
Abstract
In this study, small RNA sequencing has been performed to identify the miRNAs and explore their regulatory mechanism in T. sinensis sprouts. From HPLC analysis, the flavonoid content was higher in the BYC2 (purple toon sprout) than in the GYC2 (green toon sprout), whereas the volatile terpenoids revealed an inverse change. Therefore, the small RNA libraries of two varieties were constructed, and 331 known miRNAs and 23 novel miRNAs were discovered. Differential gene expression analysis demonstrated the upregulation of 25 miRNAs and the downregulation of 27 miRNAs between BYC2 and GYC2. qRT-PCR analysis showed that nine miRNAs involved in the biosynthetic regulation of flavonoids were downregulated in the BYC2, whereas eight miRNAs related to the regulation of the biosynthesis of terpenoids were upregulated, when compared with GYC2. The results indicated that the differential expression of the miRNAs mentioned above played a crucial role in the regulation of the formation of the flavonoids and terpenoids in T. sinensis sprouts. The present study not only fills in the paucity of knowledge regarding the T. sinensis miRNA, it also provides more valuable information for the genetic improvement of T. sinensis cultivation for the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Toona sinensis (A. Juss) Roem, commonly called the Chinese toon, is a deciduous tree of Meliaceae and is widely distributed across Asia, including China, India, and North Korea (Edmonds and Staniforth 1998). The sprouts or the young shoots of the plant are very popular in vegetarian cuisine for their unique flavor and high nutrition (Zhai and Granvogl 2019). The consumption of toon sprouts as raw material, as toon tea or toon beef sauce is favored by consumers all over the world (Zhao et al. 2019). The seeds, roots, bark, and leaves too, have been well known over a long time for their oriental medicinal properties (Chen et al. 2019). These characteristics in T. sinensis are induced by the presence of a large number of natural active compounds, such as polyphenols, flavonoids and terpenoids, etc. (Shen et al. 2017; Peng et al. 2019). Besides the nutritional value, the high concentration of secondary metabolites present in the toon sprouts showed the beneficial effects on human health, which increased the interest of the researchers to discover the details regarding the secondary metabolic pathways.
Since T. sinensis is a non-model plant and its genomic background still remains unknown, many of the reports available at present dealing with the secondary metabolic pathways of T. sinensis were based on the transcriptome sequencing analysis (Zhang et al. 2016; Sui et al. 2019; Zhao et al. 2019). In our earlier study, some dominant genes involved in the biosynthetic pathways of the flavonoids were identified in the toon bud transcriptome and the anthocyanin contents were comparatively analyzed between the purple toon (Black Youchun: BYC2) and green toon (Green Youchun: GYC2) (Zhao et al. 2017). Many putative unigenes, related to the terpenoids metabolism were also examined in the postharvest toon buds after cold storage (Zhao et al. 2019). Two terpene synthase genes related to the terpenoids biosynthesis were isolated and functionally characterized (Hsu et al. 2012). Liu et al. (2019) have accomplished the sequencing of the complete chloroplast genome of T. sinensis using the Illumina sequencing platform. Apart from these reports, no other molecular information is available on the biosynthesis and regulation of the secondary metabolites in T. sinensis.
To a large degree, plant development and the biosynthesis of its secondary metabolites are regulated by the microRNAs (miRNAs) and their targets, encoding the functional genes or transcription factors (Samad et al. 2017). As post-transcriptional regulators, the miRNAs are an endogenous group of non-coding small molecular single RNA strands, 20–24 nucleotides (nt) in length (Bulgakov and Avramenko. 2015; Singh et al. 2018Zhang et al. 2018). In plants, the miRNAs reveal high diversity and conservative properties across species, with one of their distinct features being the hairpin structure in their precursors. The plant miRNAs show high complementarity to target the transcripts through base pairing and mediate endogenous specific messenger RNA (mRNA) cleavage or translation repression at the post-transcriptional level, which results in silencing the target gene (Rogers and Chen 2013). In general, the miRNAs are a class of key regulators of physiological processes in plants.
The involvement of the miRNAs in the post-transcriptional regulation of the biosynthesis of the secondary metabolites in medicinal plants has been reported in Salvia miltiorrhiza, Catharanthus roseus, Picrorhiza kurroa, Xanthium strumarium, and Rauvolfia serpentina (Fan et al. 2015; Prakash et al. 2015, 2016; Vashisht et al. 2015; Xu et al. 2015). Many genes related to the flavonoid biosynthesis were targeted by the miRNAs, like phenylalanine ammonia lyase (PAL), dihydroflavonol 4-reductase (DFR), 4-coumarate CoA ligase (4CL), chalcone synthase (CHS) targeted by the miR1873, miR172i, and miR829.1, respectively (Biswas et al. 2016). Besides, some important genes were related to the terpenoid biosynthesis including acetyl-CoA-acetyltransferase (AACT), 3-hydroxy-3-methyl glutaryl coenzyme A reductase (HMGR), and 1-deoxy-D-xylulose-5-phosphate synthase (DXS) targeted by the miR5072 (Xu et al. 2015), miR1134 (Fan et al. 2015), and miR156 (Singh et al. 2016). Moreover, some transcription factors, such as myeloblastosis (MYB) and SQUAMOSA promoter-binding protein-like (SPL) proteins play crucial roles in the regulation of the biosynthesis of flavonoids, anthocyanins and terpenoids. Gene encoding transcription factors mentioned above are targeted by the miRNAs, just as MYB and SPL targeted by miR858, miR159, and miR156, respectively (Yu et al. 2015; Sharma et al. 2016).
Although the miRNAs have been identified in many medicinal plants, no reports are available at present on the miRNAs of T. sinensis. Toon sprouts are rich in flavonoids and anthocyanins. However, the flavonoid content varies among the different T. sinensis varieties. Our prior works showed that the total flavonoid and anthocyanin concentrations in the BYC2 were remarkably higher than those in the GYC2 (Zhao et al. 2017). Therefore, the two T. sinensis varieties provided an ideal model for a clear understanding of the miRNA-mediated regulation of the flavonoid biosynthesis at the post-transcriptional level. Besides, the unique flavor of the toon sprouts or shoots is closely related to the formation of its volatile terpenoids (Zhao et al. 2019). However, it is not clear that the miRNAs participate in the molecular events of the post-transcriptional regulation of the terpenoid biosynthesis in toon sprouts. The present study focuses on the identification of miRNAs and their targets together with our earlier transcriptome data in two toon sprout cultivars, i.e., the BYC2 vs GYC2. These results of this study revealed the expression profiles and regulation patterns of miRNAs involved in the flavonoids and terpenoids biosynthesis of toon sprouts. The miRNAs, which play potentially important roles in the formation of secondary metabolites, were identified and analyzed in toon sprouts, and their expression levels were experimentally validated by quantitative real-time PCR (qRT-PCR). To the best of our knowledge, this is the first report on sRNA high throughput sequencing in T. sinensis. Our results will be a valuable resource for the investigation of miRNA-mediated regulation of the flavonoids and terpenoids biosynthesis in toon sprouts.
Material and Methods
Plant Materials
Toon sprouts (the first and second leaves from the top sprouts) were randomly collected from two T. sinensis cultivars (BYC-2 vs. GYC-2) raised under natural environmental conditions in the T. sinensis industry demonstration zone at Taihe County, Anhui, China, in April 2019. Samples were collected according to our previous protocol (Zhao et al. 2017). The toon sprouts samples thus collected were immediately frozen in liquid nitrogen and stored at − 80 °C until future use for total RNA extraction, sRNA library construction and component analyses of the flavonoids and terpenoids.
Extraction and Determination of Flavonoid Constituents and Volatile Terpenoid
Fifteen toon buds of each cultivar were mixed as one replicate and three replicates were performed. The total flavonoid extractions and their quantification were performed per the protocol of Jiang et al. (2019). Briefly, 100 g of the toon sprout samples under refrigerator stored at − 80 °C were ground into powder with liquid nitrogen, and then immersed in 1000 mL acidified methanol (in a volume ratio of 1% HCl) for 24 h, under dark, at room temperature 20 °C. The extracts were then centrifuged at 12,000g for 20 min. The supernatant was drawn, and the residues were re-extracted twice with 300 mL of acidified methanol. Subsequently, the extracts were amalgamated together and concentrated to 30 mL under vacuum. Also, to remove the fat residue, the concentrated solution was extracted using petroleum ether (in a volume ratio of 1:2) three times; the hydrophilic phase was once dissolved in 30 mL of acidified methanol. The samples were filtered through a 0.22 μm polyethersulfone membrane filter before HPLC analysis. The flavonoid constituents were determined on a 1260 series HPLC instrument (Agilent, USA) equipped with Waters SunFire C18 Analytical Column (5 μm, 250 × 4.6 mm). All the flavonoid constituents were monitored under uniform protocol using a UV detector at 254 nm. A gradient of water and acetonitrile was used as the mobile phase at column temperature 30 °C with a flow rate of 0.8 mL/min. The time programs for gradient elution was as given: 0–15 min, 10–30% acetonitrile (A), 70-90% water (B); 15–30 min, 30-60% acetonitrile (A), 40-70% water (B); 30–35 min, 60-100% acetonitrile (A), 0-40% water (B). The chromatogram of the standard substance of the selected flavonoid constituents were purchased from Aladdin Company (Shanghai). To determined volatile terpenoids, GC–MS technology and corresponding experimental program used in our previous study were adopted and slightly modified (Zhao et al. 2019).
RNA Extraction, Quality Control, Small RNA Library Construction and Sequencing
The total RNAs of the toon sprouts were extracted from four samples (two biological repetition per cultivar) using RNAprep Pure Plant Kit (Tiangen Biotech, Beijing). To ensure the high quality of the RNA samples for sequencing, the quality of the total RNAs was assayed as follows: (1) RNA degradation and contamination was checked on 15% denaturing polyacrylamide gels. (2) RNA purity was monitored using the Thermo Scientific™ NanoDrop™ 2000 spectrophotometer (Thermo, Waltham, MA). (3) RNA concentration was assayed using a Qubit®RNA Assay Kit in Qubit®2.0 Fluorometer (Life Technologies, CA, USA). (4) RNA integrity was checked using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). After all the RNA samples were qualified, small RNA libraries were constructed using the TruSeq® Small RNA Sample Prep Kit. To accomplish this, 1 μg of total RNA per sample was used as the starting material to prepare the small RNA (sRNA) sequencing library. Briefly, 3′ adaptors were ligated to the specific 3′OH group of the small RNA followed by the 5′ adaptor ligation using the T4 RNA ligase. The ligated products were reversely transcribed by reverse transcriptase and special primers, enriched by PCR and the amplified products were electrophoresed on 15% urea polyacrylamide gel followed by sizes of the 140–200 bp fragment selected. The small RNA library was quantified using NanoDrop and validated for quality by assaying the insert size using the Agilent 2100 bioanalyzer. Finally, Q-PCR was used to quantify accurately the effective concentration of the library (the effective concentration of the Library > 2 nM) to ensure its quality. Sequencing libraries were generated for Illumina sequencing (NEB, USA) following the manufacturer’s recommendations and index codes were added to attribute sequences to each sample. Clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v4-cBot-HS (Illumina), according to the manufacturer’s instructions. After cluster generation was complete, the library preparations were sequenced on an Illumina Hiseq 2500 platform and single-end 50 nt reads were generated (Beijing Novogene Technologies Co., Ltd, Beijing, China). All small RNA raw sequences from the GYC2_1, GYC2_2, BYC2_1, and BYC2_2 of the two varieties toon sprout libraries have been deposited in the Sequence Read Archive of NCBI with the corresponding accession numbers SRR12778803, SRR12778802, SRR12778801, and SRR12778800.
Prediction of Known and Novel miRNAs
After the sequencing run was completed, the raw data were first processed through the custom perl and python scripts. Clean data were obtained by removing the reads containing ploy-N, with 5' adapter contaminants, 3' adapter reads or the insert tag, less than 18 nt sequences and other low-quality reads from the raw data. The Rfam database was used to annotate and thus predict the non-coding RNAs such as the rRNA-, tRNA-, snRNA-, and snoRNA-derived sequences, etc. Next, the remaining sRNA sequences were mapped to the T. sinensis transcriptome sequence using the Bowtie software without mismatch, to analyze their expression and distribution on the reference (Langmead et al. 2009). The mapped sRNA tags were converged onto the miRBase v22.1 (http://www.mirbase.org/) for identification of the conserved miRNAs with zero mismatches, using the miRDeep2 software. After obtaining the known miRNAs and completing the family classification, the remaining sRNA sequences without annotation were used to predict the potential novel miRNA candidates. Initially, the sRNA reads were aligned with the T. sinensis transcriptome sequence using Bowtie. The characteristics of the hairpin structure of the miRNA precursor can be used to predict the novel miRNA. The available software miREvo and miRDeep2 were integrated to predict the novel miRNA by exploring the secondary structure (Friedlander et al. 2011; Wen et al. 2012). The reference standards for identifying the novel T. sinensis miRNA precursors were as follows (Meyers et al. 2008; Axtell and Meyers. 2018): (1) secondary stem-loop structure; (2) mature miRNA located in one arm of the stem, complementary sequence portion with the miRNA located on the opposite other arm, and termed miRNA*; (3) the miRNA/miRNA* duplex had no loop or break and possessed less than 6 base mismatches; (4) higher negative MFEI values of the miRNA precursors than the other RNAs; and (5) two nucleotides 3' overhang in the miRNA: miRNA* duplex. The Dicer cleavage site and minimum free energy of the sRNA tags were identified in the former steps. At the same time, custom scripts were employed to obtain the miRNA counts identified as well as the base bias on the first position, with certain length.
T.sinensis miRNA Expression Profiles, miRNA Target Identification and Function Annotation
The expression levels of both the known miRNAs and novel miRNAs from the buds of two toon cultivars were estimated by Transcripts Per Million (TPM) and normalized through the following criteria normalization formula (Zhou et al. 2010). Normalized expression = mapped miRNA counts / total clean read counts × 106. Differential expression analysis between the BYC2 and GYC2 was performed using the DEGseq (2010) R package. The miRNAs having p < 0.05 and |log2-fold| change ≥ 1 in comparison, were set as the threshold for significantly differentially expressed miRNAs (DE-miRNAs) (Audic and Claverie. 1997; Baggerly et al. 2003). Blasting against the transcriptome data of the earlier reported two T. sinensis cultivars, a prediction of the potential target genes of the miRNA was done using the psRobot (http://omicslab.genetics.ac.cn/psRobot/) (Wu et al. 2012). Rigorous criteria were adhered to, as follows: mismatches between the miRNA/target were not to exceed four (G-U bases set as 0.5 mismatches); no more than two adjacent mismatches and 2.5 mismatches in positions 1–12 between 5' of the miRNA and target were permitted, and specifically, no adjacent mismatches were to be present in positions 2–12, as well as mismatches in positions 10–11; the minimum free energy (MFE) of the miRNA/target duplex should be not < 60% compared to the MFE of the miRNA bound to its perfect complement. Targets were also functionally annotated by BLASTX (e value 1 × e−10) against the UniProtKB/Swiss-Prot and UniProtKB/TrEMBL databases for plants in our previous transcriptome data (Zhao et al. 2017). Gene ontology (GO) annotation for the targets of DE-miRNAs was done using the WEGO software (http://wego.genomics.org.cn/) (Ye et al. 2018). The targets of DE-miRNAs were mapped to the KEGG database (http://www.genome.jp/kegg/) to identify those genes involved in the flavonoid and terpenoid pathways (Kanehisa et al. 2017).
Expression Analysis of the miRNAs and Target Genes in Toon Sprouts by qRT-PCR
The miRNAs were extracted from the toon sprouts using the miRcute miRNA Isolation Kit (Tiangen Biotech, Beijing) according to the manufacturer's instructions. The total RNA of the toon sprouts for qRT-PCR was isolated adopting the protocol similar to the one in the previous experiment for total RNA quality control for sRNA sequencing. The miRNA poly (A) tailing and RT-PCR amplifications were performed using the miRcute Plus miRNA First-Strand cDNA Kit (Tiangen Biotech, Beijing). The RT-PCR amplifications for the target RNAs were performed using Quantscript RT Kit (Tiangen Biotech, Beijing) following the manufacturer's instructions. The qRT-PCR of the miRNA and target genes were performed in Step One Plus Real-Time PCR Systems (Thermo Fisher Scientific, Shanghai, China) using the miRcute Plus miRNA qPCR Kit (SYBR Green) (Tiangen Biotech, Beijing) and GoTaq-qPCR Master Mix Kit (Promega, Beijing, China), respectively. The primers for qRT-PCR were designed using Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) and synthesized by Sunya Biotechnology Company (Hangzhou). The primer sequences for the miRNAs and their targets are listed in Supplementary file 8. The 20 μLvolume of the reaction mixture contained 2 μL of the dilute template cDNA, 2 μL of the primer pairs, 10 μL of 2 × miRcute Plus miRNA PreMix (containing 2 μL of 50 × ROX reference dye II), and 6 μL of deionized water. The setting program of the qRT-PCR reaction for the miRNA quantity was as follows: denaturation at 95 °C for 15 min, followed by 40 cycles of 94 °C for 20 s, annealing and extension at 60 °C for 34 s. For the target ungenes, the reaction system and PCR parameters were set according to our previous criterion and procedures (Zhao et al. 2017). After the reactions, primer specificity was evaluated by the melting curve analysis. The miRNAs and corresponding target genes in the qRT-PCR quantity were analyzed in three biological replicates and three technical replicates. The relative abundance of the miRNAs and target genes was determined using the 2−ΔΔCT method and normalized by U6 rRNA and β-actin as reference genes, respectively.
Statistical Analysis
The statistical analyses of the DEGs and DE-miRNA profiles were performed using SPSS 19.0 (Chicago, IL, CA). Student t-test was used to assess for the potential differences between the BYC2 and GYC2. *P < 0.05 represented the statistically significant difference. The values were expressed as mean ± SD.
Results
Determination of Quality-Related Flavonoid and Terpenoid Contents in Toon Sprouts
The contents of the flavonoid in the toon sprouts of the BYC2 and GYC2 varieties were determined by the HPLC method (Table 1). The contents of the total flavonoid in the BYC2 and GYC2 were found to be 5722.13 ± 196.80 μg/g and 4936.72 ± 249.54 μg/g, respectively. The contents of six flavonoid monomers (except for rutin) namely, myricitrin, isoquercitrin, quercetin-3-O-α-l-arabinopyranoside, kaempferol-3-O-β-d-glucopyranose, quercitrin, and kaempferol-3-O-α-l-rhamopyranoside in the BYC2 were found to be 19.6, 38.3, 25.2, 32.8, 7.9, and 18.0% higher than those in the GYC2. However, the contents of the volatile terpenoid compounds between the two cultivars displayed a various change pattern (Table 2). The contents of the monoterpenoids such as α-pinene and d-limonene contents were significantly higher in the GYC2 than in the BYC2. Some important sesquiterpenoids such as the contents of β-caryophyllene, caryophyllene, trans-β-farnesene, α-farnesene, and isolongifolene, 9,10-dehydro in GYC2 were found to be 83.0, 152.3, 51.3, 293.5, and 45.5% higher than those in the BYC2, respectively. Moreover, β-caryophyllene showed the highest accumulation in the GYC2, with an absolute value of 6215.39 ± 523.33 ng/g achieved.
High Throughput Sequencing of Small RNA Libraries of T. sinensis
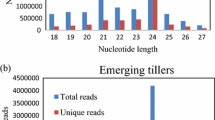
In this study, the four sRNA libraries were constructed and sequenced using two toon sprout cultivars (BYC2 vs. GYC2) with two biological replicates per cultivar. As shown in Table 3, the total raw data of 19,174,385, 23,020,318, 13,560,132, and 16,930,140 reads were generated in four libraries, namely, the GYC2_1, GYC2_2, BYC2_1, and BYC2_2, respectively. After adapter removal and low-quality data (sequence < 17 nt) filtering, 18,596,858, 22,252,682, 13,056,247, and 15,791,016 clean reads were obtained, with the total small RNA reads being in the proportion of 65.82% (12,239,853), 64.05% (14,251,756), 68.66% (8,964,927), and 51.41% (8,118,129) in each library. The read length distribution was centered mainly around 20–24 nt and contained over 60% of the total reads. As depicted in Fig. 1, the most abundant read length was 21 nt (11.03%-14.21%), followed by 24 nt (10.23%-11.99%) and 22 nt (9.28%-11.17%) (Fig. 1). Among them, 487–953 thousand unique reads corresponding to 7.54–12.58 million (about 83.95–92.66% of the total sRNA) reads were perfectly mapped to the T. sinensis transcriptome. The different types of the total sRNA sequences were further classified using the Bowtie software to blast the clean reads against the Rfam databases. The total counts of the annotated clean reads as tRNA, rRNA, snRNA, and snoRNA were 4,207,049, 4,385,655, 2,078,935, and 1,734,949 in GYC2_1, GYC2_2, BYC2_1, and BYC2_2, respectively. The remaining clean reads were aligned and edited in the Sequence Alignment Map (SAM) format and input into the miRDeep2 pipeline for the discovery of miRNA. In comparison, 818–1,277 unique reads corresponding to 40,682–76,624 reads were matched to the miRBase database. In total, 250–295 reads were found to match the known mature miRNAs collected from all plant species available in the miRBase database or be recognized as novel miRNAs in the T. sinensis transcriptome according to the miRDeep2 software.

Length distribution of clean reads of four small RNA libraries in BYC2 and GYC2
Identification of Known miRNAs in T. sinensis Sprouts
To further identify the miRNA homologs in the T. sinensis sprouts, the remaining clean reads in the four libraries were aligned to the known miRNA in the miRBase (Release 22.1). 60,900, 70,254, 35,736, and 45,105 reads from BYC2_1, BYC2_2, GYC2_1 and GYC2_2 libraries perfectly matched 243, 208, 230, and 275 known mature miRNAs, respectively. The total number of known miRNAs in the GYC2 was slightly higher than those present in the BYC2. In total, 331 known miRNAs belonging to 56 families were identified in the four libraries, with an average of about six miRNA members per family (Fig. 2A). Among these known miRNA families, only one member was identified in 29 miRNA families, while two members were identified in 11 miRNA families (Fig. 2B). However, some miRNA families possessed several members. For example, miR159 was the largest family containing 30 members, followed by miR166, miR171and miR396 with 26, 24, and 24 members, respectively (Fig. 2A). These known miRNAs were aligned to all the plant species available in the miRBase database, among which Arabidopsis thaliana, Oryza sativa, and Glycine max were the most frequent ones. The transcript numbers of these miRNAs in the toon sprouts varied greatly among the miRNA families. Some only had several sequence reads, whereas others possessed hundreds of thousands of reads. For example, most members of the miR166 family possessed more than 20,000 reads, and some of the miR166 family and most members of the miR159 family had more than 5,000 reads, and most of the miR159, miR319 and miR482 families possessed more than 2,000 read, while the majority of the other miRNAs had only a few hundred, or even fewer reads has been furnished in Supplementary file 1.

Known miRNA families and their member numbers in the toon sprouts. A The x-axis represents the members in the different miRNA families. The y-axis shows the conserved miRNA family identified in the BYC2 and GYC2. B 29 and 11 miRNA families included only one or two members
Identification of Novel miRNAs in Toon Sprouts
The remaining sRNAs that were unannotated to the miRBase were searched against the T. sinensis transcriptome, using the miRDeep2 software to identify the novel miRNAs that may be specific to the toon sprouts. Based on the recognition criteria of the potential novel miRNA precursor, a total of 23 novel miRNAs were identified in the four sRNA libraries (Table 4). The novel miRNA sequences varied in the range of 18–24 nt, with an average length of 21 nt. The pre-miRNAs lengths varied in the range of 53–293 nt, with the average being 130 nt. The average minimum folding free energy value of the hairpin structures was −45.22 kcal/mol in the toon sprouts, which is higher than that reported in Arabidopsis (−59.50 kcal/mol). The secondary structures of the 23 novel miRNA precursors have been furnished in Supplementary file 2. As shown in Table 5, the first nucleotide base analysis indicated that a large majority of the novel miRNAs started the first nucleotide with 5' uracil (U) not guanine (G) and U (73.91%) were the most dominant nucleotides in the first position, which is consistent with the typical miRNA nucleotide bias distribution patterns.
Differential Expression of the miRNAs Between the Two Toon Sprout Varieties
Pairwise analysis of the DE-miRNAs between the BYC2 and GYC2 was performed using the heat map method (Fig. 3). There were totally 52 (25 up-regulated, and 27 down-regulated) significantly DE-miRNAs between the BYC2 vs GYC2 (log2 ratio ≥ 1, adjusted P value ≤ 0.05). Among these, the miR166, miR168, miR408, and miR482 and novel miRNAs 5, 7, 13, 23, and 33 were up-regulated, while the miR156, miR171, miR172, miR394, miR396, miR403, miR858 families together with the novel miRNAs 3 and 27 were down-regulated between the BYC2 and GYC2 (Supplementary file 3). Interestingly, the levels of DE-miRNAs were found to be remarkably different among the different miRNA families. For example, the miR166, miR172, miR156 and miR858 exhibited greater differential expression than did the miR396 and miR168. Moreover, different members of the same miRNA family displayed different expression levels. For example, the miR166a and miR166d exhibited significantly reverse expression patterns. Compared with the GYC2, the miR166a was significantly up-regulated in the BYC2, whereas the miR166d was significantly down-regulated (Supplementary file 3). It is noteworthy that some DE-miRNAs were predicted to play crucial roles in the regulation of the secondary metabolism in the toon sprouts.

Clustering analysis of the DE-miRNAs was shown using heatmap. Heatmap represents the miRNAs significantly altered by the Baggerley’s test (P < 0.05) in four libraries. The blue color indicates low expression of the miRNAs, while red indicates the high expression of the miRNAs
Target Prediction and Functional Annotation
A total of 2233 target genes (or 18,224 transcripts of 2233 target genes) of miRNAs were predicted in the toon sprouts using the psRNA Target software (Supplementary file 4). Moreover, a majority of the miRNAs possessed multiple target unigenes, and the target unigenes number of miRNAs varies greatly from 1 to 1986. For instance, tsi-miR156z (belonging to the miR156 family) topped the list with 1986 target transcripts, followed by tsi-miR5658, tsi-miR414 and tsi-miR172c-3p which had 1915, 1781, and 1627 target transcripts, respectively. Among all the novel miRNAs, the novel_28 possessed the highest number of target transcripts (1377) while the lowest number of targets was found in novel_12 (36); these data have been furnished in Supplementary file 5.
To better understand the function of the miRNAs in the toon sprouts, the target unigenes of DE-miRNAs were annotated through the analysis of GO enrichment, (Supplementary file 6) and the KEGG pathway (Supplementary file 7: Table S5). The GO enrichment analysis showed that the target unigenes of DE-miRNAs appeared to be significantly enriched in the methyl indole-3-acetate esterase activity (GO: 0080030), methyl salicylate esterase activity (GO: 0080031), and methyl jasmonate esterase activity (GO: 0080032) in molecular function. Under biological process, pyrimidine ribonucleotide salvage (GO: 0010138), pyrimidine nucleotide salvage (GO: 0032262), UMP salvage (GO: 0044206), and CTP salvage (GO: 0044211) were significantly enriched. For the cellular component category, the actin cytoskeleton (GO: 0015629) and nuclear membrane (GO: 0031965) were determined to be highly represented groups (Fig. 4).

Gene ontology (GO) enrichment was classified for the potential target unigenes of the DE-miRNAs. Red, blue, and green represent the three GOs, namely, biological progress, cellular component, and molecular function, respectively
The KEGG pathway analysis demonstrated highly significant enrichment in the RNA transport, spliceosome, mRNA surveillance pathway, endocytosis, and RNA degradation (Fig. 5). Some metabolic pathways, including the other types of O-glycan biosynthesis, galactose metabolism, and purine metabolism, vitamin B6 and ascorbate metabolism, as well as essential amino acid and fatty acid biosynthesis were ranked among the top 20 enrichment pathways (Supplementary file 7). To identify a difference in the flavonoid contents between the BYC2 and GYC2, we focused on those miRNAs that may negatively regulate the unigenes associated with the flavonoid biosynthesis. In our study, there are key enzyme unigenes as potential targets regulated by T. sinensis miRNAs, such as cinnamic acid 4-hydroxylase (C4H), shikimate O-hydroxycinnamoyltransferase (HCT), dihydroflavonol-4-reductase (DFR), leucoanthocyanidin dioxygenase (LDOX), leucoanthocyanidin reductase (LAR), and anthocyanidin 3-O-glucosyltransferase (UGT) which were predicted to be targeted by the tsi-miR403-3p, tsi-miR168b, tsi-miR166a, tsi-miR395a, tsi-miR319i, tsi-miR172c, and tsi-miR396b, respectively. The novel_27 was predicted to be a mutual regulator of the targets in 4-coumarate CoA ligase (4CL), and chalcone synthase (CHS) (Table 6).

Top 20 KEGG pathways enrichment was analyzed for the potential target unigenes of the DE-miRNAs. A large enrichment factor denotes a high degree of enrichment. The lower q-value represents the more significant enrichment of the DEGs
By mapping these target unigenes of DE-miRNAs to the KEGG pathway, some targets encoding putative enzymes in the terpenoid biosynthesis were identified in T. sinensis. For example, the upstream target enzymes were located in the biosynthesis of the terpenoid backbone, including 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR), mevalonate kinase (MK), phosphomevalonate kinase (PMK), and farnesyl diphosphate synthase (FPPS) in the mevalonate (MVA) pathways in the cytoplasm and 1-deoxy-D-xylulose-5-phosphate synthase (DXS), and 4-hydroxy-3-methylbut-2-enyl-diphosphate synthase (HDS) in 2-C-methyl-D-erythritol-4-phosphate (MEP) in the plasmid. These unigenes were predicted to be targets by tsi-miR408-3p, tsi-miR171a, novel_28, tsi-miR390b-5p, tsi-miR-1260, and novel_27. The downstream targets belonged to the terpene synthases (TPS) in the terpenoid biosynthesis. For example, (R)-limonene synthase (LS) and linalool synthase (LIS) were targeted by tsi-miR395d and tsi-miR172c-3p in monoterpenoid biosynthesis. Terpene synthase 11 (TPS11), (E)-β-farnesene synthase (FAS), and (-)-germacrene D synthase (GDS) in the biosynthesis of sesquiterpenoid were targeted by the tsi-miR8155, tsi-miR482d-5p and tsi-miR395b-3p. Besides, a large number of transcription factors that could be involved in regulation of secondary metabolite biosynthesis, growth and development in the toon sprouts were predicted as the candidate targets of some conserved miRNA families. For example, the squamosa promoter-binding-like protein (SPL) transcripts (Yu et al. 2015), anthocyanin regulatory C1 protein (MYB) transcripts (Xia et al. 2012), WD40-repeat protein (WD40) transcripts, homeodomain leucine zipper family (ZIP) transcripts (Sharma et al. 2016), ethylene-responsive transcription factor (ERF) transcripts, WRKY transcription factor (WRKY) transcripts (Sun et al. 2017), and auxin response factor (ARF) transcripts were potentially targeted by the miR156, miR858, miR159, miR396, miR166, miR167, miR172, and miR160 families, respectively (He et al. 2019).
qRT-PCR Experimental Validation and Analysis of miRNAs and their Targets
The qRT-PCR was performed to validate the sRNA sequencing results and analyze the relationship between these miRNAs and their potential targets involved in the metabolic pathways of the flavonoid and terpenoid. Overall, the relative expression levels of the 40 miRNAs and 51 unigenes were quantified by the qRT-PCR. The qRT-PCR results of the miRNAs matched the expression profiles obtained by the sRNA sequencing, indicating that the sRNA sequencing data was credible and reliable. On the whole, the expression levels of the corresponding targets exhibited a trend opposite to those of the miRNAs (Figs. 6, 7, 8). In the flavonoid pathway, the tsi-miR4995, tsi-miR403-3p, novel_27, tsi-miR171c-5p, novel_34, tsi-miR172c, tsi-miR319i, tsi-miR408d, and tsi-miR396b showed significantly negative relationships with their responding target unigenes, PAL, C4H, Trans-CMO, 4CL, HCT-2, CHS, DFR, LDOX, LAR-1, LAR-2, and UGT (Fig. 6). To explore the function of the miRNAs on the terpenoid biosynthesis in the toon sprouts, the expression profiles of the miRNA-target unigene pairs were selected for qRT-PCR analysis with P value < 0.05 as the significant difference. Among these, the expression level of 8 miRNAs was found to have a remarkably negative correlation with their target unigenes including the tsi-miR-1260, novel_28, tsi-miR390b-5p, tsi-miR-107b, tsi-miR156g, tsi-miR8155, tsi-miR482d-5p, and tsi-miR395-3p, targeted for DXS-1, HDR, FPPS, GPPS, GGPPS-1, TPS11, FAS, and GDS (Fig. 7). Moreover, the other differentially expressed miRNA-target unigene pairs between the BYC2 and GYC2 were investigated in our study, and have been annotated as vital transcription factors in the regulation of secondary metabolism in plants, including SPL, MYB, WD40, WRKY, ERF, and ARF. A negative correlation was observed between the expression profiles of six miRNAs (tsi-miR156a-5p, tsi-miR858a, tsi-miR159, tsi-miR167, and tsi-miR172c-3p, and tsi-miR160b) and their targets SPL1, SPL2, MYBC1-1, MYBC1-2, MYB1R1, MYB, WD40-2, ERF, WRKY, and ARF (Fig. 8). The results stated above were consistent with our high-throughput sequencing data, which once again demonstrated that the credibility of our sRNA sequencing data.

The qRT-PCR validation of miRNAs and their target unigenes related to the flavonoid biosynthesis in BYC2 and GYC2. All the values represent an average ± SD (n = 6). The error bars with asterisks are the statistically significant differences (*P < 0.05; Student’s t test)

The qRT-PCR validation of the miRNAs and their target unigenes related to the biosynthesis of volatile terpenoids in BYC2 and GYC2. All the values represent an average ± SD (n = 6). The error bars with asterisks are the statistically significant differences (*P < 0.05; Student’s t test)

The qRT-PCR validation of the miRNAs and their transcription factor-associated target unigenes in BYC2 and GYC2. All the values represent an average ± SD (n = 6). The error bars with asterisks are the statistically significant differences (*P < 0.05; Student’s t test)
Discussion
T. sinensis is one of the most highly valuable plants classified under Medicine Food Homology (MFH) in China. The natural properties among different varieties reveal significant diversity during evolution (Chen et al. 2018). In Taihe County of the Anhui Province of China, two fine T. sinensis strains were obtained, and extensively planted after a long cultivation and domestication history. One variety was called purple toon or “Black Youchun” (BYC2) for its rich flavonoid or anthocyanin, the other named green toon or “Green Youchun” (GYC2) for its strong aromas, is known as mahogany and used for architecture or furniture. Our study confirmed that the presence of significant differences in the total flavonoid contents between BYC2 and GYC2, with the flavonoid components ranging from 254.00 to 1765.06 μg/g of fresh toon sprouts. Rutin, isoquercitrin, and quercetin-3-O-α-l-arabinopyranoside are the three main representative components, known for their high abundance. The total flavonoid content in the BYC2 was higher than that in GYC2, which is consistent with their phenotypic property. However, compared with the flavonoids, the content of the volatile terpenoid in the GYC2 was higher than in the BYC2. The difference in the abundance of volatile terpenoid compounds between the BYC2 and GYC2 may account for their respective characteristics aromas.
To explore the biosynthesis pathways of flavonoid and terpenoid, transcriptome sequencing was performed earlier to explain the difference in the anthocyanin contents between the BYC2 and GYC2. The results showed that the expression levels of some principal genes related to the flavonoid biosynthesis were up-regulated more significantly in BYC2 than GYC2 (Zhao et al. 2017). The difference in the gene expression profiles could conspicuously contribute to the difference in the secondary metabolites in the toon sprouts. The miRNAs were considered to be in control of the vital physiological processes in plants, including the regulation of the secondary metabolic pathways, mainly at the post-transcriptional level. To the best of our knowledge to date, this is the first report on attempting to investigate and explore the miRNAs and their potential targets in T. sinensis. In view of the lack of genomic information for T. sinensis, high throughput RNA sequencing could be performed to reveal the miRNA-target pairs which control the metabolic flows of the flavonoid and terpenoid. To study the miRNA-mediated regulation of the gene expression at the post-transcriptional level in the toon sprouts, sRNA sequencing combined with the previous transcriptome data was done to identify the miRNA candidates and their potential targets. In this study, the sRNA sequencing was done to characterize the miRNAs and verify their expression profiles in both the BYC2 and GYC2. In fact, 22 million or more clean reads were obtained from the four libraries. The sRNAs in the toon sprouts exhibited a wide range of variations in length, with the 21-nt RNAs being the most abundant type, followed by 24-nt and 22-nt sRNAs, representing the typical length of the Dicer-cleaved mature plant miRNAs (Moro et al. 2018). Such a distribution pattern of the sRNAs according to length, was also recorded for Moringa oleifera, Solanum tuberosum, and Glycine max (Li et al. 2015; Pirro et al. 2016; Qiao et al. 2017), implying that most miRNAs in the toon sprouts chiefly mediated the cleavage of the target genes, as well as the post-transcriptional inhibition and chromatin modeling of target genes (Axtell 2013; Lee and Carroll. 2018). Among the four sRNA libraries, a total of 331 known miRNAs classified under 56 families and 23 novel miRNAs were identified in the BYC2 and GYC2. Most of the known miRNAs of the toon sprouts exhibited a high degree of sequence conservation with the other plants in the miRBase database and a comparatively higher read number. Among the miRNAs of the toon sprouts identified, 44 known miRNAs and 8 novel miRNAs were statistically calculated as DE-miRNAs between the BYC2 and GYC2. This implied that these DE-miRNAs may be variety-specific and play crucial roles in the phenotypic determination of the two varieties. Furthermore, the majority of the plant miRNAs possessed almost perfect matches to their targets (Jones-Rhoades et al. 2006). This feature facilitated the identification of their potential targets in the toon sprout. At the same time, it could also provide certain vital information about their putative functions. In our analysis of the target unigenes, a substantial number of potential targets were aligned to the previous transcriptome data. A single miRNA has multiple target genes, whereas a target gene may be recognized and regulated by multiple miRNAs (Wei et al. 2015; Licursi et al. 2019). Further studies focusing on the miRNA-mediated the regulation of the flavonoid and terpenoid pathways are mandatory to elucidate difference in the abundance of the secondary metabolites between the BYC2 and GYC2 varieties. Several miRNAs were bioinformatically predicted to target the unigenes in the biosynthesis pathway of the flavonoid, including PAL, C4H, 4CL, CHS, DFR, and UGT, etc. A high expression level of miRNA that targets the genes related to flavonoid formation may inhibits the total flavonoid accumulation. For both the BYC2 and GYC2, the expression levels of the tsi-miR4995, tsi-miR403-3p, novel_27, tsi-miR171c-5p, novel_34, tsi-miR172c, tsi-miR319i, tsi-miR408d, and tsi-miR396b were inversely correlated with the total flavonoid content. The key enzyme genes involved in the flavonoid biosynthetic pathway that were targeted by the miRNAs were previously recorded in Podophyllum hexandrum, such as the 4CL and CHS targeted by the phe-miR172i and phe-miR829.1, respectively (Biswas et al. 2016). A similar study was reported in both genotypes of soybean, gma-miR396 and gma-miR5434, which demonstrated an inverse relationship with their corresponding targets UGT and CHI (chalcone isomerase), respectively (Gupta et al. 2019). The flavor of toon sprouts consists of diverse volatile terpenoid compounds, which were biosynthesized separately in two distinct pathways, the MVA and MEP, respectively. From the target prediction, a total of 13 miRNA-target pairs were identified, that seemed to be directly relevant to the biosynthesis of these terpenoids (Table 5). Among these miRNAs, the expression levels of 8 miRNAs were negatively correlated with the terpenoid contents in the BYC2 and GYC2. For instance, DXS-1 targeted for tsi-miR-1260 and HDR targeted for novel_28 in the MEP pathway, and FPPS targeted for the tsi-miR390b-5p, GPPS targeted for tsi-miR-107b, GGPPS -1 targeted for the tsi-miR156g, FAS targeted for the tsi-miR8155, TPS targeted for the tsi-miR482d-5p, and GDS targeted for the tsi-miR395b-3p in the downstream pathway of the terpenoid biosynthesis. These results indicated that compared with the BYC2, the high abundance of volatile terpenoid in GYC2 was due to the relatively lower levels of expression of these miRNAs. Samad discovered that DXS and DXR were targeted by pmi-miR396a and pmi-miR398f/g in the MEP pathway, respectively (Samad et al. 2019). However, in our study, these genes were targeted by the T. sinensis-specific miRNAs that were different from the reports cited above. A plausible reason is that, in different species, different miRNAs were applied to regulate their targets.
More importantly, the target prediction in this study revealed that certain miRNAs not only targeted the enzyme genes related to the metabolic pathways, they also negatively regulated several transcription factors (TFs). These TFs play a crucial regulatory role in plant growth, development and biosynthesis of secondary metabolites. For example, four MYB unigenes were also targeted by ath-miR858a and aqc-miR159. A similar study showed that ath-miR858a targeted the R2R3-MYB involved in the biosynthesis of flavonoid in Arabidopsis thaliana, and the overexpression of miR858a in transgenic Arabidopsis resulted in the down-regulation of several MYBs and inhibition of the flavonoid biosynthesis (Sharma et al. 2016). A decrease in the miR858 activity resulted in flavonoid accumulation in the Arabidopsis leaves and enhanced its resistance to pathogen infection (Camargo-Ramírez et al. 2018). The WD40 protein was reported to be an essential component of the MYB-bHLH-WD40 (MBW) transcription complex for anthocyanin biosynthesis (Sunitha et al. 2019). The WD40 transcripts, as potential targets of tsi-miR167 showed higher expression in the BYC2 than in the GYC2. The MBW complex could regulate the DFR and UFGT expressions to control the anthocyanin biosynthesis in Arabidopsis (Xu et al. 2014; Yang et al. 2018). The SPLs are the other TFs widely prevalent in plants, which play an important role in plant growth and development, primary and secondary metabolism (Gou et al. 2012; Yu et al. 2012). In Arabidopsis, the pattern of flavonoid or anthocyanin accumulation was under the regulation of miR156-targeted SPL gene. The SPLs acted as the negative regulators of anthocyanin accumulation by destabilizing the MBW complex (Gou et al. 2012). On the other hand, the SPLs could directly bind to the TPS promoters to act as positive regulators of the biosynthesis of volatile terpenoids (Yu et al. 2015). In our study, the results of the sRNA sequencing and qRT-PCR showed that the tsi-miR156a-5p significantly up-regulated, whereas the expression of its targets SPLs were down-regulated in the BYC2 compared to that in the GYC2, which was in accordance with the TPS expression level and content of volatile terpenoid as stated above. The tsi-miR172c-3p and tsi-miR160b possibly targeted the ERF, WRKY and ARF TFs to play crucial roles in sucrose signaling. The miR172 and miR160 might target the ERF and ARF to activate auxin-mediated signaling (Gao et al. 2019; He et al. 2019). All of these signal transduction processes would induce a profound impact on the activities of the structural genes, and subsequently construct a complex molecular network mechanism to regulate secondary metabolites biosynthesis in the toon sprouts.
Abbreviations
- AACT:
-
Acetyl-CoA-acetyltransferase
- ARF:
-
Auxin response factor
- BYC2:
-
Black Youchun
- CHS:
-
Chalcone synthase
- C4H:
-
Cinnamic acid 4-hydroxylase
- 4CL:
-
4-Coumarate CoA ligase
- DFR:
-
Dihydroflavonol 4- reductase
- DXS:
-
1-Deoxy-d-xylulose-5-phosphate synthase
- ERF:
-
Ethylene-responsive transcription factor
- FAS:
-
(E)-β-farnesene synthase
- F3H:
-
Flavanone 3-hydroxylase
- FLS:
-
Flavonol synthase
- FPPS:
-
Farnesyl diphosphate synthase
- GDS:
-
(-)-Germacrene D synthase
- GO:
-
Enrichment Gene Ontology enrichment
- GYC2:
-
Green Youchun
- MBW:
-
MYB-bHLH-WD40 (MBW) transcription complex
- miRNA:
-
MicroRNA
- MEP:
-
2-C-methyl-d-erythritol-4-phosphate
- MYB:
-
Myeloblastosis
- HMGR:
-
3-Hydroxy-3-methyl glutaryl coenzyme A reductase
- HCT:
-
Shikimate O-hydroxycinnamoyltransferase
- HDS:
-
4-Hydroxy-3-methylbut-2-enyl-diphosphate synthase
- KEGG:
-
Pathway kyoto encyclopedia of genes and genomes pathway
- LAR:
-
Leucoanthocyanidin reductase
- KS:
-
Ent-kaur-16-ene synthase
- LDOX:
-
Leucoanthocyanidin dioxygenase
- LIS:
-
Linalool synthase
- LS:
-
(R)-limonene synthase
- MK:
-
Mevalonate kinase
- PAL:
-
Phenylalanine ammonia lyase
- PMK:
-
Phosphomevalonate kinase
- qRT-PCR:
-
Quantitative real-time PCR
- SE:
-
Squalene epoxidase
- SQS:
-
Squalene synthase
- sRNA:
-
Small RNA
- SPL:
-
Squamosa promoter-binding protein-like
- TPS:
-
Terpene synthases
- UGT:
-
Anthocyanidin 3-O-glucosyltransferase
- WRKY:
-
WRKY transcription factor
- ZIP:
-
Homeodomain leucine zipper
References
Audic S, Claverie JM (1997) The significance of digital gene expression profiles. Genome Res 7:986–995
Axtell MJ (2013) Classification and comparison of small RNAs from plants. Annu Rev Plant Biol 64:137–159
Axtell MJ, Meyers BC (2018) Revisiting criteria for plant microRNA annotation in the era of big data. Plant Cell 30:272–284
Baggerly KA, Deng L, Morris JS, Aldaz CM (2003) Differential expression in SAGE: accounting for normal between-library variation. Bioinformatics 19:1477–1483
Biswas S, Hazra S, Chattopadhyay S (2016) Identification of conserved miRNAs and their putative target genes in Podophyllum hexandrum (Himalayan Mayapple). Plant Gene 6:82–89
Bulgakov VP, Avramenko TV (2015) New opportunities for the regulation of secondary metabolism in plants: focus on microRNAs. Biotechnol Lett 37:1719–1727
Camargo-Ramírez R, Val-Torregrosa B, Segundo BS (2018) MiR858-mediated regulation of flavonoid-specific MYB transcription factor genes controls resistance to pathogen infection in Arabidopsis. Plant Cell Physiol 59:190–204
Chen QQ, Rong LY, Shao ZJ, Liu T, Wei L, Song ZQ (2018) Genetic diversity analysis of Toona sinensis germplasms based on SRAP and EST-SSR markers. Acta Hortic Sinica 45:967–976
Chen YC, Chen HJ, Huang BM, Chen YC, Chang CF (2019) Polyphenol-rich extracts from Toona sinensis bark and fruit ameliorate free fatty acid-induced lipogenesis through AMPK and LC3 pathways. J Clin Med 8:1664
Edmonds JM, Staniforth M (1998) Toona Sinensis: Meliaceae. Curtis’s Bot Mag 15:186–193
Fan R, Li Y, Li C, Zhang Y (2015) Differential microRNA analysis of glandular trichomes and young leaves in Xanthium strumarium L. reveals their putative roles in regulating terpenoid biosynthesis. PLoS ONE 10:e0139002
Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N (2011) miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40:37–52
Gao J, Peng H, Chen FB, Luo M, Li WB (2019) Genome-wide analysis of transcription factors related to anthocyanin biosynthesis in carmine radish (Raphanus sativus L.) fleshy roots. Peer J 7:e8041
Gou JY, Felippes FF, Liu CJ, Weigel D, Wang JW (2012) Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell 23:1512–1522
Gupta OP, Dahuja A, Sachdev A, Kumari S, Jain PK, Vinutha T, Praveen S (2019) Conserved miRNAs modulate the expression of potential transcription factors of isoflavonoid biosynthetic pathway in soybean seeds. Mol Biol Rep 46:3713–3730
He LH, Tang RM, Shi XW, Wang WB, Cao QH, Liu XY, Wang T, Sun Y, Zhang HM, Li RZ, Jia XY (2019) Uncovering anthocyanin biosynthesis related microRNAs and their target genes by small RNA and degradome sequencing in tuberous roots of sweet potato. BMC Plant Biol 19:232
Hsu CY, Huang PL, Chen CM, Mao CT, Chaw SM (2012) Tangy scent in Toona sinensis (Meliaceae) leaflets: isolation, functional characterization, and regulation of TsTPS1 and TsTPS2, two key terpene synthase genes in the biosynthesis of the scent compound. Curr Pharm Biotechnol 13:1–12
Jiang XX, Zhang BB, Lei MH, Zhang JJ, Zhang JF (2019) Analysis of nutrient composition and antioxidant characteristics in the tender shoots of Chinese toon picked under different conditions. LWT-Food Sci Technol 109:137–144
Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol 57:19–53
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45(D1):D353–D361
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25
Lee CH, Carroll BJ (2018) Evolution and diversification of small RNA pathways in flowering plants. Plant Cell Physiol 59:2169–2187
Li WB, Wang PP, Li YG, Zhang KX, Ding FQ, Nie TK, Yang X, Lv QX, Zhao L (2015) Identification of microRNAs in response to different day lengths in soybean using high-throughput sequencing and qRT-PCR. PLoS ONE 10:e0132621
Licursi V, Conte F, Fiscon G, Paci P (2019) MIENTURNET: an interactive web tool for microRNA-target enrichment and network-based analysis. BMC Bioinform- 20:545
Liu BB, Zhang JF, Shi YC (2019) Complete chloroplast genome of Toona sinensis (Meliaceae), a goluptious ‘tree vegetables.’ Mitochondrial DNA Part B 4:3025–3026
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao XF, Carrington JC, Chen XM, Green PJ, Griffiths-Jones S, Jacobsen SE, Mallory AC, Martienssen RA, Poethig RS, Qi YJ, Vauchere H, Voinnet O, Watanabe Y, Weigel D, Zhu JK (2008) Criteria for annotation of plant microRNAs. Plant Cell 20:3186–3190
Moro B, Chorostecki U, Arikit S, Suarez IP, Höbartner C, Rasia RM, Meyers BC, Palatnik JF (2018) Efficiency and precision of microRNA biogenesis modes in plants. Nucleic Acids Res 46:10709–10723
Peng W, Liu YJ, Hu MB, Zhang MM, Yang J, Liang F, Huang QW, Wu CJ (2019) Toona sinensis: A comprehensive review on its traditional usages, phytochemisty, pharmacology and toxicology. Rev Bras Farmacogn 29:111–124
Pirro S, Zanella L, Kenzo M, Montesano C, Minutolo A, Potesta M, Sobze MS, Canini A, Cirilli M, Muleo R, Colizzi V, Galgani A (2016) MicroRNA from Moringa oleifera: identification by high throughput sequencing and their potential contribution to plant medicinal value. PLoS ONE 11:e0149495
Prakash P, Ghosliya D, Gupta V (2015) Identification of conserved and novel microRNAs in Catharanthus roseus by deep sequencing and computational prediction of their potential targets. Gene 554:181–195
Prakash P, Rajakani R, Gupta V (2016) Transcriptome-wide identification of Rauvolfia serpentina microRNAs and prediction of their potential targets. Comput Biol Chem 61:62–74
Qiao Y, Zhang JJ, Zhang JW, Wang ZW, Ran A, Guo HX, Wang D, Zhang JL (2017) Integrated RNA-seq and sRNA-seq analysis reveals miRNA effects on secondary metabolism in Solanum tuberosum L. Mol Genet Genomics 292:37–52
Rogers K, Chen XM (2013) Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell 25:2383–2399
Samad AFA, Sajad M, Nazaruddin N, Fauzi IA, Murad AMA, Zainal Z, Ismail I (2017) MicroRNA and transcription factor: key players in plant regulatory network. Front Plant Sci 8:565
Samad AFA, Rahnamaie-Tajadod R, Sajad M, Jani J, Murad AMA, Noor NM, Ismail I (2019) Regulation of terpenoid biosynthesis by miRNA in Persicaria minor induced by Fusarium oxysporum. BMC Genomics 20:586
Sharma D, Tiwari M, Pandey A, Bhatia C, Sharma A, Trivedi PK (2016) MicroRNA858 is a potential regulator of phenylpropanoid pathway and plant development. Plant Physiol 171:944–959
Shen YP, Xu MH, Deng PP, Gu QY, Yin HW, Xia GH, Jia XB, Yang H, Tam J (2017) Establishment of a rapid method to quantify eight flavonol glycosides for quality assessment of red toon using UPLC. Acta Chromatogr 30:1–7
Singh N, Srivastava S, Sharma A (2016) Identification and analysis of miRNAs and their targets in ginger using bioinformatics approach. Gene 575:570–576
Singh A, Gautam VS, Das SS, Verma S, Mishra V, Mukherjee S, Sarkar AK (2018) Plant small RNAs: advancement in the understanding of biogenesis and role in plant development. Planta 248:545–558
Sui JJ, Qu CQ, Yang JX, Zhang WN, Ji YT (2019) Transcriptome changes in the phenylpropanoid pathway in senescing leaves of Toona sinensis. Acta Physiol Plant 41:126
Sun YY, Qiu Y, Duan MM, Wang JL, Zhang XH, Wang HP, Song JP, Li XX (2017) Identification of anthocyanin biosynthesis related microRNAs in a distinctive Chinese radish (Raphanus sativus L.) by high-throughput sequencing. Mol Genet Genomics 292:215–229
Sunitha S, Loyola R, Alcalde JA, Arce-Johnson P, Matus JT, Rock CD (2019) The role of UV-B light on small RNA activity during grapevine berry development. G3 9:769–787
Vashisht I, Mishra P, Pal T, Chanumolu S, Singh TR, Chauhan RS (2015) Mining NGS transcriptomes for miRNAs and dissecting their role in regulating growth, development, and secondary metabolites production in different organs of a medicinal herb, Picrorhiza kurroa. Planta 241:1255–1268
Wei RC, Qiu DY, Wilson IW, Zhao H, Lu SF, Miao JH, Feng SX, Bai LH, Wu QH, Tu DP, Ma XJ, Tang Q (2015) Identification of novel and conserved microRNAs in Panax notoginseng roots by high-throughput sequencing. BMC Genomics 16:835
Wen M, Shen Y, Shi S, Tang T (2012) miREvo: an integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinformatics 13:140
Wu HJ, Ma YK, Chen T, Wang M, Wang XJ (2012) PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res 40:W22–W28
Xia R, Zhu H, An YQ, Beers EP, Liu ZR (2012) Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol 13:R47
Xu WJ, Grain D, Bobet S, Gourrierec JL, Thévenin J, Kelemen Z, Lepiniec L, Dubos C (2014) Complexity and robustness of the flavonoid transcriptional regulatory network revealed by comprehensive analyses of MYB–bHLH–WDR complexes and their targets in Arabidopsis seed. New Phytol 202:132–144
Xu XB, Jiang QH, Ma XY, Ying QC, Shen B, Qian YS, Song HM, Wang HZ (2015) Deep sequencing identifies tissue-specific microRNAs and their target genes involving in the biosynthesis of tanshinones in Salvia miltiorrhiza. PLoS ONE 9:e111679
Yang JF, Li BB, Shi WJ, Gong ZZ, Chen L, Hou ZX (2018) Transcriptional activation of anthocyanin biosynthesis in developing fruit of blueberries (Vaccinium corymbosum L.) by preharvest and postharvest UV irradiation. J Agric Food Chem 66:10931–10942
Ye J, Zhang Y, Cui HH, Liu JW, Wu YQ, Cheng Y, Xu HX, Huang XX, Li ST, Zhou A, Zhang XQ, Bolund L, Chen Q, Wang J, Yang HM, Fang L, Shi CM (2018) WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res 46(W1):W71–W75
Yu S, Galvão VC, Zhang YC, Horrer D, Zhang TQ, Hao YH, Feng YQ, Wang S, Schmid M, Wang JW (2012) Gibberellin regulates the Arabidopsis floral transition through miR156-targeted SQUAMOSA PROMOTER BINDING–LIKE transcription factors. Plant Cell 24:3320–3332
Yu ZX, Wang LJ, Zhao B, Shan CM, Zhang YH, Chen DF, Chen XY (2015) Progressive regulation of sesquiterpene biosynthesis in Arabidopsis and Patchouli (Pogostemon cablin) by the miR156-targeted SPL transcription factors. Mol Plant 8:98–110
Zhai XT, Granvogl M (2019) Characterization of the key aroma compounds in two differently dried Toona sinensis (A. Juss.) Roem. by means of the molecular sensory science concept. J Agric Food Chem 67:9885–9894
Zhang BH, Unver T (2018) A critical and speculative review on microRNA technology in crop improvement: current challenges and future directions. Plant Sci 274:193–200
Zhang X, Song ZQ, Liu T, Guo LL, Li XF (2016) De Novo assembly and comparative transcriptome analysis provide insight into lysine biosynthesis in Toona sinensis Roem. Int J Genom 3:1–9
Zhao H, Ren LP, Fan XY, Tang KJ, Li B (2017) Identification of putative flavonoid-biosynthetic genes through transcriptome analysis of Taihe Toona sinensis bud. Acta Physiol Plant 39:122
Zhao H, Feng SS, Zhou W, Kai GY (2019) Transcriptomic analysis of postharvest toon buds and key enzymes involved in terpenoid biosynthesis during cold storage. Sci Hortic 257:108747
Zhou L, Chen JH, Li ZZ, Li XX, Hu XD, Huang Y, Zhao XK, Liang CZ, Wang Y, Sun L, Shi M, Xu XH, Shen F, Chen MS, Han ZJ, Peng ZY, Zhai QN, Chen J, Zhang ZF, Yang RL, Ye JX, Guan ZC, Yang HM, Gui YT, Wang J, Cai ZM, Zhang XQ (2010) Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS ONE 5:e15224
Acknowledgements
We would like to thank Dr. Gopal Patel’s help to revise the manuscript from Laboratory of Medicinal Plant Biotechnology, Pharmacy College, Zhejiang Chinese Medicine University. We are also grateful to Postgraduate San Peng Fan, Dawei You, and Man Xu for their help to improve the experiment operation from Laboratory of Medicinal Plant Biotechnology, Pharmacy College, Zhejiang Chinese Medicine University. We thank Novogene Technologies (Beijing, China) for supplying small RNA sequencing service.
Funding
This work was supported by grants from the Natural Science Key Foundations of the Anhui Bureau of Education and institution of higher education outstanding top talent cultivation funding project of the Anhui Bureau of Education (No. KJ2019A0515, gxgwfx2020049), the Natural Science Key Foundation of Fuyang normal university (No. 2020FSKJ01ZD), the Innovation Program for College Students (No. 202010371013), the Open Fund of Advantaged and Characteristic Disciplines (Traditional Chinese Medicine of Zhejiang Chinese Medical University) for Key Construction Universities in Zhejiang Province (No. ZYAOX2018027).
Author information
Authors and Affiliations
Contributions
HZ, WZ, and GK conceived and designed this experiment. HZ drafted the manuscript. WL and CQ collected samples of toon sprouts. YC and SF extracted and assayed flavonoid components and volatile terpenoid compounds. PG, HZ and WZ carried out qRT-PCR experiments and analyzed the data. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
12374_2021_9321_MOESM2_ESM.jpg
Supplementary file2 (JPG 1392 KB) Figure S1. Predicted secondary structures of potential novel miRNAs from toon sprouts. Sequences indicated in red correspond to predicted miRNA
12374_2021_9321_MOESM3_ESM.xlsx
Supplementary file3 (XLSX 30344 KB) Table S2. Target prediction results of miRNAs from toon sprouts. Table S2. Target prediction results of miRNAs from toon sprouts.
Rights and permissions
About this article

{kind=link}
Cite this article
Zhao, H., Shi, X., Shen, C. et al. Comparative Analysis of miRNA Expression Profiles Provides Insight into Regulation of Biosynthesis of Flavonoids and Terpenoids Between Two Varieties of Toona sinensis Sprouts. J. Plant Biol. 65, 291–310 (2022). https://doi.org/10.1007/s12374-021-09321-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12374-021-09321-6