
Abstract
Introduction
Cigarette smoking remains a substantial public health problem. Nicotine replacement therapy (NRT) is an effective treatment that increases the success of a quit attempt. There are different NRT formats with no difference in efficacy, but their pharmaceutical form or route of administration may translate into individual preferences. A novel prototype mini lozenge was developed to offer smokers a new NRT option to aid in their quit attempt. Two studies were conducted to characterize the pharmacokinetic parameters and to evaluate its bioequivalence to a commercially available nicotine mini lozenge.
Methods
Two randomized, open-label, crossover studies were conducted to evaluate either the 2 or 4 mg dose level. Heavy smokers in otherwise good health were randomly assigned to one of two treatment sequences: the prototype mini lozenge followed by a commercially available mini lozenge, or the converse. After a 5 to 7 day washout period, subjects crossed over to receive the other study treatment. Blood sampling occurred pre- and post-dose nicotine and was assessed using a validated solid-phase extraction with ultra-high-performance liquid chromatography and tandem mass spectrometry. The primary endpoint was bioequivalence as determined by maximal plasma nicotine concentration (Cmax) and the extent of nicotine absorption (AUC0–t and AUC0–∞). The secondary endpoints included the time to Cmax (Tmax), half-life, the elimination constant (Kel), and safety.
Results
The prototype mini lozenge was bioequivalent to the commercially available mini lozenge, with no significant difference in Cmax, AUC0–t, or AUC0–∞ or any of the secondary outcomes. The most common treatment-emergent adverse event was throat irritation, of which all cases were mild in severity. There were no serious adverse events.
Conclusion
The prototype mini lozenge is bioequivalent to a commercially available mini lozenge and may provide smokers with a new oral NRT option to aid in smoking cessation and of tobacco dependence through the relief of nicotine withdrawal symptoms, including cravings.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Why carry out this study? |
Nicotine replacement therapy (NRT) has been demonstrated to be an effective treatment to aid in smoking cessation and of tobacco dependence through the relief of nicotine withdrawal symptoms, including cravings |
The present analysis evaluated the pharmacokinetics of the novel prototype mini lozenge and determined its bioequivalence to a commercially available mini lozenge |
What was learned from the study? |
The prototype mini lozenge was bioequivalent to a commercially available mini lozenge with no difference in pharmacokinetic parameters between the treatments |
The prototype mini lozenge may provide smokers with a new NRT option to help aid in their quit attempt |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.14605407.
Introduction
Cigarette smoking remains a substantial public health problem, causing more than 7 million deaths each year worldwide, with an additional 1.2 million deaths attributed to second-hand smoke [1]. In addition, cigarette smoking is associated with an increased risk of morbidity and mortality from cancers, diabetes mellitus, and diseases of the cardiovascular, cerebrovascular, and respiratory systems. [2] Despite these risks, an estimated 1.3 billion individuals use tobacco worldwide, including 34.3 million who smoke cigarettes in the USA [1, 3].
Nicotine is highly addictive, as it is rapidly absorbed and reaches the brain within 10–20 s, where it causes positive psychoactive and neuroadaptive effects over time [4, 5]. In addition, individuals develop anticipatory cues, such as an urge to smoke after eating a meal; positive reinforcement mechanisms such as improved mood and heightened vigilance; and negative reinforcement mechanisms such as withdrawal symptoms including irritability, anxiety, impaired concentration, and increased appetite [6,7,8]. Taken together, these effects contribute to the extreme difficulty of achieving a successful quit attempt from smoking. This difficulty was highlighted in the 2015 National Health Interview Surveys (NHIS), in which only 7.4% of the 68% of adult smokers who wanted to quit smoking were successful [9]. Furthermore, individuals required a mean of 2–6 quit attempts before they were successful [9, 10]. Therefore, although quitting smoking improves health and prolongs life-years, many individuals have difficulty successfully quitting cigarette smoking and would benefit from treatments that are indicated as an aid to smoking cessation and of tobacco dependence through the relief of nicotine withdrawal symptoms, including cravings.
Nicotine replacement therapy (NRT) is recommended by the United States Public Health Service (USPHS) guideline as a first-line treatment for smoking cessation [11]. It is an effective treatment that increases the success of a quit attempt [12]. There are currently six forms of NRT available in the USA, four of which are available over-the-counter (OTC). Among these options, there are long-acting forms, such as the nicotine patch, and short-acting forms, such as the lozenge, mini lozenge, gum, nasal spray, and inhaler. The efficacy is similar between these forms [13], but their pharmacokinetics (PKs) differ in terms of their maximal plasma nicotine levels (Cmax), the time to reach Cmax (Tmax), and their half-lives (t1/2). Short-acting NRT offers a convenient form with faster craving relief than long-acting forms. For lozenges, the main difference is visualization and taste. The multiple formulations and non-prescription status of many NRT formats enables broad access and enables smokers to choose the format that best fits their personal preferences and lifestyle, which may further improve their chance of successfully quitting smoking.
A novel mini lozenge form, referred to herein as the prototype mini lozenge, has been developed to offer smokers an innovative new short-acting NRT option with a new mint flavor that may be preferred by some individuals. In these studies, in order to receive the marketing authorization, it was necessary to demonstrate the bioequivalence to the commercially available nicotine mini lozenge. Therefore, the aim of the present study was to evaluate the bioequivalence of two different doses of a new, innovative prototype mini lozenge with a commercially available mini lozenge among smokers.
Methods
Study Design
The present analysis includes two single-center, open-label, randomized, four-treatment, two-period, crossover studies conducted between June and November 2018 and June to March 2019 in Lincoln, Nebraska. The aim was to evaluate the bioequivalence of two different doses of the prototype mini lozenge with a commercially available mini lozenge among smokers. The team involved in the analysis of nicotine in plasma samples was blinded to treatment; however, the participants and the clinical staff were aware of the treatment administered because of visual differences in the prototype and commercially available mini lozenges. The studies were reviewed and approved by an independent institutional review board (IRB) at Advarra IRB and the studies were conducted in full compliance of all relevant laws and regulations in the USA and with the requirements specified in the Declaration of Helsinki, and in accordance with the Good Clinical Practice guidelines of the International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. All subjects provided written informed consent prior to any study-specific procedures. Subjects could withdraw from either study at any time for any reason.
Study Subjects
Subjects were recruited to the study through Celerion’s subject database and recruitment campaigns that included advertisements through social media, newspaper, and booths at shopping malls and fairs. All subjects were considered heavy smokers, defined as having smoked cigarettes for at least 12 months with the first cigarette of the day smoked within 30 min of waking. Subjects were in otherwise good health, with key inclusion criteria including age 19–55 years and body mass index (BMI) of 19–28 kg/m2. Subjects were excluded if they had an acute or chronic medical or psychiatric condition or laboratory abnormality that many increase their risk of taking the study treatments; current pregnancy or breastfeeding; history of regular alcohol use of more than 14 drinks per week; a positive illicit drug screen including tetrahydrocannabinol; and, drinking more than five cups of coffee or tea per day. Subjects were not allowed to use NRT, tobacco products other than cigarettes, or electronic cigarettes within 21 days of the first study session. Other key exclusion criteria included use of prescription or experimental drugs within 2 weeks or five half-lives prior to the first dose of the study drug, except for contraception or hormone replacement therapy, and any known or suspected allergies/intolerances to the study treatment, or any medical condition that could alter the absorption, distribution, metabolism, or excretion of the nicotine.
Sexually active subjects of childbearing potential were required to agree to appropriately using a highly effective method of contraception during the study period and for at least 5 days after the last treatment dose. In addition, subjects of childbearing potential underwent a serum pregnancy test during the screening visit and at check-in prior to the first and second treatment sessions.
Treatment Periods
Subjects underwent a screening visit prior to the studies. For both studies, subjects were confined to the facility for the duration of each treatment period for a total of 60 h each. Each confinement period included a baseline check-in visit with randomization, pretreatment phase lasting 36 h, and a treatment phase lasting 24 h. During confinement, the subjects abstained from smoking, which was confirmed by an expired carbon monoxide (CO) of 10 ppm or less as measured by a calibrated Bedfont Smokerlyzer (Bedfont Scientific Ltd, Harrietsham, UK). Prespecified CO measurements were obtained at baseline, immediately before randomization, and prior to dose administration. In addition, there were four random CO measurements during each treatment period. Subjects also abstained from alcohol and strenuous exercise 48 h prior to each blood collection.
Washout
The washout period began 24 h after the study treatment was administered during the first treatment period and lasted from 5 to 7 days. Subjects were allowed to smoke cigarettes during the washout period. Smoking abstinence was required during the second treatment period as it was during the first treatment period.
Study Treatments
Subjects were randomly assigned to one of two dosing sequences in each study with the washout period between the different study treatments. The sequences included 2 or 4 mg of the prototype mini lozenge followed by 2 or 4 mg of a commercially available mini lozenge (GSK Consumer Healthcare, Aiken, South Carolina, USA), respectively, or 2 or 4 mg of the commercially available mini lozenge followed by 2 or 4 mg of the prototype mini lozenge, respectively. Study treatments were administered as a single dose given at approximately 8:00 a.m., after a 10 h overnight fast. The clinical staff provided subjects with the standard directions for mini lozenge use consistent with the product label. Subjects placed the lozenge into their oral cavity and occasionally moved the lozenge from side to side to allow it to dissolve slowly, without chewing and while minimizing swallowing.
Study Assessments
Screening Visit
All subjects participated in a screening visit at least 2 days before random assignment, during which they underwent standard screening procedures, and physical and clinical exams. Subjects’ level of nicotine dependency was measured using the Fagerstrom Test for Nicotine Dependence (FTND).
Treatment Periods
The check-in visit occurred 2 days prior to the beginning of each treatment period to ensure that subjects abstained from nicotine use 36 h prior to the first study treatment. The baseline visit occurred 1 day after the check-in visit, during which expired CO was measured and subjects were randomized at the beginning of the first treatment visit within 36 h before the study treatment was administered.
Subjects received a single dose of the study treatment at the beginning of the second treatment visit, which lasted for 24 h. Blood sampling occurred pre-dose and post-dose.
At the end of the second treatment visit, subjects underwent a washout period of 5–7 days followed by the second treatment session in which subjects received their second treatment sequence.
Sample Collection
Blood was collected for PK analyses during the first and second treatment periods of both studies. The blood specimens were collected beginning 45 min prior to and up to and including 24 h after study drug administration. Samples were collected at 45, 30, and 15 min predose, then 5, 10, 20, 30, 40, 50, 60, 75, and 90 min postdose, then 2, 3, 4, 6, 8, 10, 14, 20, and 24 h postdose. For each time point, 6 mL of blood was collected via direct venipuncture into tubes containing K2EDTA and plasma was harvested for further analyses. Plasma was split into two aliquots and stored frozen at − 20 °C until analysis.
Plasma nicotine concentration was determined by a certified bioanalytical laboratory (Celerion, Lincoln, Nebraska, USA) using a validated method of solid-phase extraction with ultra-high-performance liquid chromatography and tandem mass spectrometry (LC–MS/MS) with a lower limit of quantitation for nicotine of 0.200 ng/mL.
Safety Evaluation
The safety evaluation included documentation of all adverse events (AEs) that occurred during the study beginning at the time of signing the informed consent form to 5 days following the last treatment administration. An AE was defined as any untoward medical occurrence experienced by a study subject, regardless of whether it was considered related to the treatment drug. All treatment-emergent AEs (TEAEs) were summarized by primary System Organ Classes (SOC) and Preferred Term (PT) and coded using MedDRA Version 21.0.
Data Analyses
Sample Size
The sample sizes calculations for both studies assumed a 20% dropout and non-evaluable rate. For the study of the 2-mg prototype mini lozenge, at least 40 subjects were planned for screening to ensure that 32 subjects completed the study. This sample size would achieve 90% power at a 5% significance level, assuming the highest intrasubject coefficient of variation (CV) was 23%. The true ratio that was used in the sample size calculation was 1.05.
For the 4-mg prototype mini lozenge, at least 37 subjects were planned for screening to ensure that 29 subjects completed the study. This sample size would achieve 90% power at a 5% significance level, assuming the highest intrasubject CV of 22%. The true ratio that was used in the sample size calculation was 1.05.
Analysis Populations
There were three analysis populations. All patients who were randomly assigned, regardless whether they received study treatment or not, were part of the randomized population. The safety population included all subjects who received at least one dose of the study treatment. The PK population included all randomized subjects who completed both treatment periods and who had no major protocol deviations concerning PK.
Statistical Analyses
There were two PK analysis sets (PKAS). PKAS1 included data from all subjects in the PK population. Subjects with a baseline nicotine concentration greater than 5% of the individual Cmax in either study period were excluded. PKAS2 included only baseline-adjusted data from subjects in the PK population for which the relevant baseline-adjusted PK parameters (at least one AUC or Cmax) could be derived, including those with baseline nicotine concentrations greater than 5% of the individual Cmax in either period.
The primary endpoint was to determine the bioequivalence of the prototype mini lozenge to the commercially available mini lozenge, which was assessed by pairwise comparison of the PK parameters (AUC0–t, AUC0–∞, and Cmax) for the baseline-adjusted nicotine concentration profiles from PKAS1.
The PK parameters were calculated using Phoenix WinNonlin Version 7.0 or higher. A linear mixed-effects model was fit to the natural log (ln)-transformed PK variables (AUC0–t, AUC0–∞, and Cmax) as the dependent variable, and treatment, period, and sequence as fixed effects. Subject nested within sequence was a random effect. Least-squares estimates of treatment effects were calculated and a 90% confidence interval (CI) for the treatment difference was computed. The treatment difference and its 90% CI were exponentiated to obtain the geometric mean ratios (GMR) between the test and reference products and its 90% CI. Bioequivalence was determined if 90% CIs of the GMRs of AUC0–t, AUC0–∞, and Cmax fell completely within the 0.80–1.25 range.
The secondary endpoints included comparing PK parameters (Tmax, t1/2, Kel), and safety of the prototype mini lozenge and the commercially available mini lozenge among subjects in PKAS1 and PKAS2. A nonparametric analysis for Tmax was performed to compare treatment differences using the Wilcoxon signed-rank test. Median difference (test − reference), the Hodges–Lehmann estimator, and estimated CI were used to examine the location shift in Tmax [14]. Tmax was not ln-transformed.
Results
Subject Disposition and Demographics
2-mg Study
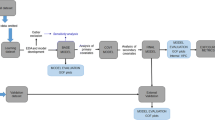
For the 2-mg study, 90 subjects were screened and 46 were included in the study. A total of 46 subjects were treated with the prototype mini lozenge and 44 subjects were treated with the commercially available mini lozenge (Fig. 1). The safety population included all 46 subjects who were randomized. Six subjects were excluded from the PK population because of major protocol deviations. All 40 subjects in the PK population were included in the PKAS2 population. An additional 15 subjects were excluded from the PKAS1 population (n = 25) because of protocol deviations.

Subject disposition
Among the safety population, 65.2% of subjects were male and 34.8% were female (Table 1). The majority of subjects were white (71.7%), followed by 23.9% who were black, 2.2% who were Asian, and 2.2% who were American Indian or Alaska native. There were 4.3% of subjects who were of Hispanic or Latino ethnicity. The median age at baseline was 38.5 years and the median body mass index (BMI) was 23.8 kg/m2.
4-mg Study
For the 4-mg study, 61 subjects were screened and 37 were included in the study. A total of 36 subjects were treated with the prototype mini lozenge and 35 subjects were treated with the commercially available mini lozenge. The safety population included all 37 subjects who were randomized. There were three subjects who were excluded from the PK population because they did not complete both treatment periods. The PKAS2 population included all 34 subjects from the PK population. There were seven subjects who were excluded from the PKAS1 population (n = 27) because of protocol deviations.
In the safety population, 67.6% of subjects were male and 32.4% were female. Similar to the 2-mg study, the majority of subjects were white (81.1%), followed by black (16.2%), or American Indian or Alaska native (2.7%). None of the subjects were of Hispanic or Latino ethnicity. The median age at baseline was 36 years and the median BMI was 24.8 kg/m2.
Pharmacokinetics
Primary Endpoints
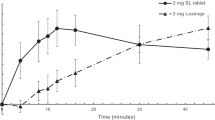
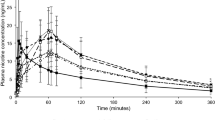
The mean baseline-adjusted nicotine plasma profile of the 2-mg prototype mini lozenge was similar to the 2-mg commercially available mini lozenge, with a mean Cmax of 4.61 ng/mL (standard deviation [SD], 1.39 ng/mL) and 5.15 ng/mL (SD 1.65 ng/mL), respectively (Fig. 2a, Table 2). The mean Cmax of the 4-mg prototype mini lozenge was 8.78 ng/mL (SD 3.21 ng/mL) compared to 8.97 (SD 2.74 ng/mL) with the 4-mg commercially available mini lozenge.

Mean linear scale following prototype mini and commercially available mini lozenges for PKAS1 (baseline-adjusted concentrations) for the a 2-mg study and b 4-mg study
AUC0–t was also similar between the prototype mini lozenge and commercially available mini lozenge (Table 2). In the 2-mg study, the baseline-adjusted mean AUC0–t was 16.56 ng·h/mL (SD 6.97 ng·h/mL) with the prototype mini lozenge compared to 17.50 ng·h/mL (SD 7.88 ng·h/mL) with the commercially available mini lozenge. The geometric mean was 15.39 ng·h/mL with a geometric CV of 39.4% with the 2-mg prototype mini lozenge and was 16.19 ng·h/mL with a geometric CV of 40.1% with the 2-mg commercially available mini lozenge. In the 4-mg study, the mean AUC0–t was 38.43 ng·h/mL (SD 18.58 ng·h/mL) with the prototype mini lozenge compared to 39.82 ng·h/mL (SD 18.83 ng·h/mL) with the commercially available mini lozenge. The geometric mean was 34.44 and 36.39 ng·h/mL, with the prototype and commercially available mini lozenges, respectively, with geometric CVs of 51.5% and 43.9%.
The prototype mini lozenge demonstrated a similar baseline-adjusted AUC0–∞ as the commercially available mini lozenge at both dose levels (Table 2). The 2-mg prototype mini lozenge had a mean AUC0–∞ of 17.80 ng·h/mL (SD 7.55 ng·h/mL) compared to 18.79 ng·h/mL (SD 8.56 ng·h/mL) with the commercially available mini lozenge. The geometric mean was 16.53 and 17.38 ng·h/mL with the prototype and commercially available mini lozenges, respectively, and the geometric CVs were 39.5% and 40.2%. The 4-mg prototype mini lozenge resulted in a mean AUC0–∞ of 40.64 ng·h/mL (SD 19.57 ng·h/mL) compared to 42.10 ng·h/mL (SD 19.53 ng·h/mL) with the commercially available mini lozenge, with geometric means of 36.39 and 38.54 ng·h/mL, respectively. The geometric CV was 52.0% with the prototype and 43.6% with the commercially available mini lozenges.
Both the 2-mg and 4-mg prototype mini lozenges were bioequivalent to their commercially available comparators for AUC0–t, AUC0–∞, and Cmax (Table 3). In the 2-mg study, the geometric least-squares mean (LSM) of the baseline-adjusted AUC0–t was 15.87 ng·h/mL with the prototype mini lozenge compared to 16.64 ng·h/mL with the commercially available mini lozenge (GMR 0.954; 90% CI 0.906–1.004; intrasubject CV% 10.19). The geometric LSM of the AUC0–∞ was 17.06 ng·h/mL and 18.08 ng·h/mL with the prototype and commercially available mini lozenges, respectively (GMR 0.944; 90% CI 0.895–0.996; intrasubject CV% 10.14). The Cmax was also bioequivalent with a geometric LSM of 4.419 ng/mL with the prototype mini lozenge and 4.950 ng/mL with the commercially available mini lozenge (GMR 0.893; 90% CI 0.817–0.976; intrasubject CV% 17.74).
In the 4-mg study, the geometric LSM of AUC0–t was 34.41 ng·h/mL with the prototype mini lozenge compared to 36.35 ng·h/mL with the commercially available mini lozenge (GMR 0.947; 90% CI 0.882–1.016; intrasubject CV% 15.25). The geometric LSM of AUC0–∞ was 36.32 ng·h/mL and 38.50 ng·h/mL with the prototype and commercially available mini lozenges, respectively (GMR 0.943; 90% CI 0.878–1.013; intrasubject CV% 15.13). The Cmax was also bioequivalent between treatments, with a geometric LSM of 8.282 ng/mL with the prototype mini lozenge and 8.527 ng/mL with the commercially available mini lozenge (GMR 0.966; 90% CI 0.897–1.041; intrasubject CV% 16.06).
Additional Secondary Endpoints
Tmax was similar between the 2-mg treatments, which was reached at 1.056 h (SD 0.387) with the 2-mg prototype mini lozenge compared with 1.065 h (SD 0.312) with the commercially available mini lozenge (Table 4). As the 95% CI of the median difference in nicotine Tmax between the 2-mg prototype mini and commercially available mini lozenges treatments contained 0, the Tmax of nicotine was similar. The median of paired differences in Tmax between the treatments was − 0.094 with a median of − 0.023 (95% CI − 0.179 to 0.129; P = 0.666; Table 5). The half-lives of the 2-mg lozenges were similar, with a mean of 3.643 h (SD 3.307 h) with the prototype mini lozenge and 3.462 h (SD 3.130 h) with the commercially available mini lozenge.
Tmax was also similar between the 4-mg treatments, which was reached in a mean of 1.423 h (SD 0.554) with the 4-mg prototype mini lozenge compared with 1.342 h (SD 0.455) with the commercially available mini lozenge. As the 95% CI of the median difference in nicotine Tmax between the 4-mg prototype mini and commercially available mini lozenges treatments contained 0, the Tmax of nicotine was similar. The median of paired differences was 0.179 between the treatments with a median of 0.117 (95% CI − 0.125 to 0.254; P = 0.246). The half-life was similar and occurred at a mean of 5.924 (SD 3.269) and 6.324 h (SD 3.827) with the 4-mg prototype and commercially available mini lozenges, respectively.
The Kel was also similar between the treatment groups in both studies. In the 2-mg study, the mean Kel was 0.276 h−1 (SD 0.118 h−1) with the prototype mini lozenge compared to 0.278 h−1 (SD 0.111 h−1) with the commercially available mini lozenge (Table 4). With the 4-mg prototype mini lozenge, the mean Kel was 0.1610 h−1 (SD 0.102 h−1) compared with 0.154 h−1 (SD 0.091 h−1) with the commercially available mini lozenge.
Safety
The 2-mg and 4-mg lozenges were well tolerated by study subjects. There were no serious adverse events (SAEs) or deaths during either study among the safety populations. There were no discontinuations due to AEs in the 2-mg study, and one subject who received the commercially available mini lozenge discontinued the study because of an AE unrelated to the study treatment.
In the 2-mg study, 50.0% of subjects experienced a treatment-emergent AE (TEAE), including 28.3% in the prototype mini lozenge arm and 34.1% in the commercially available mini lozenge arm. All of the AEs were mild in severity, except for a case of moderate pruritus in the mini lozenge arm.
The rate of suspected treatment-related TEAEs was also similar between arms, with 10.9% of subjects experiencing a treatment-related TEAE in the prototype mini lozenge arm compared to 13.6% of subjects in the commercially available mini lozenge arm (Table 6). The most common treatment-related TEAEs were throat irritation, which occurred in three subjects in the prototype mini lozenge arm and two subjects in the commercially available mini lozenge arm, and dyspepsia, which occurred in two subjects in the prototype mini lozenge arm and three subjects in the commercially available mini lozenge arm. There were no changes in the clinical laboratory or vital sign evaluations during the study.
In the 4-mg study, 43.2% of subjects developed a TEAE, including 36.1% of subjects with the prototype mini lozenge and 17.1% of subjects with the commercially available mini lozenge. All of the TEAEs were mild in severity. Suspected treatment-related TEAEs were higher with the prototype mini lozenge, with 30.6% of subjects reporting a treatment-related TEAE compared to 11.4% with the commercially available mini lozenge. Similar to the 2-mg study, the most common treatment-related TEAE was throat irritation, which occurred in five subjects during their use of the 4-mg prototype mini lozenge and two subjects when using the 4 mg commercially available mini lozenge. Other treatment-related TEAEs that occurred in at least 5% of subjects were hiccups (5.4%), nausea (10.8%), oral discomfort (5.4%), dizziness (5.4%), and dyspepsia (5.4%). There were no changes in the clinical laboratory or vital sign evaluations during the study.
Discussion
NRT is an effective treatment to aid in smoking cessation and of tobacco dependence through the relief of nicotine withdrawal symptoms, including cravings. A meta-analysis of 163 randomized trials demonstrated that NRT in any form—as a lozenge, gum, patch, inhaler, or nasal spray—significantly increased abstinence from smoking compared with placebo or no treatment (relative risk 1.55; 95% CI 1.49–1.61) [12]. Another meta-analysis of 63 randomized studies found no significant difference in efficacy between NRT forms, suggesting that efficacy is not associated with different routes of administration, such as the transdermal route with the patch, the buccal route with the gum and lozenge, and through the nasal epithelium with the nasal spray [13].
The nicotine lozenge is a form of NRT that is placed in the mouth, during which nicotine is released as the lozenge dissolves. The 2-mg and 4-mg nicotine lozenges were demonstrated to improve the rates of abstinence from smoking compared with placebo, with treatment effects lasting for at least 1 year [15]. The mini lozenge was introduced to offer another dosage form option and was shown to deliver the same amount of nicotine as the larger lozenge, but with a faster dissolution time in vitro and a greater reduction in cravings or the urge to smoke in smokers compared with placebo [16]. In addition, the mini lozenge has been shown to reduce behaviorally provoked cravings among heavy and high-dependency smokers as early as 3 min and for at least 10 min [16].
The prototype mini lozenge was developed to offer smokers a novel mini lozenge with a sweet mint flavor with visual differentiation, faster dissolution time, and an improved flavor compared with the commercially available mini lozenge. The present studies demonstrated that the prototype mini lozenge is bioequivalent to the commercially available mint mini lozenge at both the 2-mg and 4-mg doses. The PK parameters of Cmax, AUC0–t, and AUC0–∞ were bioequivalent between the prototype mini lozenge and the commercially available mini lozenge. In addition, the secondary PK parameters, including Tmax, t1/2, and Kel, were also similar between the two types of mini lozenges.
The bioequivalence of the prototype mini lozenge to the commercially available mini lozenge was determined in two separate studies, one of the 2-mg dose and one of the 4-mg dose. Comparisons cannot be made between the studies; however, the higher 4-mg dose resulted in a higher baseline-adjusted plasma nicotine concentration than the 2-mg dose. Both types of mini lozenges were well tolerated and there were no new safety signals observed. At both dosage levels, throat irritation was the most common TEAE reported by the subjects and was mild in severity. None of the subjects discontinued the study as a result of throat irritation or any other TEAE.
Limitations
The crossover designs could result in some residual nicotine from the previous treatment session; however, the washout period and the 36-h period of nicotine abstinence before the next treatment administration was implemented to avoid residual plasma nicotine.
Conclusions
The results of the two present studies demonstrate that 2 and 4 mg of a prototype mini lozenge are bioequivalent with 2 and 4 mg of a commercially available nicotine mini lozenge, respectively. Both dose levels were well tolerated and no new safety signals were identified. Therefore, the novel prototype mini lozenge may provide smokers seeking to quit with a new mini lozenge format that they may prefer to aid in smoking cessation. This potential additional new option will provide the convenience, rapid dissolution, and fast craving relief of a mini lozenge, but with an improved flavor that some individuals may prefer, and thus may help such individuals adhere to their NRT and ultimately achieve their desire to quit smoking.
References
World Health Organization. Tobacco. May 27, 2020. https://www.who.int/news-room/fact-sheets/detail/tobacco. Accessed May 10, 2021.
Christensen CH, Rostron B, Cosgrove C, et al. Association of cigarette, cigar, and pipe use with mortality risk in the US population. JAMA Intern Med. 2018;178(4):469–76.
Adhikari B, Kahende J, Malarcher A, Pechacek T, Tong V. Smoking-attributable mortality, years of potential life lost, and productivity losses—United States, 2000–2004. MMWR Weekly. 2008;57(45):1226–8.
Benowitz NL. Nicotine addiction. N Engl J Med. 2010;362(24):2295–303.
Benowitz NL, Hukkanen J, Jacob P 3rd. Nicotine chemistry, metabolism, kinetics and biomarkers. Handb Exp Pharmacol. 2009;192:29–60.
Russell MA. Nicotine replacement: the role of blood nicotine levels, their rate of change, and nicotine tolerance. Prog Clin Biol Res. 1988;261:63–94.
Svensson CK. Clinical pharmacokinetics of nicotine. Clin Pharmacokinet. 1987;12(1):30–40.
Flowers L. Nicotine replacement therapy. Am J Psychiatry Residents’ J. 2016;11(6):4–7.
Babb S, Malarcher A, Schauer G, Asman K, Jamal A. Quitting smoking among adults—United States, 2000–2015. MMWR Morb Mortal Wkly Rep. 2017;65(52):1457–64.
Borland R, Partos TR, Yong HH, Cummings KM, Hyland A. How much unsuccessful quitting activity is going on among adult smokers? Data from the International Tobacco Control Four Country cohort survey. Addiction. 2012;107(3):673–82.
Tobacco Use and Dependence Guideline Panel. Treating tobacco use and dependence: 2008 update. Rockville: U.S. Department of Health and Human Services; 2008.
Hartmann-Boyce J, Chepkin SC, Ye W, Bullen C, Lancaster T. Nicotine replacement therapy versus control for smoking cessation. Cochrane Database Syst Rev. 2018;5:CD000146.
Lindson N, Chepkin SC, Ye W, Fanshawe TR, Bullen C, Hartmann-Boyce J. Different doses, durations and modes of delivery of nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2019;4:CD013308.
Hollander and Wolf. Nonparametric statistical methods. 2nd ed. Hoboken: Wiley; 1999.
Shiffman S, Dresler CM, Hajek P, Gilburt SJA, Targett DA, Strahs KR. Efficacy of a nicotine lozenge for smoking cessation. Arch Intern Med. 2002;162:1267–76.
Nides M, Shanga GM, Bishop A, Becker WD. Nicotine lozenges in the relief of behaviorally provoked craving. Am J Health Behav. 2018;42:69–79.
Acknowledgements
We would like to acknowledge and thank the subjects for their participation in these studies.
Funding
Sponsorship for this study and Rapid Service Fee were funded by GlaxoSmithKline.
Authorship
All authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship of this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
Pamela M Lai contributed to the concept and design of the study and drafting of the manuscript; Mako Araga contributed to the concept and design of the study, the statistical analysis, and the drafting of the manuscript; Ana Hamilton contributed to the concept and design of the study and the drafting of the manuscript; Marianna Armogida contributed to the concept and design of the study and the drafting of the manuscript.
Medical Writing and/or Editorial Assistance
Editorial assistance in the preparation of this article was provided by Andrea S. Blevins Primeau, PhD, MBA, of Medica Communications Inc. Support for this assistance was funded by GlaxoSmithKline.
Disclosures
Pamela M Lai, Mako Araga, Ana Hamilton, and Marianna Armogida are employees of GlaxoSmithKline.
Compliance with Ethics Guidelines
The studies were reviewed and approved by an independent institutional review board: Advarra IRB, 6940 Columbia Gateway Drive, Suite 110, Columbia, Maryland, 21046, USA (IRB# 00000971; IORG # 0000635). The studies were conducted in full compliance of all relevant laws and regulations in the USA and with the requirements specified in the Declaration of Helsinki, and in accordance with the Good Clinical Practice guidelines of the International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. All subjects provided written informed consent prior to any study-specific procedures. Subjects could withdraw from either study at any time for any reason.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lai, P.M., Araga, M., Hamilton, A. et al. Pharmacokinetic Characterization of a Prototype Mini Nicotine Lozenge. Adv Ther 38, 3997–4012 (2021). https://doi.org/10.1007/s12325-021-01798-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-021-01798-4