
Abstract
Ascorbate peroxidase (APX) is a key enzyme for active oxygen scavenging of higher plants, and it plays an important role in plants against abiotic stresses. In this study, a thylakoidal APX (TAPX) gene was cloned from sugarcane leaves by using RT-PCR technique, which was registered in GenBenk with the accession number JQ958327. Bioinformatics analysis showed that the new gene contained a 1,422 bp open reading frame and encoded 473 amino acids, and the predicted molecular weight was 51.1 kDa, containing the typical functional area and heme binding sites of peroxidase gene family. Real-time fluorescence quantitative PCR analysis showed the TAPX gene expressed in the roots, stems and leaves of sugarcane plant, and the highest expression level was found in the leaves. The TAPX gene was expressed in three exogenous stresses, i.e. polyethylene glycol (PEG), NaCl and H2O2. The expression pattern varied due to different regulatory mechanisms and the impact of low temperature stress on the expression is not significant. The TAPX gene was successfully expressed in E. coli. The TAPX gene excessive plant expression vector was built and transformed into tobacco with Agrobacterium-mediation, and transgenic tobacco plants were obtained. In conclusion, the TAPX gene was mainly expressed in the green tissue of sugarcane plant. It is presumed that TAPX gene is related with sugarcane resistance to osmotic stress. Further investigation on the biological functions of TAPX in sugarcane are warranted.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Under stressed conditions, plants produce large amounts of reactive oxygen species (ROS) but scavenging of reactive oxygen is decreased. The reactive oxygen of enzymatic removal system is important for maintaining the normal physiological functions in plant. In higher plants, ascorbic acid (ASA)–glutathione (GSH) cycle plays an important role for ROS scavenging. Ascorbate peroxidase is a key enzyme in the ASA–GSH cycle, and substrate ASA was specific electron donor. It mainly deoxygenates H2O2 accumulated in the cytoplasm and chloroplasts to H2O and O2 for avoiding from cells poisoning (Asada 1992; Shigeoka et al. 2002; Ishikawa and Shigeoka 2008). Also known as vitamin C peroxidase, it is commonly found in higher plants, eukaryotic algae and cyanobacteria. (Foyer and Hailiwell 1997) first discovered peroxidase. Subsequently, enzymatic properties, distribution, positioning, mechanism of action, physiological function and molecular biological characteristics of APX were studied carefully, which showed APX genes exited in higher plants with the form of a gene family, they were cytoplasm APX gene (cAPX), thylakoid APX gene (tAPX), stromal APX gene (sAPX), microfossils APX gene (mAPX) and peroxisomes APX gene (pAPX) (Yoshimura et al. 2000; Chew et al. 2003; Mano et al. 1997). Up to now, APX genes have been cloned and transformed in Arabidopsis (Arabidopis thaliana) (Wang et al. 1999), tomato (Lycopersicon esculentum) (Najami et al. 2008), wheat (Triticum aestivum) (Chen et al. 2006), rice (Oryza sativa) (Teixeirra et al. 2006), soybean (Glycinne max) (Shi et al. 2008), east China grapes (Vitis pseudoreticulata) (Lin et al. 2006), aspen (Populus tomentosa) (Lu et al. 2009) and other large numbers of plants. Some results showed that APX gene expression was induced by multiple stresses. Over expression of Arabidopsis APX genes in tomato increased the endurance to salt stress (Badawi et al. 2004). The tobacco transgenic plants with Arabidopsis APX gene increased the resistance to oxidative stress (Wang et al. 1999). So cloning APX gene, studying its relationship with stress and using transformation to enhance the expression of the gene have important implications for improving the resistance of crops. Sugarcane (Saccharum officinarum L.) is an important sugar crop. Guangxi is the largest sugarcane producer in China. However more than 80 % of sugarcane in Guangxi is grown in hilly upland areas and faces varying degrees of drought threat every year. Chilling injury caused huge losses to the sugarcane production in recent years. So stress is the main factor affecting sugarcane production currently (Li and Yang 2009, Li et al. 2011; Deng et al. 2009). Our laboratory previously cloned an APX gene located in the cytoplasm in sugarcane (accession number JN591764 in GenBank, but other APX gene families in sugarcane have not been reported yet. In the present study, we designed the primers based on the thylakoid membrane TAPX gene sequences of other crops published on the NCBI, amplified the full length of sugarcane TAPX gene by RT-PCR, and cloned and analyzed the gene sequence characteristics using bioinformatics. Real-time quantitative PCR technology was employed to study the temporal spatial expression pattern and differential expressions of TAPX gene in different organs under a variety of adverse stresses, and the gene expression in E. coli was detected and the plant over expression vector was built. Using Agrobacterium-mediated tobacco leaf disc transformation, we proved the gene has relation with a variety of adverse stresses, providing references for further study on sugarcane APX gene function in sugarcane resistance to adverse stresses and the molecular mechanism of sugarcane resistance breeding.
Materials and Methods
Experimental Material
Experiment was conducted in State Key Laboratory for Conservation and Utilization of Subtropical Agro-Bioresources, Guangxi University from October 2011 to December 2012. Sugarcane variety Guitang 28 (GT 28) was chosen, and the robust and uniform cane setts with a single bud were planted in sand. After the shoots grew to 15 cm, they were transferred to nursery cups with the medium of 1:1 sand:soil to culture. When the plants grew to 5–6 leaves stage, consistent plants were selected and divided into four groups treatment as follows: For group 1, the plants were moved into a cold room of 4 °C with 16 h/8 h for light/dark with the humidity of 65 %. For the groups 2 and 3, the plants were treated with 100 mM of NaCl and 15 % PEG, respectively. For group 4, 10 mM of H2O2 was sprayed on the leaves. The samples of leaf +1 were collected at 0, 6, 12, 24, 48 and 72 h, respectively. And the samples of roots, stems and leaves were taken at the processing stage. All the samples collected were immediately fixed in liquid nitrogen, and stored in a refrigerator at −80 °C until RNA extraction.
Experiment Reagents
The Trizol reagent was purchased from Beijing Kangwei Century Company. DreamTaq DNA Plolymerase (EP0701), Xho I and EcoR I restriction enzymes were bought from Fermentas Company. M-MLV cDNA first strand synthesis reagents, PrimeScript RT Reagent Kit with gDNA Eraser, PMD18-T vector and SYBR® Premix Ex Taq™ II were from TaKaRa Company. Biospin Gel Extraction Kit was purchased from BioFlux Company. Escherichia coli DH5α, BL21 (DE3) and prokaryotic expression vector PET-30A were kept by the laboratory. Other conventional reagents were analytical grade reagents.
Total RNA Extraction and First-Strand cDNA Synthesis
Sugarcane total RNA was extracted by Trizol reagent according to the instructions. Concentration and integrity of RNA were tested by UV spectrophotometer and 1 % agarose gel electrophoresis. M-MLV reverse transcriptase was used in the cDNA synthesis for gene cloning, and the reverse tailed primer is Oligo (DT) 18: 5′-GGCCACGCGTCGACTAGTACTTTTTTTTTTTTTTTTTT-3′. Primescript RT reagent Kit With gDNA Eraser (Perfect Real Time) kit was used in the cDNA synthesis for fluorescence quantitative expression analysis, and specific steps were based on the product instructions. After the completion of the reaction, the templates of all the samples are diluted to 100 mg/μL for use.
TAPX Cloning and Bioinformatics Analysis
NCBI database was searched to select nucleotide sequences of TAPX gene from Gramineae family having high homology with sugarcane. The gene upstream degenerate primer STAPX was designed by the Primer software, and the primer is 5′-ATGGCGGAGCG (C/T/G) (A/C) TCGC (C/G) (G/T/A) CCTC-3′, and downstream primer is the reverse tailed primer 3 SIDE: 5′-GGCCACGCGTCGACTAGTAC-3′.
The total volume of the PCR reaction system for fragment amplification of TAPX target gene was 25 μL. The leaf cDNA of sugarcane was used as the template, and the Dream TaqDNA polymerase instructions were followed. The PCR reaction parameters included pre-denaturation at 95 °C for 5 min; denaturation for 40 s at 95 °C; annealing for 50 s at 60 °C, and extending for 2 min at 72 °C, 35 cycles; followed by final extension for 10 min at 72 °C. When the reaction finished, the PCR products was analyzed with 1.0 % agarose gel electrophoresis and the target bands were collected, inserted in the T vector, and then transformed into DH5α competent cells by heat shock method. Positive clones were screened out, and sequenced by Shanghai Sangon Company. The sequences were spliced for obtaining the full-length of the target gene.
Bioinformatic Analysis
The amino acid sequence of the gene was predicted by BioXM2.6; and basic physical and chemical properties of the speculated protein sequence of the gene were analyzed by ExPASy (http://expasy.org/tools/). The sugarcane TAPX gene and the homology with other species were analyzed by GenBank Blast (http://blast.ncbi.nlm.nih.gov) online analysis software. Protein blast program (http://blast.ncbi.nlm.nih.gov/Blast.cgi), SMART and Motif Scan software were used to analyze the functional domains of the TAPX protein. The phylogenetic tree of TAPX gene amino acid sequences of sugarcane and other species were constructed by MEGA software.
Real-time Fluorescence Quantitative Expression Analysis of TAPX Gene
According to the TAPX full length cDNA sequence obtained, fluorescence quantitative PCR specific primers, TAPX-PET30aF (5′-CTCCAGAACCAGAACCAGCA-3′) and TAPXR (5′-TTTGGCAGCGACGAAGG-3), were designed respectively. The target fragment length was 122 bp. GAPDH gene (NCBI accession No.: EF189713) was used as the internal control gene to design the internal control primer GAPDHF (5′-AAGGGTGGTGCCAAGAAGG-3′) and GAPDH R (5′-CAAGGGGAGCAAGGCAGTT-3′), respectively. Real-time fluorescence quantitative expression analysis was conducted by using ABI Real-time PCR analyzer 7500. The 20 μL reaction system contained 1 μL 100 mg/μL cDNA template, 0.3 μL 10 μM upstream and downstream primers, respectively, 10 μL SYB, 0.4 μL ROX, and 8 μL double distilled water. Each sample was designed for three repeats. The reaction protocol included pre-denaturation at 95 °C for 3 min; denaturation for 30 s at 95 °C, annealing for 60 s at 60 °C, 40 cycles. The melting curve was analyzed when the reaction ended. The method of 2−ΔΔCT was adopted to calculate the relative expression of the gene.
TAPX Prokaryotic Expression
Using PET-30a (+) as the expression vector, the construction primers of prokaryotic expression vector for amplification of TAPX gene open reading frame TapX-PET30aF (5′-CGGAATTCATGGCGGAGCGCATCGCCGC-3′) and TapX-PET30aR (5′-CCCTCGAGGTTCCCGACCAGAGACGTCA-3′), were designed respectively. The underlined parts were EcoR I restriction sites of N-terminal and Xho I digested sites of C-terminal. The target fragment was collected after PCR amplification and the pET-30a plasmid was extracted, and then digested with EcoR I and Xho I, finally the double digested products were collected. The target gene and the vector fragment were ligated by DNA Ligation Kit, and then transformed into the competent cells BL21 (DE3) of the expression strain by heat shock. The suspension of recombinant plasmid was detected and then sent to the Shanghai Sangon Company to sequence, and to extract the recombinant plasmid for verification with double enzyme digestion.
Both the recombinant strain and the strain containing the PET-30A empty vector were transformed into the liquid LB medium which contained Kan 100 mg L−1 for overnight shake culture at 150 rpm at 37 °C to induce gene expression. When OD600 reached 0.4–0.6, the medium was added with IPTG with the final concentration of 1.0 mM and 1.0 ml medium was collected at 0, 2, 4, 6, and 8 h. After completion of the induction, the product was centrifuged at 12,000 × g for 5 min, the supernatant and precipitate were collected respectively and then 70 μL 2× sample buffer were added, keeping in boiling water bath for 5 min, after cooling, 20 μL supernatant was taken for SDS-PAGE electrophoresis after 12,000 × g centrifugation for 5 min. The concentrations of spacer gel and separation gel for SDS-PAGE was 4 and 12.5 %, respectively. After electrophoresis, the gel was stained with Brilliant blue R-250 for imaging analysis.
Transformation of Tobacco Plants with TAPX Gene
Using PBI121 as expression vector, the construction primers of over expression vectors for amplification TAPX gene open reading frame TapX-PBI121F (5′- GGTCTAGAATGGCGGAGCGCATCGCCGC -3′) and TAPX-PBI121R (5′- TTCCCGGGGTTCCCGACCAGAGACGTCA -3′) were designed. The underlined parts were Xba l restriction sites of the N-terminal and Sma I digestion sites of C-terminal. The target fragment was collected after PCR amplification and PBI121 plasmid was extracted, and then digested with Xba l and Sma respectively, and the double digested products were collected finally. The target gene and the vector fragment were ligated with DNA Ligation Kit and then transformed into the competent cells EHA105 of the expression strain by heat shock. The suspension of recombinant plasmid was detected and then sent to the Shanghai Sangon Company to sequence, and to extract the recombinant plasmid for verification with double enzyme digestion.
The suspension of verified Agrobacterium containing recombinant plasmid was used to infect tobacco leaf discs. The MS differentiation medium containing Kan 100 mg L−1 was used to induce adventitious buds and then subcultured for 2 weeks. When adventitious buds grew to 2–3 leaves, they were cut and transferred to MS rooting medium containing Kan 75 mg L−1 for rooting. When the tobacco plants grew to 5–6 leaves, the genomic DNA was extracted from the fresh tobacco leaves. Using the extracted tobacco genome DNA as template, non-transgenic tobacco was negatively controlled, and TAPX-PBI121 plasmid as positive control, the specific primer of TAPX gene was used for PCR analysis to detect the transgenic tobacco. At the same time, the designed primers of GFP sequence in the vector GFPF (5′-CCCGGGATGGGCAAAGGAGAA-3′) and GFPR (5′-GAGCTCTTATTTGTATAGTCA-3′) were also used for PCR detection respectively, to exclude the impact of TAPX gene of tobacco itself.
Results and Analysis
Total RNA Quality Detection
Integrity of the extracted total RNA from sugarcane was checked with 1.2 % agarose gel. The brightness ratio of the two bands of 28S and 18S was 2:1, and the 5S band was visible (Fig. 1). It indicated that the integrity of the extracted RNA was good enough for the next experiment.

The total RNA of leaf of sugarcane
cDNA Cloning of TAPX Full Length
The cDNA synthesized by reverse transcription was used as template for PCR amplification with primers STAPX and 3 SIDE. A band of about 1,700 bp was obtained as shown in Fig. 2. The detected fragments were recovered, ligated, transformed, cloned and sequenced, and a fragment of 1,703 bp was obtained.

PCR product of TAPX gene from sugarcane. M: DL2000 Marker; A: PCR product of the full-length cDNA of TAPX gene
The obtained sequence was compared with those in GenBank Blast, and the result showed it to be TAPX gene. The full length of the cloned sugarcane TAPX gene sequence was 1,703 bp, including the promoter ATG and terminator TGA, and it was registered in GenBenk with the accession number JQ958327. The gene contains a 1,422 bp complete open reading frame ranged from 1 to 1,422 bp, encoding 473 amino acids. Besides, the gene contains a 281 bp 3′ non-coding region as shown in Fig. 3.

Nucleotide sequence and predicted amino acid sequence of TAPX
Bioinformatics Analysis of TAPX Gene
Using the Expasy online website (http://expasy.org/tools/), it was predicted that the amino acid sequence encoded by TAPX gene has an isoelectric point of 5.71, and a protein molecular weight of 51.1 kDa. The composition of amino acid reveal that Ala, Pro, Leu, Gly, and they accounted for 14.6, 8.7, 8.2 and 7.8 % of total amino acids respectively. The protein formula was C2263H3521N619O697S13, and the position of 463 showed the maximum hydrophobicity of 2.933. On the contrary, the position of 290 showed the smallest hydrophobicity of −3.033. It was speculated that the protein was unstable. The subcellular localization revealed that the protein was located in chloroplast. SOSUI signal software analysis predicted that the TAPX protein contains 5 transmembrane regions, and two signal peptides located in 1–14 and 451–473 amino acids, respectively, and the protein is a transmembrane protein. Moreover, the results predicted 10 sites of serine phosphorylation, 4 sites of threonine phosphorylation and 5 sites of the tyrosine phosphorylation. SOPMA was used to predict the secondary structure of TAPX, and the results indicated that the protein contains 180 α-helices, accounting for 38.05 %; 41 extended strands, accounting for 8.67 %; 30 β-reverse turn, accounting for 6.34 %; and 222 random coils, accounting for 46.93 %. Protein blast program predicted that the TAPX protein has a variety of specific binding sites, and they are heme binding site, substrate-binding sites and K+ binding sites (Fig. 4).

Conserved domains of TAPX
In addition, SMART and Motif Scan software was used to analyze protein functional domains of TAPX, and the results showed that site located in 20–23 for CAMP and CGMP protein kinase phosphorylation, sites 157–160, 168–171, 188–191, 349–352, 400–403 were for casein kinase II phosphorylation sites; sites 119–124, 134–139, 155–160, 261–266 for N-myristoylation; 18–20, sites 38–40, 53–55, 124–125, 309–311, 429–431 for protein kinase C phosphorylation sites; site 224–239 for peroxidase heme binding and 58–473 were the typical function region of chloroplast ascorbate peroxidase. The sequence alignment results of amino acid sequences of sugarcane TAPX gene and other plant species are given in Fig. 5, which reveal TAPX is highly conserved gene in the functional region.

Multialignment of amino acid sequences of the TAPX proteins isolated from seven plants. JQ958327 (Saccharum officinarum), XM_002453931.1 (Sorghum bicolor), NM_001156037.1 (Zea mays), AB114856.1(Oryza sativa), XM_003575081.1(Brachypodium distachyon), AK356057.1 (Hordeum vulgare), EF184291.1 (Triticum aestivum)
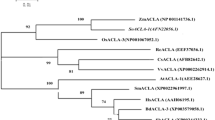
The phylogenetic tree for TAPX genes of sugarcane and 15 other plant species was built by MEGA software (Fig. 6). The TAPX gene of sugarcane was clustered into one category with corn, rice, Brachypodium distachyon and sorghum, indicating that they may have evolved from the same ancestor but through different pathways. Among them, the homology of TAPX gene between sugarcane and rice was 89 %, the highest among all.

Phylogenetic relationship of amino acid sequences between sugarcane TAPX and other TAPX proteins
Analysis of TAPX Gene Expression Levels
With real-time quantitative PCR technology and GAPDH gene as an internal control, we analyzed the sugarcane TAPX gene expressions in the roots, stems, leaves (Fig. 7), and the expression differences in leaves in four different stresses and at different time (Fig. 8).

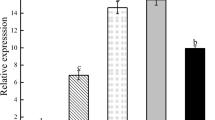
Tissue-specific expression of TAPX gene in root, stalk and leaf of sugarcane

Expression profiles of sugarcane TAPX gene in different treatments
As shown in Fig. 7, the leaves, roots and stems all had TAPX gene expression, but there were significant differences in expression levels. If the expression in the stems was set as 1, it was 0.02 times only in the roots, however, in the leaves it reached to 14.9 times.
As shown in Fig. 8, the expression of TAPX gene showed almost no change under low-temperature stress. The expression trend was from low to high under hydrogen peroxide stress, which showed decrease during the early stage of the treatment but later increase to 3.16 times of that in the control indicating significant induced expression. Under NaCl stress, the expression level was inclined firstly and then declined, but again increased 6 h after treatment and was 1.77 times of that of the control at 12 h and then declined. The expression level was significantly induced under PEG stress and reached the maximum expression at 48 h when the expression level was 2.79 times of that of control followed by a slight decrease. Based on the above quantitative results, it could be concluded that TAPX gene showed up-regulated expression under the stresses of PEG, hydrogen peroxide and NaCl, but there was no obvious change under low temperature stress.
Construction of Prokaryotic Expression Plasmid and Identification of Recombinant
After the TAPX gene was recovered and purified, and the pET-30a (+) plasmid extracted, they were double digested with EcoR I and Xho I, respectively. The recovered target fragments were ligated with the expression vector pET-30a (+) and transformed into competent cells BL21 (DE3). The recombinant plasmid pET30a-TAPX was double digested with EcoR I and Xho I, and amplified by PCR. The agarose gel electrophoresis results showed that all of them were able to get the target fragment of about 1,430 bp, indicating that the TAPX gene was inserted into the vector plasmid. As shown in Fig. 9, the prokaryotic expression vector of sugarcane TAPX gene pET30a-TAPX had been built successfully.

Restriction enzyme digestion and PCR analysis of pET30a-TAPX. 1 Digestion of pET30a-TAPX; 2 PCR analysis; M1 DL 10000 Marker; M2 DL 2000 Marker
SDS-PAGE Analysis
The plasmid expression strain BL21 which had been identified containing pET30a-TAPX was induced with IPTG at a final concentration 1.0 mM at 37 °C for 4, 6, and 8 h, respectively. The pET30a-TAPX recombinant plasmid without IPTG induction and empty pET30a vector were used as the controls, and the expression products were collected for SDS-PAGE analysis. As shown in Fig. 10, the target protein started expression at about 2 h after IPTG induction. A strip of fusion protein expression was induced at the position of approximately 58 kDa. The theoretical molecular weight of the fusion protein was 58.1 kDa, which was consistent with the theoretical 51.1 kDa molecular weight of the sugarcane TAPX protein coding region plus 7 kD molecular weight of the vector His. Tag protein. There was no target protein expression in the controls, revealing that the prokaryotic expression vector had been constructed correctly.

Expression of recombinant TAPX protein. M protein marker; 1 pET-30a without induction; 2 pET-30a induced for 6 h; 3 pET30a-TAPX without induction; 4–7 pET30a-TAPX induced with 1 mM IPTG for 2, 4, 6, 8 h, respectively
Construction of Excessive Plant Expression Vector and Identification of Transgenic Tobacco
After the TAPX gene was recovered and purified, and PBI121 plasmid extracted, they were doubly digested with Xba l and Sma I, respectively. The recovered target fragment was ligated with the expression vector PBI121 and transformed into competent cells DH5α. The recombinant plasmid was extracted and transformed into the expression strain EHA105 by freeze–thaw method. The obtained recombinant plasmid PBI121-TAPX was doubly digested with Xba l and Sma I and amplified by PCR. The agarose electrophoresis results showed that the target fragment of about 1,430 bp was got, indicating the TAPX gene had been inserted into the vector plasmid. It proved that the eukaryotic expression vector PBI121-TAPX of sugarcane TAPX protein gene had been successfully constructed (Fig. 11).

Restriction enzyme digestion and PCR analysis of PBI121-TAPX. 1 Digestion of PBI121-TAPX; 2 PBI121-TAPX; 3 PCR analysis; M1 DL 10000 Marker; M2 DL 2000 Marker
Agrobacterium which had been checked containing a recombinant plasmid was used to infect tobacco leaf discs by culturing The infected tobacco leaf discs were cultured in the MS differentiation medium containing Kan 100 mg L−1, they were subcultured in half a month, and adventitious buds were induced in 20 days. The adventitious buds were cut when they grew to 2–3 leaves, and transferred into the rooting MS medium containing Kan 75 mg L−1. Resistant regenerants were obtained in about 30 days, and the plantlets were transplanted after hardening treatment (Fig. 12).

The obtained TAPX transgenic tobacco. a Resistant callus or buds; b Tobacco regenerated seedlings; 1 Control 0 (Non-transgenic tobacco without antibiotic pressure); 2 Control 1 (Non-transgenic tobacco with antibiotic pressure); 3 Transgenic tobacco buds or plants
Genomic DNA was extracted from the fresh leaves of 5–6 leaved tobacco plants and was used as template and that from non-transgenic tobacco plant as negative control and TAPX-PBI121 plasmid as positive control, 15 transgenic tobacco plants were randomly selected for PCR detection using TAPX gene specific primers. As shown in Fig. 13, nine plants were detected to have a single target band, suggesting that TAPX transgenic tobacco plants had been obtained, and the transformation efficiency of approximately 60 %.

PCR assay of TAPX transgenic plants
Discussion
Previous studies have shown that the destruction of reactive oxygen enzymatic clearance mechanism was one of the important reasons for physiological function decline or even death in plant (Hernandez et al. 1995; Bandeoğlu et al. 2004). Chloroplast is the vital organ for photosynthesis in plant, where superoxide anion radicals are disproportionate to H2O2 by superoxide dismutase, and the H2O2 is scavenged mainly by ASA–GSH cycle pathway which needs the participation of APX and other enzymes (Asada 1999). But catalase (CAT), peroxidase (POD), cytochrome C and pyridine nucleotides were not found in chloroplasts, and APX had high affinity for H2O2. Therefore, APX is the key enzyme for clearance of H2O2 in chloroplast (Chen and Asada 1989).
In this study, TAPX gene was cloned from the sugarcane, and sequence analysis proved it had a complete open reading frame. Bioinformatics analysis showed that the protein has a signal peptide which is a transmembrane protein located in the chloroplast. So it is speculated that after the protein synthesis, it would be transported to the chloroplasts where it functions, and it was located in the thylakoid membrane ascorbate peroxidase. The encoded protein had a high homology with other Gramineae species. Functional analysis showed TAPX gene had many peroxidase-specific binding sites. They were the heme binding sites, substrate binding sites, K+ binding sites and typical functional region of ascorbate peroxidase. The TAPX gene was very conservative in all reported ASA gene sequences (Jiang et al. 2012; Hou et al. 2009; Guan et al. 2009). Up to now, there have been many reports about plant resistance to oxidative stress and TAPX gene in plants. However, it has not been found any report about TAPX gene involved in sugarcane resistance to stress. In this study, the TAPX gene expression differences in sugarcane tissue were analyzed by RT-PCR. It was found that TAPX gene was constitutively expressed in root, stem, leaf of sugarcane plant, and had specific expression site. The highest expression was found in leaves, followed by stems and rarely in roots. It was in line with the results mentioned before that the TAPX gene was mainly located in chloroplast. In addition, the expression of sugarcane TAPX gene was analyzed by RT-PCR under the exogenous stresses of low temperature, H2O2, PEG and NaCl. The results showed TAPX genes did not change obviously under low temperature stress, while the cAPX was found significantly upregulated under low temperature stress in our pre-experiment. From this phenomenon, it can be concluded that the different members of APX gene family located in different regions of the cell, and had different regulation patterns, thus the sensitivity and expression levels were different under various abiotic stresses. D’Arcy-Lameta et al. (2006) indicated that the cell structure was destroyed under drought stresses, so it is impossible to accurately determine the APX enzyme activity in different cell intervals. Therefore, in order to analyze the role of the APX gene family better, it is necessary to clone the genes of different isozymes, and to study their expression levels under different stresses. It is believed that the adverse stresses cause plant damage and thus lead to increase of membrane lipid peroxidation level, and decrease of protective enzyme system activity. In this experiment, the expression of APX gene of sugarcane was down-regulated by H2O2 stress in a short time before significant expression was induced. This may be due to the dramatic increase of H2O2 in the intracellular of plants at the early stage of oxidant H2O2 treatment, resulting in rapid accumulation of active oxygen in the plants which led to minor injury of cell structures. As a result, the level of the expression of the APX decreased slightly. However, H2O2 in a certain concentration range can serve as a signaling molecule inducing gene expression alteration, and usually the concentration of H2O2 played a signal role in the intracellular was 1–700 nM (Stone 2004; Yamaguchi-Shinozaki and Shinozaki 2006). This may be why the sugarcane TAPX gene expression increased with 10 mM H2O2 treatment in this experiment. Sun et al. (2011) also proved that excessive expression of tomato StAPX gene in tobacco can effectively remove H2O2 to improve the plant resistance to oxidative stress. Murgia et al. (2004) indicated that over expression of tAPX gene of Arabidopsis can increase adversity resistance which was caused by paraquat. NaCl and PEG stresses were significantly induced the expression of the TAPX gene in a short time. There have been reports showed that over expression of TAPX gene can improve plant resistance to salt stress in many plants (Badawi et al. 2004). Over expression of LetAPX in tobacco can increase the activity of APX under salt stress, thereby reducing H2O2 accumulation and damage of membranes which were helpful of improving the antioxidant capacity and tobacco tolerance to salt (Sun et al. 2009). Owing to the effect of drought stress, osmotic stress was similar to the stresses of PEG, NaCl and H2O2 to plants. So it can be speculated the TAPX genes play a role in sugarcane resistance to drought, osmotolerance and oxidative stresses, but the regulation models and the degrees might be different, and further tests would be needed to determine the specific role. Also, under biotic and abiotic stresses, the reactive oxygen increase in plants when they were hurt, and the reactive oxygen serves as a second messenger that can induce TAPX gene expression, therefore, TAPX gene plays a key role in plant resistance to adversity.
In this study, the TAPX gene from sugarcane was ligated with the expression vector pET-30a (+) and the expression strain BL21 (DE3) to induce fusion protein expression. The analysis results showed that the sugarcane TAPX protein combined with the tagged protein to form a fusion protein which was successfully expressed in a prokaryotic expression system. This lays the foundation for further preparation of polyclonal antibodies. Besides, the over-expression vector of TAPX gene was constructed and transformed into tobacco by Agrobacterium-mediation, and transgenic tobacco plants were obtained. The transgenic tobacco obtained provides a theoretical basis for further researches on functions of TAPX genes in response to adverse stresses, signal transduction, regulatory pathways, and sugarcane resistance breeding.
Conclusion
TAPX gene was cloned and registered in GenBank with accession number JQ958327 in this study. The gene contains a 1,422 bp open reading frame encoding 473 amino acids. It is predicted the protein molecular weight is 51.1 kDa. Bioinformatics analysis showed that the gene contains typical functional areas of peroxidase as well as heme binding sites. Phylogenetic analysis showed that the gene was located in the same evolutionary branch with other gramineous plants and had the highest homology with that of rice. Prokaryotic expression results showed that the gene was expressed in fusion protein which relative molecular weight is 58.0 kDa approximately. Quantitative PCR results showed that the TAPX gene expressed in roots, stems and leaves of sugarcane, but the most in leaves. Several abiotic stresses induced the expression of the gene, suggesting that it plays a role in sugarcane response to drought, salt and other stresses.
References
Asada, K. 1992. Ascorbate peroxidase—A hydrogen peroxide-scavenging enzyme in plants. Physiologia Plantarum 85(2): 235–241.
Asada, K. 1999. The water-water cycle in chloroplasts: scavenging of active oxygens and dissipation of excess photons. Annual Review of Plant Physiology and Plant Molecular Biology 50: 601–639.
Badawi, G.H., N. Kawano, Y. Yamauchi, E. Shimada, R. Sasaki, A. Kubo, and K. Tanaka. 2004. Over-expression of ascorbate peroxidase in tobacco chloroplasts enhances the tolerance to salt stress and water deficit. Physiologia Plantarum 121(2): 231–238.
Bandeoğlu, E., F. Eyidoğan, M. Yücel, and H.A. Öktem. 2004. Antioxidant responses of shoots and roots of lentil to NaCl-salinity stress. Plant Growth Regulation 42: 69–77.
Chen, G.X., and K. Asada. 1989. Asorbate peroxidase in tea leaves occurrence of two isoenzymes and their differences in enzymatic and molecular properties. Plant and Cell Physiology 30(7): 987–998.
Chen, Y., H. Wang, X. Wang, A. Cao, and P. Chen. 2006. Cloning and expression of peroxisomal ascorbate peroxidase gene from wheat. Molecular Biology Reporters 33(3): 207–213.
Chew, O., J. Whelan, and A.H. Millar. 2003. Molecular definition of the ascorbate-glutathione cycle in Arabidopsis mitochondria reveals dual targeting of antioxidant defenses in plants. Journal of Biological Chemistry 278(47): 46869–46877.
D’Arcy-Lameta, A., R. Ferrari-Iliou, D. Contour-Ansel, P.T. Anh-Thu, and Y. Zuily-Fodil. 2006. Isolation and characterization of four ascorbate peroxidase cDNAs responsive to water deficit in cowpea leaves. Annals of Botany 97(1): 133–140.
Deng, Z.Y., H.B. Liu, G.M. Zhang, F.X. Fang, H. He, Y.R. Li, R.Z. Yang, M. Li, G.L. Chen, C.X. Liang, and S.L. Bi. 2009. Survey of frost occurred regular harm in sugarcane. Sugar Crops of China 1: 47–50.
Foyer, C.H., and B. Hailiwell. 1997. Purification and properties of dehydroascorbate reductase from spinach leaves. Phytochemistry 16(9): 1347–1350.
Guan, Q.J., L. Li, T. Takano, and S.K. Liu. 2009. Cloning of an ascorbate peroxidase gene from Puccinellia tenuiflora and its expression analysis. Genomics and Applied Biology 28(4): 631–639.
Hernandez, J.A., E. Olmos, F.J. Corpas, F. Sevilla, and L.A. del Rio. 1995. Salt-induced oxidative stress in chloroplasts of pea plants. Plant Science 105(2): 151–167.
Hou, Y.X., H.R. Tang, Y. Zhang, Y. Luo, and Q. Chen. 2009. Cloning and expression analysis of ascorbate peroxidase gene during fruit development and ripening in Fragaria. World Journal of Agricultural Science 5(6): 675–679.
Ishikawa, T., and S. Shigeoka. 2008. Recent advances in ascorbate biosynthesis and the physiological significance of ascorbate peroxidase in photosynthesizing organisms. Bioscience, Biotechnology, and Biochemistry 72(5): 1143–1154.
Jiang, M., Z.X. Zhang, and L.G. Yuan. 2012. Cloning and expression analysis of an ascorbate peroxidase gene BoAPX2 from Brassica oleracea var. italic. Aata Phytopathologica Sinica 42(4): 374–380.
Li, Y.R., and L.T. Yang. 2009. New developments of sugarcane industry and technology in China since 1990s. Southwest China Journal of Agricultural Sciences 22(5): 1469–1476.
Li, Y.R., F.X. Fang, J.M. Wu, X. Li, R.H. Zhang, X.H. Liu, H. He, R.Z. Yang, L.T. Yang, G.L. Chen, S.Q. Sun, J.L. Xie, X.Y. Liu, W.H. Huang, W.X. Duan, W.Z. He, and M. Wang. 2011. Survey and defensive of production cold damage on sugarcane in Guang Xi in 2010/2011. Journal of Southern Agriculture 42(1): 37–42.
Lin, L., X. Wang, and Y. Wang. 2006. cDNA clone, fusion expression and purification of the novel gene related to ascorbate peroxidase from Chinese wild Vitis pseudoreticulata in E. coli. Molecular Biology Reporters 33(3): 197–206.
Lu, H., R.L. Han, and X.N. Jiang. 2009. Heterologous expression and characterization of a proxidomal ascorbate peroxidase from Populus tomentosa. Molecular Biology Reporters 36(1): 21–27.
Mano, S., K. Yamaguchi, M. Hayashi, and M. Nishimura. 1997. Stromal and thylakoid-bound ascorbate peroxidases are produced by alternative splicing in pumpkin. FEBS Letters 413(1): 21–26.
Murgia, I., D. Tarantino, C. Vnanini, M. Bracale, S. Carravieri, and C. Soave. 2004. Arabidopsis thaliana plants overexpressing thylakoidal ascorbate peroxidase show increased resistance to Paraquat-induced photooxidative stress and to nitric oxide-induced cell death. Plant Journal 38(6): 940–953.
Najami, N., T. Janda, W. Barriah, G. Kayam, M. Tal, M. Guy, and M. Volokita. 2008. Ascorbate peroxidase gene family in tomato: its identification and characterization. Molecular Genetics and Genomics 279(2): 171–182.
Shi, F., R. Yamamoto, S. Shimamura, S. Hiraga, N. Nakayama, T. Nakamura, K. Yukawa, M. Hachinohe, H. Matsumoto, and S. Komatsu. 2008. Cytosolic ascorbate peroxidase 2 (cAPX 2) is involved in the soybean response to flooding. Phytochemistry 69(6): 1295–1303.
Shigeoka, S., T. Ishikawa, M. Tamoi, Y. Miyagawa, T. Takeda, and Y. Yabuta. 2002. Regulation and function of 85 ascorbate peroxidase isoenzymes. Journal of Experimental Botony 53(372): 1305–1319.
Stone, J.R. 2004. An assessment of proposed mechanism for sensing hydrogen peroxide in mammalian systems. Archives of Biochemistry and Biophysics 422: 119–124.
Sun, W.H., X.Y. Chen, B. Du, W. Song, Y. Zhou, and Y. Wang. 2011. Tobacco seedlings overexpressing StAPX enhanced resistance to oxidative stress. Plant Physiology Journal 47: 613–618.
Sun, W.H., F. Li, D.F. Shu, X.C. Dong, X.M. Yang, and Q.W. Meng. 2009. Tobacco plants transformed with tomato sense LetAPX enhanced salt tolerance. Scientia Agricultura Sinica 42(4): 1165–1171.
Teixeirra, F.K., L. Menezes-Renavente, V.C. Galvão, R. Margis, and M. Margis-Pinheiro. 2006. Rice ascorbate peroxidase gene family encodes functionally diverse isoforms localized in different subcellular compartments. Planta 224(2): 300–314.
Wang, J., H. Zhang, and R.D. Allen. 1999. Overexpression of an Arabidopsis peroxisomal ascorbate peroxidase gene in tobacco increases protection against oxidative stress. Plant and Cell Physiology 40(7): 725–732.
Yamaguchi-Shinozaki, K., and K. Shinozaki. 2006. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annual Review of Plant Biology 57: 781–803.
Yoshimura, K., Y. Yabuta, T. Ishikawa, and S. Shiqeoka. 2000. Expression of spinach ascorbate peroxidase isoenzymes in response to oxidative stresses. Plant Physiology 123(1): 223–234.
Acknowledgments
This work was supported in part by National “863” Program project (2013AA102604), National Program for International Scientific Exchange projects (2013DFA31600), National Natural Foundation of China (31360293), Guangxi Special Funds for Bagui Scholars and Distinguished Guangxi R&D Research Program projects (Gui Ke He 1347004-2, Gui Ke Neng 14121008-2-1), Guangxi Natural Science Foundation projects (2011GXNSFF018002, 2012GXNSFDA053011, 2013NXNSFAA019073), Guangxi Governor Fund (11166-02) and Guangxi Academy of Agricultural Sciences project (Gui Nong Ke 2012YT01).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Wang, S., Zhang, KK., Huang, X. et al. Cloning and Functional Analysis of Thylakoidal Ascorbate Peroxidase (TAPX) Gene in Sugarcane. Sugar Tech 17, 356–366 (2015). https://doi.org/10.1007/s12355-014-0354-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12355-014-0354-x