representing cluster as a candidate species. Additionally, even a greater genetic distance of 17% was calculated for our Pseudorhombus sp. from the South African Pseudorhombus elevatus (JF494305, JF494307).

RaxML tree of our sequences of Flatfishes and those of related species from GenBank database. Bootstrap and posterior probability values from a Bayesian tree of identical topology are indicated at nodes
Four sequences in the family Soleidae were generated. Among them, three sequences lumped into genera of Brachirus (54808-09) and Phyllichthys (55326), and a single sequence confirmed the presence of Dagetichthys commersonnii (55258). COI sequences of our two Brachirus specimens were 100% similar to Brachirus orientalis from Saudi Arabia, Arabian Gulf (KU499833) whereas differed from specimens from Bangladesh (MF611618) and China (EF607337) by the divergence of 17%. Our Brachirus specimens diverge in several morphological characters (unpublished data) from the closely related B. orientalis which was originally described from Sri Lanka. This further supposes that Brachirus in North Arabian Sea is a potential candidate species. The morphological data of one of our specimens correspond to the genus Phyllichthys but deviate from any of the known Phyllichthys species (unpublished data). This genus is represented by three species namely, Phyllichthys punctatus, Phyllichthys sclerolepis, and Phyllichthys sejunctus, all distributed in Western Pacific and Eastern Indian Ocean. Although the genus has never been reported from the West Indian Ocean, and we found no sequence data of the members of genus Phyllichthys in the GenBank for our analysis, given the morphological differences and distant distribution, a potential candidate species in Phyllichthys from the Indian Ocean is warranted in future. Our single specimen of D. commersonnii corresponded with D. commersonnii from Bangladesh (MH429299) with only 1.0% genetic deviations from the Pakistani sequence.
A total number of 20 sequences (10 GenBank and 10 present study) in the family Cynoglossidae were used in the present BI/ML analysis. Our Cynoglossus sequences formed four distinct clusters. BI/ML tree shows that Cynoglossus sp.1 (55299-55301) is a sister clade to C. bilineatus (EF607350 China) with a high nodal support value (1.0/100) and a p distance of 12%. Three sequences of Cynoglossus sp.2 (55151-55153) formed a separate cluster from all other Cynoglossus species with nodal support of 0.62/62(BI/ML) and a p distance of 19–22%. The available Indian sequences of C. macrostomus in GenBank (FJ347911-FJ347912) showed more than 99% resemblance with our C. macrostomus (55252-55253). The type locality of C. macrostomus is in India. Two COI sequences of C. arel (54649 and 55150) in our collection are similar (100%) to specimens from Saudi Arabia (KU499774) and Iran (MG052949) but diverge from Indian (KT323970) and Myanmar (MH235626) sequences in GenBank by 4% and 12%, respectively. The type locality of C. arel is from India.
Spiny flathead
DNA barcoding of spiny flathead fishes confirms the occurrence of Cociella punctata (55249-55250), C. heemstrai (55032-55034), and Grammoplites suppositus (55270) in Pakistani waters. To compare and verify, available sequences of the same species from the nearby regions were obtained from GenBank and a BI/ML tree was created (Fig. 7). In the present study, the nodal support for spiny flathead fishes was high with a BI/ML value of 1.0/100. The intraspecific distances for C. heemstrai (55032-34) from other spiny flathead fishes ranged from 11 to 21 % for K2P and 10 to 18% for p distances. Nearly similar interspecific distances were recorded for our C. punctata (55249-50) and G. suppositus (55270).

RaxML tree of our sequences of Spiny-Flathead and those of related species from GenBank database. Bootstrap and posterior probability values from a Bayesian tree of identical topology are indicated at nodes
Cociella heemstrai is described from Kenya, the West Indian Ocean. COI sequence divergence of our C. heemstrai (55032-55034) was 3% from the South African clade (JF493229, JF493230, JX488290), suggesting a possibility for the presence of an unknown population of heemstrai in the North Arabian Sea. On the other hand, G. suppositus was found to exhibit 100% sequence similarity with the available GenBank sequences from Saudi Arabia (KU499610, KU499611).
Lizardfish (Saurida) and Trachinocephalus
Twelve sequences of Saurida species were successfully amplified and five additional sequences were obtained from GenBank including S. undosquamis (FJ347930, FJ347931, India; KP266852, South China Sea) and S. longimanus (KR105861, KR105862, India). The BI/ML analysis produced well-supported (>0.90/80) deep divergent clades (Fig. 8). Seven sequences of our S. tumbil exhibited 100% (BI/ML= 0.99/99) similarity with a specimen from India (EF609600). Sequences from our two specimens (55336, 55337) corresponded 99% and 100% with S. lessepsianus from Turkey (KY176610, KY176611) and Saudi Arabia (KU499724), respectively with high nodal support values (BI/ML=0.99/76). COI sequences from our three specimens of Saurida sp. (54654, 54655, 54646) formed a distinct clade, deviating with an average genetic distance (uncorrected p distance) of 15% from all Saurida species used in this study. BI/ML tree shows that our Saurida sp. is a sister clade to S. lessepsianus and S. undosqamis from China (KP266852) and India (FJ347931). Keeping in view the deviation in morphological (unpublished data) and COI data, this Saurida sp. is a potential candidate species from Pakistan.

RaxML tree of our sequences of Lizardfish (Saurida spp.) and those of related species from GenBank database. Bootstrap and posterior probability values from a Bayesian tree of identical topology are indicated at nodes
Trachinocephalus myops is historically known to be a circumtropical species. Based on molecular studies, Polanco et al. (2016) validated three recognized species including T. myops (restricted to the Atlantic Ocean), T. gauguini (described from the Marquesas Islands), T. trachinusis (from the Indo-West Pacific Ocean) and four undescribed lineages including our T. myops (Fig. 2) placed as sp. 4 of T. aff. trachinus.
Sillaginids
Nine COI sequences of sillaginids were amplified in this study whereas 15 sequences were obtained from GenBank for comparison and use in the construction of BI/ML tree (Fig. 9). Our sequences appeared in the tree in four separate clusters. The phylogenetic cluster of Sillago sp.1 (54637, 55266, 55329) was closely nested with the sequence of morphospecies S. panhwari (55328) with an average uncorrected p distance of 16% and the high nodal support values for BI (0.89) whereas ML support was below the significance level. The tree shows that two sequences from GenBank (KU051788, KU051787) referred to S. sihama from Indo West Pacific (originated from the Pakistani coast) clustered together with our S. panhwari with an average uncorrected p distance of 1% and high nodal support (BI/ML= 1.0/100), both the former specimens are proposed misidentified S. panhwari. Although morphological characteristics are overlapping in typical S. sihama and S. panhwari, both can be differentiated by analyzing swim bladder shape, head length, average number of lateral-line scale counts, number of transverse rows of scales counts, and gill-raker counts. Interspecific values (K2P/p distance) were ranging between 17–21/15–18 for Sillago sp.1 and 17–21/15–18 for S. panhwari from all other Sillago species used in this study. Our single sequence of S. sihama (54634), perfectly matched (100%) with two available sequences from India (EF609616, EF609617), India being the type locality of S. sihama (Bauchot et al. 1983). Another sister lineage to our S. sihama, referred in GenBank entry as S. sihama from Indo West Pacific (KU051813, KU051881, KU051857) seems to be a different species-representing cluster (BI/ML= 1.0/92) with an average genetic distance of 4.95%. Four sequences of our Sillago sp.2 (54635, 54636, 55024, 55327) and a sequence (MF571912) from Indo-West Pacific, identified as Sillago sihama were clustered together. These sequences of Sillago sp.2 and S. sihama (MF571912) diverge in p distance of 16 to 20% from all others Sillago species used in this analysis. Based on morphological data (unpublished data) and COI sequences, our Sillago sp.1 and Sillago sp.2 are seen as candidate species from Pakistan.

RaxML tree of our sequences of Sillaginids and those of related species from GenBank database. Bootstrap and posterior probability values from a Bayesian tree of identical topology are indicated at nodes
Grunts
A total of 14 sequences were used in BI/ML analysis comprising of eight sequences generated in the present study while 6 sequences were obtained from the GenBank. Three distinct clusters appeared in the BI/ML tree (Fig. 10). Our two sequences of Plectorhinchus sordius (54439, 55181) perfectly matched with a sequence of the same species from Saudi Arabia (KU499678). The second phylogenetic cluster comprised of our sequences from Pomadasys stridens (n=4; 55155, 55158, 55159, 54755) that matched 100% with a sequence from Saudi Arabia (KU499636). In both of these clusters, BI/ML support was 1.0/99 and 1.0/100 for P. sordius and P. stridens, respectively. In the third cluster, six sequences of Pomadasys species (P. olivaceus, P. incises) were grouped with a high BI/ML nodal support (0.99/89); 3% p distance was calculated between our two sequences of P. olivaceus (54340, 54970) and sequences of this taxon from South Africa (F494253) and Indo West Pacific (HQ676798)-IWP). Furthermore, our sequences of P. olivaceus diverge by only 2% from P. incises from Portugal (KJ768283) and Spain (KM017063). The differences in certain morphological characters and the genetic divergence of our P. olivaceus to those of South Africa (JF494253) and Indo-West Pacific (HQ676798) suggest the presence of cryptic diversity in the form of an undescribed species.

RaxML tree of our sequences of Grunts (Haemulidae) and those of related species from GenBank database. Bootstrap and posterior probability values from a Bayesian tree of identical topology are indicated at nodes
Stargazers
Two COI sequences of Uranoscopus species (stargazers) were generated and matched with three available GenBank sequences from South Africa (GU805069), Arabian Gulf (KU317874), and India (KJ093279). BI/ML tree (Fig. 11) showed that U. archionema from South Africa was placed separately with high nodal support (1.0/100) and genetic divergence of 12 % (p distance) from specimens from Pakistan, India, and Saudi Arabia. The sequences of Uranoscopus species from the Arabian Gulf and India matched perfectly with Pakistani Uranoscopus.

RaxML tree of our sequences of Stargazer (Uranoscopidae) and those of related species from GenBank database. Bootstrap and posterior probability values from a Bayesian tree of identical topology are indicated at nodes
BIN Analysis Reports
BIN analysis led to the recognition of 141 BIN clusters. Among them, 46 BIN clusters were taxonomically concordant with the other barcode data that were BOLD-assigned to the same species. A total of 91 BIN clusters were discordant with the morphological species or consisted of different species. Among these, discordant BIN clusters the rank of conflict occurred in 5 families, 31 genera, and 55 species. In five families, a total number of BIN members in BOLD were 299, among them, 279 (93%) were concordant to our identification and 7% were discordant. At the genus level, BIN members were 1,039 and 77% (806) were concordant to our identification whereas 2,691 (74%) records of species were concordant to our results in total BIN members of 3660 species. In particular, 4 BINs records (Ablennes hians, Dussumieria elopsoides, Phyllichthys sp., and Narke sp.) were indicated as a singleton.
Discussion
The Indo-Pacific region is known as a biodiversity hot-spot (Tittensor et al. 2010), comprising of around 17,435 fish species (marine 72%; freshwater 33%); this high diversity is explained because of relatively old (5.3–34 million years ago) colonization (Miller et al. 2018). There are gaps in the data of fish fauna of different geographical areas found in the Indo-West-Pacific region (Hourston 2010). The Northern Arabian Sea is also a part of the Indo-Pacific region and is highly diverse in natural habitats, providing an ideal home for rich biodiversity. Let alone lacking complete fish species inventory, the fish diversity of the Northern Arabian Sea has not been explored through DNA barcoding. The only available reference of DNA barcoding of fishes from the nearby region of Pakistan comes from Lakra et al. (2011) who barcoded 115 species from the Indian Ocean. In the present study, 303 specimens were successfully sequenced, representing 138 single-species representing clusters through NJ, BI/ML tree, and ABGD, whereas 141 single-species representing clusters through BIN report, representing about 22% of species reported in a recent publication “FAO fish identification guide” (Psomadakis et al. 2015). It is also expected that the actual fish diversity will be higher than the number mentioned in Psomadakis et al. (2015) because several fish groups were not included in their identification guide, for example, Syngnathidae, Blenniidae, and others. Our study of barcoding of coastal fishes of Pakistan revealed an addition of at least fourteen finfish species that were not previously recorded by Psomadakis et al. (2015), including Planiliza sp., Pterocaesio chrysozona Cuvier, 1830, Psammogobius biocellatus Valenciennes, 1837, Halichoeres timorensis Bleeker, 1852, Leiognathus robustus Sparks & Dunlap, 2004, Pempheris rhomboidea, Abudefduf septemfasciatus Cuvier, 1830, Saurida lessepsianus, Siganus sutor Valenciennes 1835, Sphyraena pinguis Günther, 1874, Cynoglossus macrostomus Norman, 1928, Scorpaenopsis possi Randall & Eschmeyer 2002, Arius arius Hamilton, 1822, and Chiloscyllium burmensis.
In this study, the average GC content was 47.03% while the average transition and transversion ratio was 1.31; similar results were obtained by Lakra et al. (2011) from the Indian Ocean. Keskin and Atar (2013) and Ward et al. (2005) reported average GC content of 46.74% and 47.1% for their studies, respectively, the results similar to our findings. We found the highest GC content (52.8%) for Chirocentrus dorab (Osteichthyes) and lowest (37.3%) for Chiloscyllium burmensis (Chondrichthyes) while Lakra et al. (2011) recorded the highest GC content (51.20%) for family Carangidae and the lowest (44.7%) for family Leognathidae.
Our overall mean Kimura 2-parameter genetic distance (K2P) was 0.25% that was higher than reported by Zhang and Hanner (2012) for China (0.18%), similar to Canadian Pacific (0.25%; Steinke et al. 2009) and lower from India (0.3%; Lakra et al. 2011), Japan (0.3%; Zhang and Hanner 2011), and Sao Paulo State in Brazil (0.31%; Ribeiro et al. 2012). Ward et al. (2005) reported the highest overall mean intraspecific distance of 0.39% for their study on Australian fishes. In our study, the highest genetic distance (0.365%) was calculated between Narcine timlei (55296) and Otolithes sp.2 (55077). Among 18 previously unknown taxa discernible by single-species representing clusters, five had no previous submission records in GenBank (Cociella punctata, Epinephelus stolizkae, Pomadasys ahenius, Scorpaenopsis possi, and Sillago panhwari). According to Ward (2009), 98–99% similarity in COI sequences of fishes, there is a probability that these are from the same species.
The Clupeiforms is a large group of fishes distributed worldwide. In Pakistan, Clupeiformes is comprised of five families including Clupeidae, Dussumieriidae, Engraulidae Pristigasteridae, and Chirocentridae. Phylogenetic relationships among each family in the clupeformes have been studied and remain poorly understood (Bloom and Lovejoy 2014). The species diversity of clupeoid fishes is highest in the Indo-West Pacific region; species distribution generally follows two patterns of species richness, the longitudinal and latitudinal gradients whereas regional endemism is also important at the species and genus levels (Lavoué et al. 2013). In the present study, BI/ML analysis showed 12 distinct clusters including an unknown single-species representing cluster of Thryssa sp. (55314) diverging at a genetic distance of 14–26% (p distance) from all other Clupeiform species. Detail studies on Thryssa sp. will be carried out separately in future work. In our collection of Clupeiforms, two sequences of Chirocentrus dorab were present, whereas C. nudus is also reported from Pakistani waters but not represented in our study (Psomadakis et al. 2015; Bianchi 1985). Both species are nearly similar in morphological characters and identification of either species is challenging. In a recent study, Lavoué et al. (2019) examined both Chirocentrus species from Indo-West Pacific for their genetic divergences and found a significant p distance (6.3%) among them.
Croakers and drums are members of the family Sciaenidae, comprising more than 290 species in 66 genera. They produce sounds using sonic muscles and swim bladder (Ramcharitar et al. 2006). Sciaenids are highly commercial and are found abundantly in Pakistani waters, contributing towards a major portion of daily fish landing. The systematics and phylogenetic relationship of sciaenids based on swim bladder and otolith was discussed by several authors (Chao 1978; Sasaki 1989) but more recent studies focus on molecular techniques to resolve taxonomic issues in the family Sciaenidae (Lo et al. 2015). In the present study, three of our single-species representing clusters (Otolithes sp.1, Otolithes sp.2, and Johnius sp.) appeared in three distinct clades which reflects that the sciaenids fauna of Pakistan is more productive in terms of diversity but need to be comprehensively examined. Chao et al. (2019) recently described a new species, Johnius taiwanensis Chao et al., 2019, from the west coast of Taiwan. This species has been misidentified for decades, as J. sina Cuvier, 1830, J. macrorhynus Lal Mohan 1976, and J. belangerii Cuvier, 1830 (Chu et al. 1963; Lin et al. 2007).
Flatfishes belong to the order Pleuronectiformes, receiving attention in evolutionary biology from Darwin’s time because of its asymmetry (both eyes lying on the same side of the head (Campbell et al. 2013). This is the species-rich group with more than 800 recognized species in 14 families. Flatfishes are represented in Pakistan by six families: Psettodidae, Paralichthyidae, Bothidae, Samaridae, Soleidae, and Cynoglossidae. Based on morphological analysis, several researchers have done taxonomic studies on flatfishes of Pakistan including Day (1878), Fisher and Bianchi (1984), Ramanathan et al. 1977, Qureshi (1960), Ahmad and Niazi (1988), and Hoda (1988). In the present study, we could not morphologically identify the species falling in five single-species representing clusters (Pseudorhombus sp. (n=5), Cynoglossus sp.1 (n=3), Cynoglossus sp.2 (n=3), Brachirus sp. (n=2), and Phyllichthys sp. (n=1)). The BI/ML analysis shows that these clusters do not correspond to any sequences available in the GenBank nor their morphology corresponds to the known forms. Recently, a new species Cynoglossus nanhaiensis Wang, Munroe and Kong 2016, has been described from the coastal waters of the South China Sea.
The taxonomy of spiny-flatheads has a long history of confusion, with approximately 150 proposed species names, but only 77 are regarded as valid. Cryptic species of spiny-flatheads have been recognized only recently, mostly in large expanses of the tropical Indo-West Pacific (Cheng et al. 2019). A spiny-flathead Cociella crocodilus Cuvier, 1829 was reported by Bianchi (1985) from Pakistani waters. Psomadakis et al. (2015) regard C. crocodilus as extralimital for Pakistan, while restricting this species to the South China Sea. Our present study reveals two distinct Cociella taxons from Pakistani waters namely Cociella punctata Cuvier, 1829, and Cociella heemstrai Knapp, 1996. The genetic divergence between Cociella heemstrai from South Africa (JF493229, JF493230) and Madagascar (JX488290) from near the type locality (Kenya) of this species and those from the Pakistani waters is greater than 2.7% and is suggestive of the presence of a cryptic genetic population. A grunt species P. olivaceus is available in GenBank but not from the type locality i.e., Balochistan coast, Pakistan. The GenBank P. olivaceus from South Africa (JF494253, HQ676798) is proposed as an undescribed genetic population of P. olivaceus with a p distance of more than 3% from the Pakistani P. olivaceus. The stargazer Uranoscopus archionema Regan, 1921 was described from 15–20 miles off Umvoti River, KwaZulu-Natal, South Africa, southwestern Indian Ocean. Another stargazer U. dollfusi Brüss, 1987 was described from the Gulf of Suez, Red Sea, and later on recorded from the Gulf of Oman and Persian Gulf (Fricke 2018). Ronald Fricke (personal communication) did not find trenchant differences in the descriptions of both these Uranoscopus species and showed a possibility of U. dollfusi as a junior synonym of U. archionema. Our Uranoscopus COI sequences did not match with the material of U. archionema from South Africa. At the same, we found contrasting morphological characters differing from the description assigned to U. dollfusi and U. archionema. Based on our morphological data (not presented here) and the present phylogenetic inference, the existence of a potential genetic population in the genus Uranoscopus from the north Arabian Sea is warranted in the future.
Far from their close congeners, several species were placed at different positions in the NJ, BI/ML trees (Fig. S1, Fig. 2). This type of situation has been reported by Keskin and Atar (2013) who reported the taxonomic deviation at genus, family, and order level and explained this type of placement in the NJ and BI/ML trees due to differences in genetic distances. In our study, the genetic distance (K2P) between Mugil cephalus (Mugiliformes) and Abudefduf septemfaciatus (Perciformes) was above the threshold level of confamilials (17.9%) and was found lowest (0.208) as compared to M. cephalus with other mugil species. For example, the genetic divergence between M. cephalus and other mugil species was Ellochelon vaigiensis Quoy & Gaimard, 1825 (0.224), Osteomugil seheli (0.223), Planiliza sp. (0.233), Osteomugil sp. (0.224), and Planiliza klunzingeri Day 1888 (0.226).
DNA barcodes are highly reliable for delineating fish species, species validation, description of new species, and, more importantly, clarifying ambiguities in species complexes and cryptic taxa. In the present study, a high level of K2P/p distances was observed in distinct lineages for several taxa among 138 distinguishable single-species representing clusters through NJ and BI/ML tree-based approaches (Figs. 2 and 3). Our analysis reflects that geographic distance plays an important role in the deep divergence of cryptic species (Zemlak et al. 2009). Furthermore, it is required to establish a DNA barcode reference library for each species inhabiting the aquatic environment of Pakistan for their proper management and conservation. Except for some sporadic work, this is the first initiative to document the fish fauna of Pakistan through DNA barcoding.
References
Ahmad MF, Niazi MS (1988) Edible fishes of Pakistan. Karachi, Pakistan: Zoological Survey Department, Block 61, Pakistan Secretariat, Government of Pakistan, p 31
Aquilino SVL, Tango JM, Fontanilla IKC, Pagulayan RC, Basiao UZ, Ong PS, Quilang JP (2011) DNA barcoding of the ichthyofauna of Taal Lake, Philippines. Mol Ecol Resour 4:612–9. https://doi.org/10.1111/j.1755-0998.2011.03000.x
Basheer VS, Chowdhury LM, Mohitha C, Bineesh KK (2016) Barcoding of Indian marine fishes: for identification and conservation. In: Trivedi S, Ansari A, Ghosh S, Rehman H (eds) DNA barcoding in marine perspectives. Springer International Publishing, Switzerland, pp 243–270. https://doi.org/10.1007/978-3-319-41840-7
Bauchot ML, Desoutter M, McKay RJ (1983) Catalogue critique des types de poissons du Muséum national d’Histoire naturelle. (Suite) (Famille des Haemulidae et des Sillaginidae). Bulletin du Museum National d’Histoire Naturelle, Série 4: Section A: Zoologie, Biologie et Écologie Animales 5(2 Suppl):27–61
Bianchi G (1985) FAO species identification sheets for fishery purposes. Field guide to the commercial, marine and brackish water species of Pakistan. Rome, Italy: Food and Agriculture Organization of the United Nations, p 200
Bloom DD, Lovejoy NR (2014) The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B Biol Sci 281(1778):20132081. https://doi.org/10.1098/rspb.2013.2081
Campbell MA, Chen WJ, López JA (2013) Are flatfishes (Pleuronectiformes) monophyletic? Mol Phylogenet Evol 69(3): 664–673. https://doi.org/10.1016/j.ympev.2013.07.011
Chao NL (1978) A basis for classifying Western Atlantic Sciaenidae (Teleostei: Perciformes). NNOA Technical Report Circular 415:1-65
Chao NL, Chang CW, Chen MH, Guo CC, Lin BA, Liou YY, Shen KN, Liu M (2019) Johnius taiwanensis, a new species of Sciaenidae from the Taiwan Strait, with a key to Johnius species from Chinese waters. Zootaxa 4651 (2): 259–270. https://doi.org/10.11646/zootaxa.4651.2.3
Cheng J, Wang Z, Song N, Yanagimoto T, Gao T (2019) Phylogeographic analysis of the genus Platycephalus along the coastline of the northwestern Pacific inferred by mitochondrial DNA. BMC Evol Biol 19:159. https://doi.org/10.1186/s12862-019-1477-1
Chu YT, Lo YL, Wu HL (1963) A study on the classification of the Sciaenoid fishes of China, with description of new genera and species. Shanghai Science and Technology Press, Shanghai, pp 1–140
Cuvier G, Valenciennes A (1837) Histoire naturelle des poissons. Tome douzième. Suite du livre quatorzième. Gobioïdes. Livre quinzième. Acanthoptérygiens à pectorales pédiculées 12: i-xxiv + 1-507 + 1 p., Pls. 344-368.
Díaz J, Villanova GV, Brancolini F, del Pazo F, Posner VM, Grimberg A, Arranz SE (2016) First DNA barcode reference library for the identification of South American freshwater fish from the Lower Paraná River. PLoS ONE 11(7): e0157419. https://doi.org/10.1371/journal.pone.0157419
Dibattista JD, Choat JH, Gaither MR et al (2016) On the origin of endemic species in the Red Sea. J Biogeogr 43:13–30. https://doi.org/10.1111/jbi.12631
Fisher W, Bianchi G (1984) FAO species identification sheets for fishery purpose. Western Indian Ocean (Fishing Area 51), vol 1–6. Danish International Development Agency (DANIDA), Rome
Fricke R (2018) Two new species of stargazers of the genus Uranoscopus (Teleostei: Uranoscopidae) from the western Pacific Ocean. Zootaxa 4476 (1): 157–167. https://doi.org/10.11646/zootaxa.4476.1.15
Froese R, Pauly D (2020, June 25) FishBase. World Wide Web electronic publication. www.fishbase.org. Accessed 25 June 2020
Harrison LR, Dulvy NK (eds) (2014) Sawfish: a global strategy for conservation. IUCN Species Survival Commission’s Shark Specialist Group, Vancouver
Hebert PDN, Cywinska A, Ball SL, deWaard JR (2003) Biological identifications through DNA barcodes. Biol Sci 270(1512):313–21. https://doi.org/10.1098/rspb.2002.2218
Hoda SMS (1988) Fish from the coast of Pakistan. Biologia 33:1–38
Hou G, Chen WT, Lu HS, Cheng F, Xie SG (2018) Developing a DNA barcode library for perciform fishes in the South China Sea: species identification, accuracy, and cryptic diversity. Mol Ecol Resour 18(1):137–146. https://doi.org/10.1111/1755-0998.12718
Hourston M (2010) Review of the exploitation of marine resources of the Australian Indian Ocean Territories: the implications of biogeographic isolation for tropical island fisheries. Department of Fisheries, Western Australia. Fisheries Research Report 208:1–49
Jordan DS, Seale A (1905) List of fishes collected at Hong Kong by Captain William Finch, with description of five new species. Proceedings of the Davenport Academy of Sciences (Iowa) 10:1-17, Pls. 1–13
Kekkonen M, Mutanen M, Kaila L, Nieminen M, Hebert PDN (2015) Delineating species with DNA barcodes: a case of taxon dependent method performance in moths. PLoS ONE 10(4):e0122481. https://doi.org/10.1371/journal.pone.0122481
Keskin E, Atar HH (2013) DNA barcoding commercially important fish species of Turkey. Mol Ecol Resour 13(5):788–797. https://doi.org/10.1111/1755-0998.12120
Khamees NR, Adday T, Abed JM (2018) Occurrence and redescription of Thryssa setirostris (Broussonet, 1782) (Clupiformes, Engraulidae) from Iraqi Marine water. Bull Iraq Nat Hist Mus 15(2): 123–130. https://doi.org/10.26842/binhm.7.2018.15.2.0123
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16(2):111–120. https://doi.org/10.1007/BF01731581
Kossmann R, Räuber H (1877) Fische. Wissenschftliche Reise in die Küstengebiete des Rothen Meeres. Verhandlungen des Naturhistorisch-Medizinischen Vereins zu Heidelberg 1:378–420, Pl. 3
Kottelat M, Britz R, Tan HH, Witte K-E (2006) Paedocypris, a new genus of southeast Asian cyprinid fish with a remarkable sexual dimorphism, comprises the world's smallest vertebrate. Proc R Soc Edinb B 273(1589):895–9. https://doi.org/10.1098/rspb.2005.3419
Lakra WS, Verma MS, Goswami M et al (2011) DNA barcoding Indian marine fishes. Mol Ecol Resour 11(1):60–71. https://doi.org/10.1111/j.1755-0998.2010.02894.x
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B (2017) PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol 34 (3):772–773. https://doi.org/10.1093/molbev/msw260
Lavoué S, Miya M, Musikasinthorn P, Chen WJ, Nishida M (2013) Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS ONE 8(2):e56485. https://doi.org/10.1371/journal.pone.0056485
Lavoué S, Bertrand JAM, Shen K, Ratmuangkhwang S, Sado T, Miya M, Azizah MNS (2019) Deep genetic differentiation between two morphologically similar species of wolf herrings (Teleostei, Clupeoidei, Chirocentridae). J Appl Ichthyol 36(3): 693–700. https://doi.org/10.1111/jai.13886
Lim H, Zainal-Abidin M, Pulungan CP, deBruyn M, Mohd-Nor SA (2016) DNA Barcoding reveals high cryptic diversity of the freshwater halfbeak genus Hemirhamphodon from Sundaland. PLoS ONE 11(9): e0163596. https://doi.org/10.1371/journal.pone.0163596
Linnaeus C (1758) Systema Naturae, Ed. X. (Systema naturae per regna tria naturae, secundum classes, ordines, genera, species, cum characteribus, differentiis, synonymis, locis. Tomus I. Editio decima, reformata.) Holmiae 1: i-ii + 1-824
Lin YC, Mok HK, Huang BQ (2007) Sound characteristics of big-snout croaker Johnius macrorhynus (Sciaenidae). J Acoust Soc Am 121(1):586–93. https://doi.org/10.1121/1.2384844
Liu L, Panhwar SK, Gao T, Han Z, Li C, Sun D, Song N (2017) New genetic evidence from three keel-backed Liza species based on DNA barcoding confirms morphology-based identification. Pak J Zool 49(5):1901–1907. https://doi.org/10.17582/journal.pjz/2017.49.5.1901.1907
Lo PC, Liu SH, Chao NL, Nunoo FKE, Mok HK, Chen WJ (2015) A multi-gene dataset reveals a tropical New World origin and Early Miocene diversification of croakers (Perciformes: Sciaenidae). Mol Phylogenet Evol 88:132–43. https://doi.org/10.1016/j.ympev.2015.03.025
Locke SA, Mclaughlin D, Marcogleise DJ (2010) DNA barcodes show cryptic diversity and a potential physiological basis for host specificity among Diplostomoidea (Platyhelminthes: Digenea) parasitizing freshwater fishes in the St. Lawrence River, Canada. Mol Ecol 19(13):2813–27. https://doi.org/10.1111/j.1365-294X.2010.04713.x
Miller EC, Hayashi KT, Song D, Wiens JJ (2018) Explaining the ocean’s richest biodiversity hotspot and global patterns of fish diversity. Proc Royal Soc B 285:20181314. https://doi.org/10.1098/rspb.2018.1314
Mohsin M, Mu Y, Memon AM, Kalhoro MT, Shah SBH (2017) Fishery stock assessment of Kiddi shrimp (Parapenaeopsis stylifera) in the Northern Arabian Sea Coast of Pakistan by using surplus production models. Chin J Oceanol Limnol 35(4):936–946. https://doi.org/10.1007/s00343-017-6096-3
Miranda Ribeiro A de (1915) Fauna brasiliense. Peixes. Tomo V. [Eleutherobranchios aspirophoros]. Physoclisti. [A summary and index appeared in Miranda-Ribeiro 1918, ref. 20859; Serranidae, Haemulidae and Sciaenidae published in 1913]. Arquivos do Museu Nacional de Rio de Janeiro 17:[1-679] or 755 pp
Panhwar SK, Farooq N, Qamar N, Shaikh W, Mairaj M (2018) A new Sillago species (family Sillaginidae) with description of six Sillaginids from the northern Arabian Sea. Mar Biodivers 48:2225–2231. https://doi.org/10.1007/s12526-017-0710-7
Pathak V, Shashi B, Rajan K, Shardul G, Jaiswar AK (2016) Discrimination of species of genera Grammoplites and Cociella (Family: Platycephalidae) occurring in Indian waters, based on multi-variate analysis. Indian J Ecol 43:58–64
Polanco FA, Acero PA, Betancur-R R (2016) No longer a circumtropical species: revision of the lizardfishes in the Trachinocephalus myops species complex, with description of a new species from the Marquesas Islands. J Fish Biol 89(2) 1302–1323. https://doi.org/10.1111/jfb.13038
Psomadakis PN, Osmany HB, Moazzam M (2015) Field identification guide to the living marine resources of Pakistan. FAO species identification guide for fishery purposes. Rome, Italy: Food and Agriculture Organization of the United Nations [i–x +] 386 pp. [+ pls. 1–42]
Qamar N, Panhwar SK, Siddiqui G (2016) Fishery status and taxonomy of the Carangids (Pisces) in the northern Arabian Sea coast of Pakistan. In: Mikkola H (ed) Fisheries and Aquaculture in the Modern World. IntechOpen 212. https://doi.org/10.5772/62627
Qureshi MR (1960) Fishes of the order Pleuronectiformes found in Pakistani waters. Proc. 4th PAN Indian Ocean Science Congress Seat B. Biological Sciences, Karachi, 235–241
Rabaoui L, Yacoubi L, Sanna D, Casu M, Scarpa F, Lin YJ, Shen KN, Clardy TR, Arculeo M, Qurban MA (2019) DNA barcoding of marine fishes from Saudi Arabian waters of the Gulf. J Fish Biol 95(5):1286–1297. https://doi.org/10.1111/jfb.14130
Ramanathan N, Vijaya P, Ramaiyan V, Natarajan R (1977) On the biology of the large scaled tongue sole Cynoglossus macrolepidotus (Bleeker). Indian J Fish 24:83–89
Ramcharitar J, Gannon DP, Popper AN (2006) Bioacoustics of fishes of the family Sciaenidae (croakers and drums). Trans Am Fish Soc 135(5):1409–1431. https://doi.org/10.1577/T05-207.1
Randall JE, Eschmeyer WN (2002) Revision of the Indo-Pacific scorpionfish genus Scorpaenopsis, with descriptions of eight new species. Indo-Pacific Fishes No 34:1–79
Ratnasingham S, Hebert PD (2007) BOLD: The Barcode of Life Data System. (http://www.barcodinglife.org). Mol Ecol Notes 7(3): 355–364. https://doi.org/10.1111/j.1471-8286.2007.01678.x
Ratnasingham S, Hebert PDN (2013) A DNA-based registry for all animal species: the Barcode Index Number (BIN) system. PLoS ONE 8(7): e66213. https://doi.org/10.1371/journal.pone.0066213
Ribeiro AO, Caires RA, Mariguela TC, Pereira LHG, Hanner R, Oliveira C (2012) DNA barcodes identify marine fishes of São Paulo State. Brazil. Mol Ecol Resour 12(6):1012–20. https://doi.org/10.1111/1755-0998.12007
Ronquist F, Teslenko M, van der Mark P et al (2012) MRBAYES 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61(3):539–542. https://doi.org/10.1093/sysbio/sys029
Russell BC, Golani D, Tikochinski Y (2015) Saurida lessepsianus a new species of lizardfish (Pisces: Synodontidae) from the Red Sea and Mediterranean Sea, with a key to Saurida species in the Red Sea. Zootaxa 3956(4):559–568. https://doi.org/10.11646/zootaxa.3956.4.7
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4(4):406–25. https://doi.org/10.1093/oxfordjournals.molbev.a040454
Sasaki K (1989) Phylogeny of the family Sciaenidae with notes on its zoogeography (Teleostei, Perciformes). Mem Fac Fish, Hokkaido Univ 36:1–137
Steinke D, Dewaard JR, Gomon MF et al (2017) DNA barcoding the fishes of Lizard Island (Great Barrier Reef). Biodivers Data J 5:e12409. https://doi.org/10.3897/BDJ.5.e12409
Sonet G, Snoeks J, Nagy ZT et al (2019) DNA barcoding fishes from the Congo and the Lower Guinean provinces: assembling a reference library for poorly inventoried fauna. Mol Ecol Resour 19(3): 728–743. https://doi.org/10.1111/1755-0998.12983
Steinke D, Zemlak TS, Boutillier JA, Hebert PDN (2009) DNA barcoding of Pacific Canada’s fishes. Mar Biol 156:2641–2647. https://doi.org/10.1007/s00227-009-1284-0
Thongtam na Ayudhaya P, Muangmai N, Banjongsat N, Singchat W, Janekitkarn S, Peyachoknagul S, Srikulnath K (2017) Unveiling cryptic diversity of the anemonefish genera Amphiprion and Premnas (Perciformes: Pomacentridae) in Thailand with mitochondrial DNA barcodes. Agric Nat Resour 51(3):198–205. https://doi.org/10.1016/j.anres.2017.07.001
Tittensor DP, Mora C, Jetz W, Lotze HK, Ricard D, Berghe EV, Worm B (2010) Global patterns and predictors of marine biodiversity across taxa. Nature 466:1098–1101. https://doi.org/10.1038/nature09329
Wang L, Wu Z, Liu M, Liu W, Zhao W, Liu H, You F (2018) DNA barcoding of marine fish species from Rongcheng Bay. China. Peer J 6: e5013. https://doi.org/10.7717/peerj.5013
Wang Z, Munroe TA, Kong X (2016) A new species of tongue sole (Pisces: Pleuronectiformes: Cynoglossidae: Cynoglossus) from coastal waters of the South China Sea. Proc Biol Soc Wash 129(1):129–143. https://doi.org/10.2988/0006-324X-129.Q2.129
Ward RD (2009) DNA barcode divergence among species and genera of birds and fishes. Mol Ecol Resour 9(4):1077–85. https://doi.org/10.1111/j.1755-0998.2009.02541.x
Ward RD, Zemlak TS, Innes BH, Last PR, Hebert PD (2005) DNA barcoding Australia’s fish species. Philos Trans R Soc Lond B Biol Sci 360(1462):1847–57. https://doi.org/10.1098/rstb.2005.1716
Xiao JG, Song N, Gao TX, McKay RJ (2016) Redescription and DNA barcoding of Sillago indica (Perciformes: Sillaginidae) from the coast of Pakistan. Pak J Zool 48:317–323
Zemlak TS, Ward RD, Connell AD, Holmes BH, Hebert PDN (2009) DNA barcoding reveals overlooked marine fishes. Mol Ecol Resour Suppl s1:237–42. https://doi.org/10.1111/j.1755-0998.2009.02649.x
Zhang JB, Hanner R (2012) Molecular approach to the identification of fish in the South China Sea. PLoS ONE 7(2):e30621. https://doi.org/10.1371/journal.pone.0030621
Zhang JB, Hanner R (2011) DNA barcoding is a useful tool for the identification of marine fishes from Japan. Biochem Syst Ecol 39(1):31–42. https://doi.org/10.1016/j.bse.2010.12.017
Acknowledgements
The first author greatly acknowledges the support of the Chinese Government as a visiting scientist for 6 months under a program of the President’s International Fellowship Initiative (PIFI), Chinese Academy of Sciences (CAS). The research work (DNA barcoding of coastal fishes of Pakistan) was carried out at the Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China. The first author of the paper is much thankful for help from all the lab members including Xue Dong Xiu, Baidong Zhang, Li Yu Long, Li Yu Qiang, Zong Shas Bing, Yu Zhe, Xing TengFei, and Li MengYu. We greatly acknowledge the anonymous reviewers for their valuable comments. The support of Daniel Jablonski is greatly appreciated.
Funding
This project was supported under the CAS President’s International Fellowship Initiative (PIFI), China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for animal testing, animal care, and the use of animals were followed by the authors.
Sampling and field studies
All necessary permits for sampling and observational field studies have been obtained by the authors from the competent authorities.
Data availability
DNA sequences were uploaded to NCBI, with the following GenBank Accessions: MN511846–MN512146, MN530052, and MN530053 (see Table 1). All collecting and sequence data are available on the Barcode of Life Data System (BOLD) under the project “Barcoding of Coastal Fishes of Pakistan (BCFPA).”
Author contribution
Shabir Ali Amir carried out the major research of this paper including field sampling, laboratory analysis, manuscript writing, and revisions; Baidong Zhang contributed mainly on laboratory barcoding analysis; Li Yu Long and Xue Dong Xiu contributed to the laboratory protocols; Rafaqat Masroor carried out part of field sampling and helped in the manuscript preparation; Safia Mushtaq and Shahnaz Rashid mainly helped in field collection of some problematic fish groups; Naveed Ahmed helped in acquiring a collection of flatfishes and morphometric identification of fishes; Jean-Dominique Durand helped in preparing Fig. 1 and in the discussion section of the manuscript; Jinxian Liu designed and advised this research, and made a major contribution to data analyses, manuscript writing, and its revision.
Additional information
Communicated by R. Thiel
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 81 kb)
Rights and permissions
About this article

Cite this article
Amir, S.A., Zhang, B., Masroor, R. et al. Deeper in the blues: DNA barcoding of fishes from Pakistani coast of the Arabian Sea reveals overlooked genetic diversity. Mar. Biodivers. 52, 37 (2022). https://doi.org/10.1007/s12526-022-01272-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12526-022-01272-6