
Abstract
The present study was aimed to determine the genetic variability of sugarcane smut pathogen (Sporisorium scitamineum) in Sri Lanka to support an effective crop improvement program for sugarcane smut disease resistance. Eighty-three isolates of S. scitamineum were collected from 15 different sugarcane varieties grown in seven sugarcane cultivating areas in Sri Lanka. DNA sequencing of PCR products obtained by specific (i.e., bE 4 and bE 8) and universal (i.e., ITS 1 and ITS 4) primers showed 99% sequence similarity among the collected S. scitamineum isolates. Genetic diversity was determined using 16 inter-simple sequence repeat (ISSR) primers. It produced 104 amplified DNA fragments which are 100% polymorphic. The polymorphic information content (PIC) value of ISSR markers varied from 0.25 to 0.46 with an average of 0.37. The results revealed a moderate degree of genetic diversity among the S. scitamineum isolates. The genetic differentiation coefficient (Gst) was estimated to be 0.241 in the studied fungal population, suggesting 24% of the total genetic variation originates between the populations, and 76% originates within the populations. Geneflow (Nm) was calculated as 1. 572, indicating that gene flow occurs across populations at a faster rate. The collected isolates clustered into five genetically distinct groups having no relationship with their geographical origin. The results confirmed a considerable genetic variation among the collected isolates from major sugarcane growing areas in Sri Lanka. Therefore, it is proposed to use a mixture of S. scitamineum isolates collected from all sugarcane growing areas when screening for smut-resistant sugarcane varieties.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Sugarcane (Saccharum spp. hybrid) is an essential agro-industrial crop in tropical and subtropical regions of the world (Patel et al. 2018). Various microorganisms infect sugarcane; among them, Sporisorium scitamineum (Syn. Ustilago scitaminea) is a major pathogen (Vicente et al. 2021). Barnabas et al. (2017) have reported smut disease as one of the most conspicuous sugarcane diseases. Life cycle of S. scitamineum has genetically and morphologically different phases, namely haploid yeast-like sporidia phase, the dikaryotic hyphal phase and diploid teliospores. Infection of smut pathogen results in the formation of a whip-like structure from the plant meristem (Benevenuto et al. 2016). The emerging whip is long and curved and contains a large number of melanized teliospores (Comstock 2000).
Smut disease causes significant reductions of stalk yield and sucrose recovery, hence could cause yield losses between 20 and 50% and sugar production losses up to 75% (Su et al. 2013; Ramesh Sundar et al. 2012). Severity of the disease is influenced by cultivar resistance and environmental conditions (Ramesh Sundar et al. 2012). The elimination of susceptible varieties is often needed to manage the disease (Ferreria and Comstock 1989). Resistant varieties are the only useful and environmentally friendly solution for managing smut in plantations (Croft and Braithwaite 2006). Further, cultivation of smut-resistant varieties is a more economical and efficient approach compared to chemical treatments and agronomic practices (Ramesh Sundar et al. 2012; Xu et al. 2004). Therefore, development of smut-resistant varieties is the main disease management objective in sugarcane crop improvement programs in almost all sugarcane-producing countries, including Sri Lanka. It is imperative when screening for disease resistance, to understand the genetic variability and evolutionary prospective of the smut pathogen (McDonald and Linde 2002).
Morphological and genetic variability of S. scitamineum has been investigated in several sugarcane growing countries. Zekarias et al. (2010) have studied the morphological variation of 11 different smut isolates in Ethiopia and Luzaran et al. (2012) have investigated the morphological variation of 96 different smut isolates from the Philippines. Research has been undertaken on the genetic diversity of smut pathogens infecting a wide range of crops in different countries around the world, in conjunction with morphological studies. Molecular marker technology has been incorporated into this research; multilocus molecular markers are highly suited for evaluating the genetic structure of plant-pathogenic fungi (Chen et al. 2002; Milgroom et al. 2003; Mishra et al. 2003). Braithwaite et al. (2004) studied the genetic differences among 38 different S. scitamineum isolates collected from 13 countries using Amplified Fragment Length Polymorphism (AFLP). Xu et al. (2004) studied the genetic differences among 18 S. scitamineum isolates from six sugarcane growing regions in China using random amplified polymorphic DNA (RAPD). Xu et al. (2004) have suggested that molecular variation observed among these isolates was associated in part with geographical origin. Raboin et al. (2007) have used a pool of 142 different single teliospores of S. scitamineum from 15 different countries for polymorphism studies targeting 17 microsatellite loci. This report concluded that the genetic variation was lower among the smut pathogen isolates from America and Africa, with the highest genetic variation among the isolates from Asian populations, mainly those from the Philippines.
Luzaran et al. (2012) used SSR markers to examine the genetic diversity among 96 isolates from diverse growing regions of the Philippines; pathogen variation was higher within, rather than between the populations. Xu et al. (2017) suggested that Inter-Simple Sequence Repeats (ISSR) is the best suited molecular marker for relationship and population studies. Much information has been collected on the genomic diversity of fungi using ISSR markers (Jing et al. 2017). ISSR and RAPD markers were used to determine the genomic diversity among 35 smut pathogen isolates from mainland China (Shen et al. 2012a, b); the study clustered the isolates into two major groups. Que et al. (2012) analyzed the molecular differences in 23 smut isolates representing six major sugarcane growing regions of mainland China and concluded that the molecular variations were associated with the geographic region of origin of the isolates. However, this study did not provide any evidence on the variation of the S. scitamineum within populations. The biogeographic variation among 100 S. scitamineum isolates was studied by Xu et al. (2014) using ISSR and single primer sequence-related amplified polymorphism (SP-SRAP) markers. Zhang et al. (2015) concluded that internal transcribed spacer (ITS) regions are suitable for defining the genus and species but not for the molecular variation analysis among S. scitamineum isolates. Shen et al. (2016) have used highly polymorphic Start Codon-Targeted (SCoT) markers to analyze the gene diversity among 90 S. scitamineum isolates from mainland China. In their study, the isolates grouped into three major clusters, with the clustering based on the geographical region of origin. Benevenuto et al. (2016) studied the genetic diversity among 41 haploid sporidial strains from Brazil and six isolates from Argentina using AFLP and telomere-associated restricted fragment length polymorphism (tel-RFLP) and ITS sequencing. Barnabas et al. (2018) studied the variation of 30 S. scitamineum isolates collected from different regions in India using ISSR primers. Based on the above research findings, it is evident that there is huge genomic diversity among the S. scitamineum isolates in Asia. There are no reports or information available on the genetic diversity of the S. scitamineum population in Sri Lanka. This study aimed to identify the genetic diversity of a representative population of S. scitamineum in Sri Lanka with a view of planning an effective breeding program for sugarcane smut resistance.
Material and Methods
Collection of Samples and Preparation of Smut Teliospores for the Study
Collection of whips was done from commercial and research sugarcane fields in seven cultivation regions of Sri Lanka, namely Bandarawela, Ethimale, Hingurana, Kantale, Pelwatta, Sevanagala and UdaWalawe. The locations from where the whips were collected, represented five agroecological regions of Sri Lanka, namely IU3, DLIa, DL2a, DL1c, DL1b and DL1a (https://esdac.jrc.ec.europa.eu/images/Eudasm/Asia/images/maps/download/LK2007_CL.jpg), which are mapping units determined by the agro-climate, soil type and terrain of a given region. Newly emerged smut whips were selected from different sugarcane varieties and whips were cut approximately 10 cm below the top visible dewlap. The whips were placed in a tray and kept at ambient temperature for nearly 5 days to dry. Spores were removed from the dried whips by fingers and collected spores were sieved (1 mm aperture) to remove plant material attached to the spores. Spores were transferred to sealed containers and maintained at 4 °C. Details of the collected isolates are summarized in Table 1.
Confirmation of the S. scitamineum Identities Using Specific and Universal Primers
Fungal colonies developed from single teliospores were maintained on potato dextrose agar (PDA). Genomic DNA was extracted using 0.1 g of each fungal colony by a modified CTAB method (Abu Almakarem et al. 2012). The mycelia were ground in CTAB extraction buffer, followed by incubation at 60 ℃ for 1 h with periodical rocking, and centrifuged at 12,000 rpm for 15 min at 4 ℃. The upper layer was clarified with chloroform: isoamyl alcohol (24:1), then the DNA was precipitated with 0.8 volumes of isopropanol. The precipitate was washed with 70% ethanol and the air-dried DNA pellet was dissolved in 30–50 µl of nuclease-free water, depending on the size of the DNA pellet. Quantification of extracted DNA (ng/µl) was done spectrophotometrically (BioSpec-Nano, Shimadzu, Japan). Quality of extracted DNA was checked using ratios of OD 260/280 and OD 260/230 values and integrity was determined by running DNA on a 1.0% agarose gel.
Polymerase Chain Reaction (PCR) was done using universal (i.e., ITS 1 (3’CGCTCTGGTTCATCAA 5’) and ITS 4 (3’ CTGCCGACCGTGCTGC 5’)) and specific primers (i.e., bE 8 (3’ CGCTCTGGTTCATCAA 5’) and bE 4 (3’ CTGCCGACCGTGCTGC 5’)). The cycling protocols used for the PCR were: 5 min at 94 ℃, followed by 35 cycles with 30 s at 94 ℃, 45 s at 55 ℃ and 55.5 ℃, respectively, for specific and universal primers and 2 min at 72 ℃. The final extension step was 8 min at 72 ℃. The amplified PCR products were detected by electrophoresis using a 1.5% agarose gel and visualized by a Gel documentation system (QUANTUM CX 5, France). Amplified PCR products from representative samples were sequenced. In the phylogenetic analysis, Geneious software (Ocenar et al. 2019) was used to construct the phylogenetic tree by Neighbor-join Nucleotide alignment.
PCR Reaction with ISSR Primers
PCR was performed according to the method outlined by Xu et al. (2014), with slight modifications using a Veriti™ 96-Well Thermal Cycler (Applied Biosystem). Sixteen S. scitamineum ISSR primers (Integrated DNA Technology, USA) were used for PCR. Each PCR reaction was done with a 25 µl reaction mixture, consisting of 2 µl of genomic DNA (25 to 30 ng), 12.5 µl of master mix (Promega, USA), 3 µl of ISSR random primer (5 µM) and 7.5 µl of nuclease-free water (Shen et al. 2012a). The cycling protocol was initiated with 4 min at 95 ℃ followed by 35 cycles, each completed with 30 s at 94 ℃ C, 45 s at 50 ℃ C and 2 min at 72 ℃. The final extension step was done for 8 min at 72 ℃. The amplified PCR products were analyzed by running in a 2.0% agarose gel with 0.5 X TBE buffer with the Promega 1 Kb DNA size markers. The amplified DNA fragments were visualized and photographed by a Gel documentation system (QUANTUM CX 5, France). The experiment was repeated twice.
Analysis of Data Generated by ISSR Primers
All gel pictures showing amplicons by ISSR-PCR were scored to obtain binary matrixes. In the scoring process “1” was given to the samples resulted in DNA bands and “0” was given to the samples which did not show any band on agarose gel. Popgene version 1.31 software was used to determine genetic diversity among the studied populations; using population genetic parameters: the observed numbers of alleles (Na), the effective number of alleles (Ne), Nei’s gene diversity index (h), Shannon’s information index (i), and gene flow (Nm). These parameters were used to assess the genetic diversity in each S. scitamineum population (Yeh et al. 1999). The NTSYS-pc2.10 (Numerical Taxonomy System, Applied Biostatistics, Inc.) analytical software was used to determine the Jaccard similarity coefficient and unweighted pair-group method with arithmetic mean (UPGMA) cluster analyses (Xu et al. 2017). Polymorphic information content (PIC) was determined using an equation prearranged by Roldan-Ruiz et al. (2000). GenAIEx version 6.5 was used for the analysis of molecular variance (AMOVA) and to assess the components of genetic variance within and among populations (Peakall and Smouse 2006).
Results
Confirmation of S. scitamineum Isolates Using Specific and Universal Primers
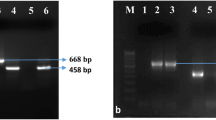
The OD260/OD230 ratios were between 1.80 and − 2.10 while the ratios of OD260/OD280 ratios ranged between 1.70 and − 2.20. PCR study of S. scitamineum using specific primer pair bE 4 and bE 8 showed a fragment of 459 bp, which was present in all the tested 83 samples. PCR products obtained using the universal primer pair ITS 1 and ITS 4, targeting the rDNA-ITS sequence resulted in a single 755 bp amplicon for all the tested fungal DNA samples of S. scitamineum isolates. Sequence analysis of the 83 amplified PCR products, separately obtained for specific primers (bE 4 and bE 8) and universal primers (ITS 1 and ITS 4), confirmed the identity of amplicons as S. scitamineum. Sequence similarity ranged from 83.13% to 100.00% for the PCR products amplified by specific primers (bE 4 and bE 8) and the same parameter ranged from 8% to 100.00% for the PCR products amplified by universal primers (ITS 1 and ITS 4).
PCR Reactions with ISSR primers
Based on a preliminary trial, 16 ISSR primers were selected out of 20, to examine the variation among the 83 different isolates belonging to the seven populations in Sri Lanka (i.e., locations). Details on the selected primers are summarized in Table 2. There were a total of 104 reproducible fragments and the average number of fragments per primer was 6.5. The fragment size of the PCR products varied from 150 to 3000 bp. All of these amplified bands were polymorphic (100%). In general, the PIC values of the 16 ISSR primers ranged between 0.25 and − 0.46 and the highest PIC value of 0.46 was recorded by ISSR 859 and P 10 primers. The ISSR 815 had the lowest PIC value of 0.25 indicating that the ISSR 859 and P 10 are most suitable to evaluate the genetic variation of S. scitamineum under Sri Lankan conditions. The details of the amplified bands with each primer and the amount of polymorphism shown by each primer are included in Table 2.
Genetic Diversity (GD) and Structure Analysis of the S. scitamineum Isolates
Values for the parameters related to population genetics (i.e., Na, Ne, h, I, pl and r) for the 83 isolates collected from different sugarcane growing areas in Sri Lanka are summarized in Table 3.
Higher values for the Na, Pl and r revealed a higher genetic difference in Sevanagala and Hingurana populations of S. scitamineum and a moderate gene diversity in the Pelwatta population (Table 3). The lowest values for Na, Pl, and r were given by the S. scitamineum population from UdaWalawe showing that it has the lowest genetic diversity.
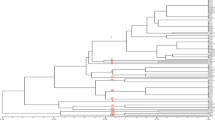
The total gene diversity (Ht) and gene diversities between subpopulations (Hs) were calculated using the ISSR data. The total gene diversity (Ht) and the gene diversities between subpopulations (Hs) values were 0.3553 and 0.2696, respectively, for this study. The genetic differentiation coefficient (Gst) was 0.2413. The Gst value revealed that total genomic variation between populations was lower than the same parameter within a population. The genetic Gst between populations and within populations are 24% and 76%, respectively. The gene flow (Nm) was 1.572, showing that a faster rate of gene flow takes place among populations. A dendrogram was produced for the collected isolates using the unweighted pair-group method with arithmetic average (UPGMA) cluster analysis. It revealed that the isolates were clustered into five main groups at the level of 0.006. The results of the cluster analysis are shown in Fig. 1.

Dendrogram generated for collected S. scitamineum isolates (83 isolates) based on ISSR data
Genetic Distances Between Populations
The genomic distances between the seven populations of S. scitamineum varied from 0.0225 to 0.2628 (Table 4). The genetic distance was smallest between samples collected from Pelwatta and Sevanagala which are in the same agroecological region, DL1. The genetic distance was largest between the UdaWalawe and Hingurana populations which are in two different agroecological regions (i.e., DL1 and DL2, respectively). The study concludes that within-population genetic differentiation among the tested 83 diploid teliospores of S. scitamineum isolates in Sri Lanka is high.
A considerable genetic difference was evident among populations (Table 5) based on the results of the AMOVA. However, the results revealed that the variance within the separate populations is higher (94%) than the genetic variance among populations (6%).
Discussion
According to the findings of Braithwaite et al. (2004) and Raboin et al. (2007), the genetic diversity among the S. scitamineum populations representing Asia was comparatively high. Although there is significant information on the genomic variation in S. scitamineum isolates from China and India, there are no reports on the genetic diversity among S. scitamineum isolates from Sri Lanka.
Benevennuto et al. (2016) studied the molecular variability and genetic relationship of Brazilian S. scitamineum isolates using the ITS region and concluded to have no polymorphism among the isolates of Brazilian collection. Xu et al. (2014) used the ITS sequences for genetic diversity studies and found that isolates shared similarities with an average of 96.34%. Moreover, the same study by Xu et al. (2014) proposed that the rDNA-ITS region is not ideal for examining the genetic variation of S. scitamineum due to its lower detected sequence variation. The present study agrees with Xu et al. (2014) as Sri Lankan isolates shared 83.13% to 100% sequence similarity, which indicates a lower sequence variation. Barnabas et al. (2018) studied the genomic variation of the smut pathogen using the specific primers bE 4 and bE 8 and revealed no noticeable difference in sequence similarity in the bE gene sequences of S. scitamineum isolates, confirming the results of the present study.
Xu et al. (2014) found that the ISSR molecular markers provide an effective and inexpensive method to analyze molecular data to determine genome diversity in S. scitamineum isolates. The present study generated 100% polymorphic bands when used 83 S. scitamineum isolates from different populations in Sri Lanka. This is a comparatively higher value than the previously reported values by several other researchers worked on the gene diversity of S. scitamineum in the Asian region. For example, studies by Xu et al. (2014) using 100 S. scitamineum isolates from the mainland China reported 86.8% polymorphism and a subsequent study by Xu et al. (2017) reported 71.0% polymorphism among the S. scitamineum isolates from China. These findings are identical to the results of Shen et al. (2012a) who used ISSR to evaluate the genetic diversity using mating-type isolates of S. scitamineum from Southern China and identified a polymorphism of 72.7%. The differences in polymorphic rate in these studies might be due to the variation in the template DNA source since Xu et al. (2014) have used template DNA, extracted from diploid spores of S. scitamineum (teliospores) while Xu et al. (2017) have used DNA samples from a single mating-type haploid of S. scitamineum. Furthermore, the environmental conditions of the sampling locations are highly correlated with the genetic differentiation of the isolates (Xu et al. 2017) and it may be another reason for the observed genetic differences. The present study also has used diploid teliospores to extract template DNA for amplification using ISSR primers. Generally, a much richer genetic variation can be expected in dikaryotic teliospores than in monosporidial mating-type isolates of S. scitamineum. The reason for the higher polymorphism in this study may be the use of dikaryotic teliospores to study the genetic variation.
Conclusion
The findings of the present study provide new information on the genetic variability in the S. scitamineum genome in Sri Lanka. All the 83 tested isolates clustered into five major genetically distinct groups independent of their geographic origin. Therefore, we propose to use a composite of S. scitamineum isolates collected from different sugarcane cultivating regions to screen for smut-resistance varieties in crop improvement programs essential for commercial sugarcane production in Sri Lanka.
References
Agro-ecological regions of Sri Lanka.1979. https://esdac.jrc.ec.europa.eu/images/Eudasm/Asia/images/maps/download/LK2007_CL.jpg.
Almakarem, A.S.A., K.L. Heilman, H.L. Conger, Y.M. Shtarkman, and S.O. Rogers. 2012. Extraction of DNA from plant and fungus tissues in situ. BMC Research Notes 5: 266.
Barnabas, L., N.M.R. Ashwin, K. Kaverinathan, A.R. Trentin, M. Pivato, A.R. Sundar, P. Malathi, R. Viswanathan, P. Carletti, and G. Arrigoni. 2017. In vitro secretomic analysis identifies putative pathogenicity-related proteins of Sporisorium scitamineum–The sugarcane smut fungus. Fungal Biology 121: 199–211.
Barnabas, L., N.M.R. Ashwin, K. Nalayeni, A. Ramesh Sundar, P. Malathi, and R. Viswanathan. 2018. Genetic and pathogenic variability among the Indian isolates of Sporisorium scitamineum causing sugarcane smut. Journal of Sugarcane Research 8 (2): 138–154.
Benevenuto, J., D.P. Longatto, G.V. Reis, N. Mielnichuk, A.C. Palhares, G. Carvalho, S. Saito, M.C. Quecine, A. Sanguino, M.L.C. Vieira, L.E.A. Camargo, S. Creste, and C.B. Monteiro-Vitorello. 2016. Molecular variability and genetic relationship among Brazilian strains of the sugarcane smut fungus. FEMS Microbiology Letters 363 (24): 277. https://doi.org/10.1093/femsle/fnw277.
Braithwaite, K.S., G. Bakkeren, B.J. Croft, and S.M. Brumbley. 2004. Genetic variation in a worldwide collection of the sugarcane smut fungus Ustilago scitaminea. Proceedings of the Australian Society Sugar Cane Technologists 26: 1–9.
Chen, F., P.H. Goodwin, A. Khan, and T. Hsiang. 2002. Population structure and mating-type genes of Colletotrichum graminicola from Agrostis palustris. Canadian Journal of Microbiology 48: 427–436.
Comstock, J.C. 2000. Smut. In CIRAD and ISSCT, ed. A. Guide and to Sugarcane Diseases. P. Rott, R. A. Bailey, J. C. Comstock, B. J. Croft, and A. S. Saumtally, 181–185. Montpellier: France.
Croft, B.J., and K.S. Braithwaite. 2006. Management of an incursion of sugarcane smut in Australia. Australasian Plant Pathology 35: 113–122.
Ferreira, S.A., and J.C. Comstock. 1989. Smut. In. Ricaud C, Egan BT, Gillaspie Jr AG, Hughes CG (eds) Disease of Sugarcane: Major Diseases. Elsevier Press, Amsterdam, Netherlands. pp. 211–229.
Jing, Y., M. Peng, L. Yang, and Q. Wang. 2017. Evaluation of genetic diversity among Piptoporus betulinus as revealed by inter simple sequence repeat markers. Biotechnology and Biotechnological Equipment 31 (2): 333–338. https://doi.org/10.1080/13102818.2016.1276413.
Luzaran, R.T., F.M. Cueva, C.J.R. Cumagun, L.R.I. Velasco, and T.U. Dalisay. 2012. Variability of sugarcane smut pathogen, Ustilago scitaminea Sydow in the Philippines. Philippine Journal of Crop Science 37: 38–51.
McDonald, B.A., and C. Linde. 2002. Pathogen population genetics, evolutionary potential, and durable resistance. Annual Review of Phytopathology 40: 349–379.
Milgroom, M.G., and P.T. Peever. 2003. Population biology of plant pathogens: The synthesis of plant disease epidemiology and population genetics. Plant Dis 87:608–617
Mishra, P.K., R.T.V. Fox, and A. Culham. 2003. Inter-simple sequence repeat and aggressiveness analyses revealed high genetic diversity, recombination and long-range dispersal in Fusarium culmorum. Annals Appl Biol 143:291–301
Ocenar, J.D., Arizala, G. Boluk, U. Dhakal, S. Gunarathne, S. Paudel, S. Dobhal, and A. Mohammad. 2019. Development of a robust, field-deployable loop-mediated isothermal amplification (LAMP) assay for specific detection of potato pathogen Dickeya dianthicola targeting a unique genomic region. PLoS ONE 14(6):e0218868. https://doi.org/10.1371/journal.pone.0218868
Patel, P., B.K. Rajkumar, P. Parmar, R. Shah, and R. Krishnamurthy. 2018. Assessment of genetic diversity in Colletotrichum falcatum Went accessions based on RAPD and ISSR markers. Journal of Genetic Engineering and Biotechnology 16: 153–159.
Peakall, R., and P.E. Smouse. 2006. Genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes 6: 288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.
Que, Y., L. Xu, J. Lin, R. Chen, and M.P. Grisham. 2012. Molecular variation of Sporisorium scitamineum in Mainland China revealed by RAPD and SRAP Markers. Plant Disease 96: 1519–1525.
Raboin, L.M., A. Selvi, K.M. Oliveira, F. Paulet, C. Calatayud, M.F. Zapater, and A. D’Hont. 2007. Evidence for the dispersal of a unique lineage from Asia to America and Africa in the sugarcane fungal pathogen Ustilago scitaminea. Fungal Genetic Biology 44: 64–76.
Ramesh Sundar, A., E. Leonard Barnabas, P. Malathi, and R. Viswanthan. 2012. A mini-review on smut disease of sugarcane caused by Sporisirium scitamineum, In: Botany, ed. J. Mworia Rijeka: In Tech. ISBN 978–953–51–0355–4.
Rampersad, S.N. 2011. Molecular and phenotypic characterization of Colletotrichum species associated with anthracnose disease of papaya in Trinidad. Plant Disease 95: 1244–1254.
Ruiz, R., J. Dendauw, E. Van Bockstaele, A. Depicker, and M. De Loose. 2000. AFLP markers reveal high polymorphic rates in ryegrasses (Lolium spp.). Molecular Breeding 6: 125–134.
Shen, W., P. Xi, M. Li, R. Liu, L. Sun, Z. Jiang, and L. Zhang. 2012a. Genetic diversity of Ustilago scitaminea Syd in Southern China revealed by combined ISSR and RAPD analysis. African Journal of Biotechnology 11 (54): 11693–11703.
Shen, W.K., P.G. Xi, M.H. Li, R. Liu, L.H. Sun, Z.D. Jiang, and L.H. Zhang. 2012b. Genetic diversity of Sporisorium scitamineum in Southern China revealed by combined ISSR and RAPD analysis. African Journal of Biotechnology 11: 11693–11703.
Shen, W., G. Xu, M. Luo, and Z. Jiang. 2016. Genetic diversity of Sporisorium scitamineum in mainland China assessed by SCoT analysis. Tropical Plant Pathology 41: 288–296.
Su, Y.C., S.S. Wang, J.L. Guo, B.T. Xue, L. Xu, and Y. Que. 2013. A TaqMan real-time PCR assay for detection and quantification of Sporisorium scitamineum in sugarcane. Science World Journal 942682: 9. https://doi.org/10.1155/2013/942682.
Vicente, C., M.E. Legaz, and E. Sánchez-Elordi. 2021. Physiological basis of smut infectivity in the early stages of sugar cane colonization. Journal of Fungi 7: 44. https://doi.org/10.3390/jof7010044.
Xu, L., Y. Que, and R. Chen. 2004. Genetic diversity of Ustilago scitaminea in Mainland China. Sugar Tech 6: 267–271.
Xu, L.P., Y.H. Lu, Q. You, X.L. Liu, M.P. Grisham, Y.B. Pan, and Y.X. Que. 2014. Biogeographical variation and population genetic structure of Sporisorium scitamineum in Mainland China: insights from ISSR and SP-SRAP markers. Science World Journal 296020 (1): 13.
Xu, G.H., Q.Q. Deng, W.K. Shen, S. Chen, and X.M. Wu. 2017. Assessment of genetic diversity and structure of Sporisorium scitamineum from China using inter-simple sequence repeat (ISSR) markers. African Journal of Biotechnology 16 (14): 727–737. https://doi.org/10.5897/AJB2016.15859.
Yeh, F.C., R.C. Yang, T.B.J. Boyle, Z.H. Ye, and J.X. Mao. 1999. POPGENE, version 1.32: the user friendly software for population genetic analysis. Edmonton: Molecular Biology and Biotechnology Centre, University of Alberta.
Zekarias, Y., G. Mashilla Dejene, and F.Y. Tegegn. 2010. Importance and status of sugarcane smut (Ustilago scitaminea) in the Ethiopian sugar estates. Ethiopian Journal of Agricultural Sciences 21: 35–46.
Zhang, Y.Y., N. Huang, X.H. Xiao, L. Huang, F. Liu, W.H. Su, and Y.X. Que. 2015. Molecular variation of Sporisorium scitamineum in Mainland China revealed by internal transcribed spacers. Genetics and Molecular Research 14: 7894–7909.
Acknowledgements
This research was funded by Sugarcane Research Institute, Sri Lanka. Assistance given by the staff of the Crop Protection Division in Sugarcane Research Institute, Sri Lanka, is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
ANWST: Design the experiment, sample collection, carried out molecular work, data analysis and writing of the manuscript. DMDC: Directed the study and corrected the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
Cite this article
Thushari, A.N.W.S., De Costa, D.M. Molecular and Genetic Variability of Sporisorium scitamineum (Sugarcane Smut Pathogen) in Sugarcane Plantations in Sri Lanka. Sugar Tech 25, 797–804 (2023). https://doi.org/10.1007/s12355-022-01239-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12355-022-01239-8