
Abstract
Background
Biliary atresia (BA) is the most common cause of obstructive jaundice in infants. Although the Kasai procedure has greatly improved the prognosis, most patients still need liver transplantation (LT) for long-term survival. The pathogenesis of BA has not been fully clarified, and liver fibrosis in BA is far beyond biliary obstructive cirrhosis.
Data sources
Literature reviews were underwent through PubMed. Persistent inflammation, immune response, biliary epithelial–mesenchymal transition, matrix deposition, decompensated angiogenesis, and unique biliary structure development all contribute to the fibrosis process. Observed evidences in such fields have been collected and form the backbone of this review.
Results
Interactions of the multiple pathways accelerate this process.
Conclusions
Understanding the mechanisms of the liver fibrosis in BA may pave the way to improved survival after the Kasai procedure.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
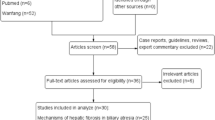
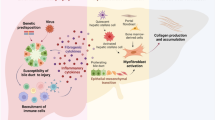
Biliary atresia (BA) is characterized by progressive inflammation and fibrotic sclerosis of the biliary tree. It is the most common cause of obstructive jaundice in infants. Children who are not treated usually die before age 2 years because of cholestasis and progressive liver fibrosis [1]. Although the Kasai procedure has greatly improved the prognosis, most patients still need liver transplantation (LT) for long-term survival, which accounts for over half of the LTs in children [2, 3]. Liver fibrosis in BA progresses is more rapidly than any other hepatic or biliary diseases among adults and children. The underlying pathogenic factors of this disease have not been fully clarified. Considering the complicated etiologies behind BA, its liver fibrosis is far beyond biliary cirrhosis secondary to obstruction [4]. Understanding the pathogenesis of liver fibrosis in BA may improve long-term outcome after operation and decrease needs for LT (Fig. 1). Different from congenital hepatic fibrosis, alpha 1 antitrypsin deficiency, citrin deficiency, progressive familial intrahepatic cholestasis, and Alagille syndrome, BA has its own liver fibrosis features. Much research has been done in the fields of ductal plate malformation, virus infection, immune reaction, and epithelial–mesenchymal transition (EMT). Long-term interactions between different factors result in connective tissue proliferation and diffuse extracellular matrix (ECM) deposition (Fig. 2). Observed evidences in such fields above have been collected and form the backbone of this review, hope it will benefit the further studies.

Possible mechanisms participate in the liver fibrosis of biliary atresia. Virus infection initiates the injury and inflammation around the perinatal period based on the knowledge from mice model. Ductal plate malformation and other associated congenital abnormalities have the constant impacts since gestation. Bacterial cholangitis will play an important role after Kasai procedure. Both virus and bacterial injuries evoke inflammation under immature immune disorders. Epithelial–mesenchymal transition, matrix collagen deposition and angiogenesis are important contributors to the process of liver fibrosis in biliary atresia

Virus infection and bacterial cholangitis cause injuries to the bile duct epithelium, which evoke the immune response and activate the HSC. Bacteria’s LPS, virus’ double-stranded DNA and TGF-β could induce EMT and transfer the bile duct epithelium into fibrogenic myofibroblasts (red arrow). MB and HSC secrete abundant collagens and increase ECM deposition (green arrow), causing the liver parenchyma’s hypoxia and inducing angiogenesis. Because of the immaturity and inflammatory of neo-vessels with high permeability, extravasation serum protein will deteriorate the liver fibrosis consequently (green arrow). ECM extracellular matrix, EMT epithelial–mesenchymal transition, HSC hepatic stellate cell, LPS lipopolysaccharides, MB myofibroblasts, TGF-β transforming growth factor-β
Bile duct development and congenital abnormalities
Human embryological studies have found that extrahepatic bile duct arises from an outpouching of foregut endoderm starting at about 20 days of gestation, while intrahepatic biliary network develops later until at least 49 days, and successful union from both sides is expected around 11–12 weeks. The intrahepatic bile duct is formed from the hepatic stem cells near the hilar portal vein and its intrahepatic branches to a double-layered epithelial structure, so-called ductal plate (DP), forms from the hepatic portal to the peripheral region [5]. Bile duct development and remodeling are regulated by genetic, immune, and micro-environmental factors. Maldevelopment of intrahepatic bile ducts is regarded to occur by failure of the DP remodeling process between 11 and 13 week gestation and becomes clinically evident after birth, as ductal plate malformation (DPM) [6]. BA patients with DPM have more severe fibrosis, inflammatory infiltration, and bridging necrosis [6] will have a shorter native liver survival [7].
Bile duct proliferation is seen in liver sections of neonatal infants with obstructive jaundice, especially in BA. The improper ductal plate resembling the DP network was reconstructed by computer-generated 3D reconstructions study from serial BA liver sections [8]. Jose and colleagues have found that hepatocytes undergo metaplasia and ductular proliferation instead of replication to form ductular cells in BA livers [9]. Such proliferation ductal reactions are typically present within the regions of bridging fibrosis, where collagen-producing myofibroblasts origin from the portal fibroblasts [10]. These ductal reactive cells express both epithelial and mesenchymal markers [11, 12]. Progenitor/stem cell marker expressing cells within and adjacent to the ductular reactions can also stimulated collagen-1α expression and periportal fibrosis [13]. All patients with DP maldevelopment and malfunction have worse bile drainage, which occurs diversely from 15.4 to 69.2% in BA [6, 7].
As we all know, BA patients have malformation beyond hepatic and biliary system. There are certain BA infants with biliary atresia splenic malformation (BASM) and have splenic abnormalities, vascular anomalies, situs inversus, and cardiac anomalies [14]. This group accounts 10% of whole BA population, featuring with an earlier onset of jaundice and extrahepatic bile ducts absence [15]. CFC-1 missense mutations have been showed in around 50% BASM cases indicating a genetic context in this subgroup [16]. It is reported that 13% BA patients had associated congenital abnormalities, with 3.6% portal vein anomaly, 6.0% inferior vena cava interruption, and 7.9% cardiac malformations [17]. Vascular malformations mentioned above could cause portal cirrhosis and liver fibrosis with long-term increasing pressure within the portal vein or inferior vena cava [18].
Virus infection, immune disorders, and inflammation
Liver inflammatory injury with different causes finally activates hepatic stellate cells (HSCs), Kupffer cells, and sinusoidal endothelial cells, initiating liver fibrosis.
The most discussed hypothesis of BA is virus infection in the perinatal period triggers immune–inflammatory injury. Inappropriate innate immune response to double-stranded RNA viruses resulted in a progressive, inflammatory, and sclerosing cholangiopathy in biliary atresia [19, 20]. For decades, viruses have been implicated as a potential trigger for aggressive auto immune response to the infection [21, 22]. Hepatotropic viruses caused an inflammatory reaction damaging the bile duct via blood flow to the liver. Nevertheless, the debate about whether the viruses are simply innocent bystanders or integral to the pathogenesis still continues. A group from Hannover, Germany looked at multiple possible viruses using polymerase chain reaction and found nucleic acid evidence of reovirus (RV) in 33%, cytomegalovirus (CMV) in 11%, enterovirus in 1.5%, and adenovirus in 1% [23]. Maybe, such viruses were just selected by the absence of bile salts [24]. The absence of dominant active virus detected in BA patients and multiple virus positive trending in older cases leave the hypothesis pending, and further investigations are needed.
Bacterial cholangitis is a common scenario in BA after Kasai procedure, which affects more than 50% of patients [25]. Recurrent cholangitis results in bile flow obstruction with a subsequent deterioration of the hepatic function, bile flow and ongoing cirrhosis [26], increasing mortality, and worsening prognosis [27]. The possible explanation is that ascending bacterial infection originating from the enteric conduit, and further blood cultures included Klebsiella pneumoniae, Enterococcus, Escherichia coli, and Pseudomonas aeruginosa [25]. Bacterial components including lipopolysaccharides induce and maintain the activation of chronic inflammation immune cells in the periductal area [20]. CMV IgM +ve BA group still has a decreased clearance of jaundice and a significantly increased mortality [28], which shows hepatotropic virus also played an important role in the post-operatively disease progress. Inflammation and fibrosis persisted in the CMV infection BA patients’ livers, and severe hepatocyte necrosis and portal area inflammation were observed even after postoperative relief of jaundice [28].
Hepatotropic virus infections also evoke inflammatory pathways and mediators, causing liver and bile duct injury and inflammatory further reaction. Biliary epithelial antigen exposure and inflammatory reactions trigger and promote liver fibrosis even after when virus could not be detected. Virus antigens activate CD4 +/CD8+ T cells and Kupffer cells via interferon (IFN)-γ, while T helper 1 cell cytokine profile is present in the portal tracts of patients with BA [29]. The over reactivated T cells express CD14, inducible nitric oxide synthase, perforin, IFN, interleukin (IL), granzyme, and tumor necrosis factor (TNF) [30]. Such cytokines, viruses, and necrotic decompositions evoke immune cells and continue to increase bile duct epithelial and liver cells necrosis and apoptosis. The imbalanced environment and inflammatory pathways finally activate HSCs, facilitate fibrogenic myofibroblasts, deposit collagen, and promote biliary fibrosis [31]. Plenty of virus induced animal models supported the proof of the abnormal inflammation and autoimmunity in BA [5, 32,33,34]. RV targeting biliary epithelia caused tissue-specific inflammatory injury in mice [34], and some RV antigens triggered an autoimmune reaction [32]. Bile duct defects were induced in zebrafish through DNA hypomethylation, and elevated IFN-γ level was shown to be the common pathway of multiple inflammatory injuries in BA [35]. Such cross-reactivity loop continues even after virus clearance and Kasai procedure.
Evidence of humoral and cellular immune attack has been found in BA models. Small amounts of bacterial toxins and lipopolysaccharide can make liver macrophages produce cytokines like TNF-α, transforming growth factor (TGF), and platelet-derived growth factor to activate HSCs. Contamination and retrograde infection by intestinal bacteria after Kasai procedure continuously activate CD14 macrophages in the BA liver to promote the inflammatory reaction, bile duct damage, and fibrogenesis [36]. T cells activated by the bile duct antigens induce biliary epithelial necrosis and apoptosis through perforin, granzyme, Fas/FasL, and locally released cytokines [30]. RV-primed CD8 + cells were proven cytotoxicity to duct epithelium in both coculture and BA mouse model [37]. Lu et al. found that α-enolase from Rhesus rotavirus (RRV) activated humoral autoimmunity in BA animals and patients [32]. The cross-talk between bile duct epithelium and parenchymatous HSC is one of the events chain in BA liver fibrosis [29, 30, 38, 39].
The cholangiocytes of extrahepatic bile ducts secreted IL-33 to increase proliferation after RRV infection. The elevation of IL-33 was mediated by upper-adjust type 2 innate lymphoid cells (ILC2s) to release IL-13 [38]. This paracrine system of IL-33-ILC2s-IL-13 axis regulated adjacent bile duct epithelial cells’ proliferation. Furthermore, IL-33 activates HSC and promotes hepatic fibrosis [39]; such biliary epithelium-humoral/cellular immune-HSC axis may also play a role in the liver parenchyma fibrosis.
Regulatory T (Treg) cells play important roles in the infectious and autoimmune diseases. An absence of Treg cells may open the time window for the biliary inflammation in murine model without inhibiting NK cell expansion [40]. It is important to maintain a stable immune environment through regulatory cytokines with an immunosuppressive effect. Th17 cells function through IL-17α, which can regulate inflammatory infiltration to activate the nuclear-factor-κB-mediated inflammatory response and induce the expression of IL-6, TNF, monocyte chemoattractant protein-1, and macrophage inflammatory protein-2 to damage liver and bile duct tissue [41]. The inflammatory pathway proliferates Th17 cells also inhibits immunosuppressive Treg cells. One study has found increased Th17 and decreased Treg cells in BA blood samples, and the imbalance is more obvious with aggravating fibrosis [41]. The infiltration of Th-17 in the liver was associated with a worse surgical outcome in BA [42]. Loss of adequate numbers of Treg cells in BA could be responsible for decreased inhibition of inflammation or autoreactivity [43]. Sustained immune disorders after the Kasai procedure have contributed to the development of liver fibrosis in BA.
Matrix collagen deposition after Kasai procedure
Biliary epithelia, Kupffer cells, HSCs, and hepatocytes secrete abundant collagen, increase ECM deposition, and facilitate liver fibrosis [44]. Activated HSCs can transform into myofibroblasts and fibroblasts, which are both effector cells secreting matrix proteins in fibrosis [31] (Fig. 2). Such effector cells synthesizing collagen in BA liver are up-regulated by microRNA (miR)-29, TNF-α, TGF-β, and IL-17 [45,46,47]. Compared to other cholestatic liver disease, BA livers had increased integrin alpha 3 and decreased laminin beta 1 in matrix [48].
Matrix metalloproteinases (MMPs) are a family of proteolytic enzymes that target the ECM. Most MMPs mainly degrade types I and III collagen, preventing matrix deposition and fibrosis in the liver. Recent study found that MMP-7 had the greatest increased fold in BA liver among MMPs [49]. Tissue inhibitors of metalloproteinase (TIMPs) are specific suppressors of MMPs. It has been found that MMPs and TIMPs are regulated in BA patients [50]. The synthetic action of MMPs and TIMPs affects the quantity and pattern of ECM. When targeted by different etiological factors, the ongoing imbalance between MMPs and TIMPs may be the initial step in collagen deposition.
Epithelial–mesenchymal transition
Epithelial–mesenchymal transition (EMT) is a basic pathophysiological phenomenon that plays an important role in embryonic development and tissue reconstruction. Polarized epithelial cells change phenotype into fibrogenic myofibroblasts through EMT. Recent research has found EMT undergoing in BA characteristic with bile ductular proliferation, obliteration and liver fibrogenesis [51], which could be developed by innate immunity against double-stranded RNA [19] (Fig. 2).
TGF-β could induce EMT and collagen deposition in cultured mouse hepatocytes [47]. There is strong staining of TGF-β mRNA in biliary epithelia of BA, which triggers the fibrosis [52] and is affected significantly by miR-200b [53]. The Wnt pathway was expressed in murine HSCs by bile duct ligation [54], which could activate the HSCs, induce fibrogenic myofibroblasts, and lead to hepatic fibrosis through EMT. Moreover, the cadherin, Snail/Slug, Shh, and Notch pathways also mediate and transform the biliary epithelial EMT [55]. All these pathways interact and create a complicated cell signaling coordination network in BA fibrosis. EMT is one of the BA liver fibrosis features, which form the cornerstone of characteristic portal fibrosis in patients and animal models’ liver [4, 7, 28, 45, 51].
Angiogenesis
Angiogenesis is a hypoxia-stimulated and dynamic process. Fibrosis and angiogenesis are found coexisting in many diseases [56]. Some studies have found a significant increasing in angiogenesis in liver cirrhosis [57,58,59]. Diffuse fibrosis decreases the blood flow and induces liver cell hypoxia in cirrhosis. Increased permeability of the sinusoidal endothelial cell worsens hypoxia by intrahepatic shunts formation, vascular contraction, and embolism. Hypoxia as a strong stimulating factor also promotes the expression of IL-1, IL-8, and TGF-β, which elevate the mRNA and protein level of vascular endothelial growth factor (VEGF) [57]. VEGF is strongly stained in hepatocytes, liver sinusoidal endothelial cells, and intrahepatic vascular endothelial cells [58, 60], which could induce HSC proliferation and activation [61].
Elevated VEGF can relieve hypoxia and maintain liver cell function by inducing liver sinusoidal endothelial cell division, hepatic sinus formation, and intrahepatic angiogenesis [62]. However, the anatomic and fluid changes in chronic liver diseases increased resistance to blood flow and oxygen delivery from the sinusoids to the parenchyma [63]. Decompensation cannot improve the blood supply in BA eventually, because of the immaturity and inflammatory of new VEGF-induced neo-vessels with high permeability and the blockage of the lumen with exosmic plasma fibrinogen (Fig. 2). Persistent hypoxia continually activates the HSCs and promotes liver fibrosis through VEGF, which might be a potential therapeutic target for BA liver fibrosis [64].
Conclusions
In summary, the complicated etiological mechanisms in BA, unique biliary structure development, persistent post-operatively inflammation and immune response, biliary EMT, matrix deposition, and decompensated angiogenesis play important roles in the liver fibrosis of BA. Interactions of the multiple pathways accelerate this process. Understanding the mechanisms of the liver fibrosis in BA may pave the way to improved survival after the Kasai procedure. Research in this field may find potential targets to relieve the liver fibrosis.
References
Bates MD, Bucuvalas JC, Alonso MH, Ryckman FC. Biliary atresia: pathogenesis and treatment. Semin Liver Dis. 1998;18:281–93.
Bessho K. Complications and quality of life in long-term survivors of biliary atresia with their native livers. J Pediatr. 2015;167:1202–6.
Shneider BL, Mazariegos GV. Biliary atresia: a transplant perspective. Liver Transpl. 2007;13:1482–95.
Gibeli NE, Tanuri U, de Mello ES, Rodrigues CJ. Bile duct ligation in neonatal rats: is it a valid experimental model for biliary atresia studies? Pediatr Transplant. 2009;13:81–8.
Mack CI, Tuker RM, Lu BR, Sokol RJ, Fontenot AP, Ueno Y, et al. Cellular and humoral autoimmunity directed at bile duct epithelia in murine biliary atresia. Hepatology. 2006;44:1231–9.
Shimadera S, Iwai N, Deguchi E, Kimura O, Ono S, Fumino S, et al. Significance of ductal plate malformation in the postoperative clinical course of biliary atresia. J Pediatr Surg. 2008;43:304–7.
Safwan M, Ramachandran P, Vij M, Shanmugam N, Rela M. Impact of ductal plate malformation on survival with native liver in children with biliary atresia. Pediatr Surg Int. 2015;31:837–43.
Vijayan V, El Tan C. Computer-generated three-dimensional morphology of the hepatic hilar bile ducts in biliary atresia. J Pediatr Surg. 2000;35:1230–5.
Cocjin J, Rosenthal P, Buslon V, Luk L Jr, Barajas L, Geller SA, et al. Bile ductule formation in fetal, neonatal, and infant livers compared with extrahepatic biliary atresia. Hepatology. 1996;24:568–74.
Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–18.
Desmet VJ. Ductal plates in hepatic ductular reactions. Hypothesis and implications. I. Types of ductular reaction reconsidered. Virchows Arch. 2011;458:251–9.
Stamp LA, Braxton DR, Wu J, Akopian V, Hasegawa K, Chandrasoma PT, et al. The GCTM-5 epitope associated with the muciun-like glycoprotein FCGBP marks progenitor cells in tissues of endodermal origin. Stem Cells. 2012;30:1999–2009.
Mavila N, James D, Shivakumar P, Nguyen MV, Utley S, Mak K, et al. Expansion of promnin-1-expressing cells in association with fibrosis of biliary atresia. Hepatology. 2014;60:941–53.
Shneider BL, Brown MB, Haber B, Whitington PF, Schwarz K, Squires R, et al. A multicenter study of the outcome of biliary atresia in the United States, 1997 to 2000. J Pediatr. 2006;148:467–74.
Schwarz KB, Haber BH, Rosenthal P, Mack CL, Moore J, Bove K, et al. Extrahepatic anomalies in infants with biliary atresia: results of a large prospective North American multicenter study. Hepatology. 2013;58:1724–31.
Davit-Spraul A, Baussan C, Hermeziu B, Bernard O, Jacquemin E. CFC1 gene involvement in biliary atresia with polysplenia syndrome. J Pediatr Gastroenterol Nutr. 2008;46:111–2.
Guttman OR, Roberts EA, Schreiber RA, Barker CC, Ng VL. Canadian pediatric hepatology research group. Biliary atresia with associated structural malformations in Canadian infants. Liver Int. 2011;31:1485–93.
Wells ML, Fenstad ER, Poterucha JT, Hough DM, Young PM, Araoz PA, et al. Imaging findings of congestive hepatopathy. Radioqraphics. 2016;36:1024–37.
Nakanuma Y, Sasaki M, Harada K. Autophagy and senescence in fibrosing cholangiopathies. J Hepatol. 2015;62:934–45.
Harada K, Nakanuma Y. Biliary innate immunity: function and modulation. Mediators Inflamm. 2010;2010:373878.
Zani A, Quaglia A, Hadzić N, Zuckerman M, Davenport M. Cytomegalovirus-associated biliary atresia: an aetiological and prognostic subgroup. J Pediatr Surg. 2015;50:1739–45.
Mack CL. The pathogenesis of biliary atresia: evidence for a virus-induced autoimmune disease. Semin Liver Dis. 2007;27:233–42.
Rauschenfels S, Krassmann M, Al-Masri AN, Verhagen W, Leonhardt J, Kuebler JF, et al. Incidence of hepatotropic viruses in biliary atresia. Eur J Pediatr. 2009;168:469–76.
Kobayashi A, Kawai S, Ohbe Y, Benno Y. Fecal flora of infants with biliary atresia: effects of the absence of bile on fecal flora. Am J Clin Nutr. 1988;48:1211–3.
Lee JY, Lim LT, Quak SH, Prabhakaran K, Aw M. Cholangitis in children with biliary atresia: health-care resource utilisation. J Paediatr Child Health. 2014;50:196–201.
Lien TH, Bu LN, Wu JF, Chen HL, Chen AC, Lai MW, et al. Use of Lactobacillus casei rhamnosus to prevent cholangitis in biliary atresia after Kasai pperation. J Pediatr Gastroenterol Nutr. 2015;60:654–8.
Bu LN, Chen HL, Chang CJ, Ni YH, Hsu HY, Lai HS, et al. Prophylactic oral antibiotics in prevention of recurrent cholangitis after the Kasai portoenterostomy. J Pediatr Surg. 2003;38:590–3.
Zani A, Quaqlia A, Hadzić N, Zuckerman M, Davenport M. Cytomegalovirus-associated biliary atresia: an aetiological and prognostic subgroup. J Pediatr Surg. 2015;50:1739–45.
Kotb MA, El Henawy A, Talaat S, Aziz M, El Tagy GH, El Barbary MM, et al. Immune-mediated liver injury: prognostic value of CD4 + , CD8 + , and CD68 + in infants with extrahepatic biliary atresia. J Pediatr Surg. 2005;40:1252–7.
Feldman AG, Mack CL. Biliary atresia: cellular dynamics and immune dysregulation. Semin Pediatr Surg. 2012;21:192–200.
Vejchapipat P, Poomsawat S, Chongsrisawat V, Honsawek S, Poovorawan Y. Elevated serum IL-18 and interferon-gamma in medium-term survivors of biliary atresia. Eur J Pediatr Surg. 2012;22:29–33.
Lu BR, Brindley SM, Tucker RM, Lambert CL, Mack CL. α-enolase autoantibodies cross-reactive to viral proteins in a mouse model of biliary atresia. Gastroenterology. 2010;139:1753–61.
Hertel PM, Crawford SE, Bessard BA, Estes MK. Prevention of cholestasis in the murine rotavirus-induced biliary atresia model using passive immunization and nonreplicating virus-like particles. Vaccine. 2013;31:5778–84.
Mohanty SK, Donnelly B, Lobeck I, Walther A, Dupree P, Coots A, et al. The SRL peptide of rhesus rotavirus VP4 protein governs cholangiocyte infection and the murine model of biliary atresia. Hepatology. 2017;65:1278–92.
Matthews RP, Eauclaire SF, Mugnier M, Lorent K, Cui S, Ross MM, et al. DNA hypomethylation causes bile duct defects in zebrafish and is a distinguishing feature of infantile biliary atresia. Hepatology. 2011;53:905–14.
Chou MH, Chuang JH, Eng HL, Chen CM, Wang CH, Chen CL, et al. Endotoxin and CD14 in the progression of biliary atresia. J Transl Med. 2010;21(8):138.
Shivakumar P, Sabla G, Mohanty S, McNeal M, Ward R, Stringer K, et al. Effector role of neonatal hepatic CD8 + lymphocytes in epithelial injury and autoimmunity in experimental biliary atresia. Gastroenterology. 2007;133:268–77.
Li J, Rzaumilava N, Gores GJ, Walters S, Mizuochi T, Mourya R, et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J Clin Invest. 2014;124:3241–51.
Marvie P, Lisbonne M, L’helgoualc’h A, Rauch M, Turlin B, Preisser L, et al. Interlecukin-33 overexpression is associated with liver fibrosis in mice and humans. J Cell Mol Med. 2010;14:1726–39.
Tucker RM, Feldman AG, Fenner EK, Mack CL. Regulatory T cells inhibit Th1 cell-mediated bile duct injury in murine biliary atresia. J Hepatol. 2013;59:790–6.
Brindley SM, Lanham AM, Karrer FM, Tucker RM, Fontenot AP, Mack CL. Cytomegalovirus-specific T-cell reactivity in biliary atresia at the time of diagnosis is associated with deficits in regulatory T cells. Hepatology. 2012;55:1130–8.
Hill R, Quaqlia A, Hussain M, Hadzic N, Mieli-Vergani G, Vergani D, et al. Th-17 cells infiltrate the liver in human biliary atresia and are related to surgical outcome. J Pediatr Surg. 2015;50:1297–303.
Yang Y, Liu YJ, Tang ST, Yang L, Yang J, Cao GQ, et al. Elevated Th17 cells accompanied by decreased regulatory T cells and cytokine environment in infants with biliary atresia. Pediatr Surg Int. 2013;29:1249–60.
Schulze F, Schardt K, Wedemeyer I, Konze E, Wendland K, Dirsch O, et al. Epithelial–mesenchyal transition of biliary epithelial cells in advanced liver fibrosis. Verh Dtsch Ges Pathol. 2007;91:250–6 (in German).
Ogawa T, Lizuka M, Sekiya Y, Yoshizato K, Ikeda K, Kawada N. Suppression of type I collagen production by microRNA-29b in cultured human stellate cells. Biochem Biophys Res Commun. 2010;39:316–21.
Amara S, Lopez K, Banan B, Brown SK, Whalen M, Myles E, et al. Synergistic effect of pro-inflammatory TNFα and IL-17 in periostin mediated collagen deposition: potential role in liver fibrosis. Mol Immunol. 2015;64:26–35.
Kaimori A, Potter J, Kaimori JY, Wang C, Mezey E, Koteish A. Transforming growth factor-betal induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem. 2007;282:22089–101.
Whitby T, Schroeder D, Kim HS, Petersen C, Dirsch O, Baumann U, et al. Modifications in integrin expression and extracellular matrix composition in children with biliary atresia. Klin Padiatr. 2015;227:15–22.
Huang CC, Chuang JH, Chou MH, Wu CL, Chen CM, Wang CC, et al. Matrilysin (MMP-7) is a major matrix metalloproteinase upregulated in biliary atresia-associated liver fibrosis. Mod Pathol. 2005;18:941–50.
Kerola A, Lampela H, Lohi J, Heikkilä P, Mutanen A, Hagström J, et al. Increased MMP-7 expression in biliary epithelium and serum underpins native liver fibrosis after successful portoenterostomy in biliary atresia. J Pathol Clin Res. 2016;2:187–98.
Deng YH, Pu CL, Li YC, Zhu J, Xiang C, Zhang MM, et al. Analysis of biliary epithelial–mesenchymal transition in portal tract fibrogenesis in biliary atresia. Dig Dis Sci. 2011;56:731–40.
Li FB, Zhao H, Peng KR, Gao ZG, Huang SJ, Tou JF, et al. Expression of transforming growth factor-β1 and connective tissue growth factor in congenital biliary atresia and neonatal hepatitis liver tissue. Genet Mol Res. 2016. https://doi.org/10.4238/gmr.15017217.
Xiao Y, Zhou Y, Chen Y, Zhou K, Wen J, Wang Y, et al. The expression of epithelial–mesenchymal transition-related proteins in biliary epithelial cells is associated with liver fibrosis in biliary atresia. Pediatr Res. 2015;77:310–5.
Miao CG, Yang YY, He X, Huang C, Huang Y, Zhang L, et al. Wnt signaling in liver fibrosis: progress, challenges and potential directions. Biochimie. 2013;95:2326–35.
Kurioka K, Wato M, Iseki T, Tanaka A, Morita S. Differential expression of the epithelial mesenchymal transition factors Snail, Slug, Twist, TGF-β, and E-cadherin in ameloblastoma. Med Mol Morphol. 2017;50:68–75.
Paternostro C, David E, Novo E, Parola M. Hypoxia, angiogenesis and liver fibrogenesis in the progression of chronic liver diseases. World J Gastroenterol. 2010;16:281–8.
Corpechot C, Barbu V, Wendum D, Kinnman N, Rey C, Poupon R, et al. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology. 2002;35:1010–21.
Lee HC, Chang TY, Yeung CY, Chan WT, Jiang CB, Chen WF, et al. Genetic variation in the vascular endothelial growth factor gene is associated with biliary atresia. J Clin Gastroenterol. 2010;44:135–9.
Yang L, Kwon J, Popv Y, Gajdos GB, Ordog T, Brekken RA, et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterology. 2014;146:1339–50.
DeLeve LD. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology. 2015;61:1740–6.
Liu Y, Wang Z, Wang J, Lam W, Kwong S, Li F, et al. A histone deacetylase inhibitor, largazole, decreases liver fibrosis and angiogenesis by inhibiting transforming growth factor-β and vascular endothelial growth factor signaling. Liver Int. 2013;33:504–15.
Yu ZY, Bai YN, Luo LX, Wu H, Zeng Y. Expression of microRNA-150 targeting vascular endothelial growth factor-A is downregulated under hypoxia during liver regeneration. Mol Med Rep. 2013;8:287–93.
Thabut D, Shah V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: new targets for the treatment of portal hypertension? J Hepatol. 2010;53:976–80.
Nakamura l, Zakharia K, Banini BA, Mikhail DS, Kim TH, Yang JD, et al. Brivanib attenuates hepatic fibrosis in vivo and stellate cell activation in vitro by inhibition of FGF, VEGF and PDGF signaling. PLoS One. 2014;9:e92273.
Funding
This work was supported by the Natural Science Foundation of China (No. 81400576).
Author information
Authors and Affiliations
Contributions
WJS and MW reviewed and wrote the draft, and SZ and GC directed and edited this scientific work. All the authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethical approval
This work had the permit from IRB of the Children’s Hospital of Fudan University.
Conflict of interest
The authors have no conflicts of interest or ties to disclose.
Rights and permissions
About this article

Cite this article
Shen, WJ., Chen, G., Wang, M. et al. Liver fibrosis in biliary atresia. World J Pediatr 15, 117–123 (2019). https://doi.org/10.1007/s12519-018-0203-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12519-018-0203-1