
Abstract
Spinocerebellar ataxia type 10 (SCA10) is an autosomal dominant cerebellar ataxia accompanied by extracerebellar signs and other neurological disorders. It is caused by an expansion of the ATTCT pentanucleotide repeat in intron 9 of ATXN10. Cases of SCA10, formerly confined to America, have been reported in Europe and Asia. In the present study, we aim to report an atypical SCA10 family in China and provide a reference for the diagnosis of SCA10 in Asia by comparing their clinical and genetic features with former SCA10 pedigrees. Genomic DNA was extracted from patients and subjected to RP-PCR (repeat-primed PCR), Southern blotting, and haplotype analysis to determine the genetic pathogenesis. Patients with SCA10 in this pedigree demonstrated atypical SCA10 manifestations, including the absence of seizures and ocular abnormalities. Magnetic resonance imaging (MRI) showed cerebellar atrophy in five patients with available data. RP-PCR and Southern blotting revealed abnormal expansion. Analysis of single nucleotide polymorphisms (SNPs) surrounding the SCA10 locus in the proband and other affected family members revealed the “C-expansion-G-G-C” haplotype, consistent with former studies. These findings imply that the SCA10 mutation may have occurred before the Amerindian migration from East Asia to North America. It also suggested that SCA10 should be taken into account during differential diagnosis in patients of Asian ancestry, even if they do not present with typical features such as epilepsy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Spinocerebellar ataxia type 10 (SCA10) is an autosomal dominant neurodegenerative disease with genetic and clinical heterogeneities. Progressive cerebellar ataxia combined with epilepsy is the hallmark of SCA10; other clinical manifestations, including dysarthria, dysphagia, diplopia, cognitive impairment, and additional non-motor symptoms, may also be accompanied [1]. Purkinje cell loss is a typical pathological feature during disease development that results in symmetrical atrophy of the hemispheres [2].
SCA10 is caused by repeated amplification of the pentanucleotide sequence ATTCT in intron 9 of ATXN10 on chromosome 22q13.3 [3,4,5]. Normal individuals typically have 9–32 repeats in this locus, whereas the number of repeats in SCA10 patients can range from 800 to 45,000. As SCA10 is a highly heterogeneous disease, patients with SCA10 may exhibit various clinical manifestations. Although ataxia accompanied by seizures is commonly observed in patients from Mexico, Venezuela, Argentina [6], Peru [7], China [8], and Japan [9], pure ataxia has been reported on occasion [10]. Intention tremor is another clinical manifestation described in Brazil [10] and Peru [7], and patients with tremors have all been women thus far with exceptionally early onset. However, the clinical manifestations of these two cases are not entirely consistent, as the Peruvian patient exhibited epilepsy, whereas the Brazilian patient [7, 10] did not. Repeated interruption of pentanucleotide expansion is considered one of the reasons for clinical heterogeneity and can cause specific presentations, such as epilepsy [5, 11, 12]. As yet, SCA10 mostly occurs on the American continent, and previous studies have suggested a founder effect among the Amerindian population, hinting that patients with SCA10 share a common ancestor. Cases were reported in China and Japan [8, 9], however, indicating that SCA10 mutation may have occurred before Amerindians diverged from their Asian forefathers.
This report presents the third SCA10 case in Asia with atypical manifestations, confirming the theory mentioned above. Together with the former two cases, these findings indicate that SCA10 should be taken into account during differential diagnosis in patients of Asian ancestry, even those lacking typical SCA10 manifestations.
Materials and Methods
Ethics Statement
Genomic DNA was extracted from leukocytes after blood samples were drawn with informed consent from the participant. The present study was approved by the ethics committee of the First Affiliated Hospital of Zhengzhou University.
Family History and Clinical Examination
A detailed family history was collected, and clinical examination was performed.
SCA10 Expansion Testing
Genomic DNA was extracted from peripheral blood leukocytes using conventional methods, and qualitative and quantitative analyses were conducted using agarose gel electrophoresis and UV spectrophotometry. Repeat-primed PCR (RP-PCR) was used to verify the abnormal repeat expansion [13]. RP-PCR-positive samples were subjected to Southern blotting to assess the size of the expanded range [3].
SCA10 Haplotype Analysis
SCA10 haplotype analysis of the pedigree was performed via PCR to evaluate SNPs. SNPs flanking SCA10 expansions were examined in a previous study, and the C (rs5764850)-expansion-G (rs72556348)-G (rs72556349)-C (rs72556350) haplotype was identified. Details, including PCR primers and restriction fragment length polymorphisms, were described previously [12, 14].
Results
Family History and Clinical Examination
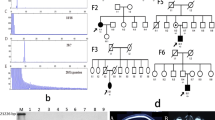
The family being described lives in central China and is of Northern Han descent. Most patients in this pedigree had gait instability as the first symptom, which was accompanied by dysarthria and dysphagia. Disease severity was measured using the Scale for the Assessment and Rating of Ataxia (SARA) and International Cooperative Ataxia Rating Scale (ICAR). The general clinical features and scores are summarized in Table 1. The pedigree is depicted as below (Fig. 1a).

Clinical information of the Chines Han spinocerebellar ataxia type 10 (SCA10) family. a Pedigree chart of the Chinese Han SCA10 family Squares, male; circles, female; black fill, affected; white fill, unaffected; gray fill, asymptomatic. lower left corner, pedigree numbering within the generation; arrow, proband; b brain MRI of the proband (IV-2) and his affected relatives (III-3) T1WI sagittal brain MRI image showings cerebellar atrophy in the proband (i, IV-2, 52 years old), his mother (ii, III-2, 73 years old), maternal aunts (iii and iv, III-7,72 years old; III-10, 53 years old), and cousin (v, IV-7, 46 years old)
The Proband and His Children
Proband (IV-2)
A 44-year-old Chinese man visited our hospital in September 2013 with a progressive unsteady gait first noted at 42 years of age, which was aggravated by alcohol consumption. He had begun to develop dysarthria and wide-based ataxic gait 1 year prior. The patient presented with slow walking, aggravated instability, and dysphagia 2 months after the initial visit. He had no significant medical history and no medications were taken.
Upon examination, he had dysphagia, slurred speech, mild limb ataxia, widened gait ataxia with impaired tandem walking and slow speed. His motor and sensory nerve functions were generally preserved, except for bilateral hyperreflexia, and the muscle tonus was normal. The finger-nose-finger and heel-to-shin tests revealed impaired coordination, and dysdiadochokinesia was detected. Seizures and ocular abnormalities were absent in this case. Brain MRI (Fig. 1b–i) showed mild white matter demyelination in the bilateral frontal lobes and cerebellar atrophy involving the vermis and both hemispheres. Electromyography (EMG), motor evoked potential (MEP), and spinal somatosensory evoked potential (SSEP) results remained normal.
Proband’s Children (V-1, V-2)
The proband’s children underwent genetic testing despite them not exhibiting suspicious manifestations. The results showed a repeated expansion in the proband’s son.
Sisters and Other Maternal Relatives of the Proband
Sister of the Proband (IV-4)
The 49-year-old sister of the proband. She presented with unbalanced gait since the age of 47, accompanied by dysarthria and dysphagia that had worsened 2 years prior. With the aggravation of ataxia, the patient had difficulty in walking up and down stairs.
Sister of the Proband (IV-5)
The 47-year-old sister of the proband. She had gait ataxia coupled with slow walking for more than 5 years. Clinical examinations manifested memory deterioration, clumsy fine hand movements, slow speech speed, and weakness of lower limbs. The symptoms had exacerbated 3 years prior and the patient developed diplopia.
Cousin of the Proband (IV-7)
The 46-year-old cousin of the proband. She had slurred speech followed by limb ataxia. Brain MRI revealed cerebellar atrophy (Fig. 1b–v).
Cousin of the Proband (IV-8)
The 43-year-old cousin of the proband. He had noted canning speech and mild gait ataxia with a widened base for 1 year. Detailed information was not obtained as the patient declined clinical examination.
All affected members of this pedigree exhibited representative SCA symptoms, including cerebellar ataxia, dysarthria, dysphagia, cognitive impairment, and other extracerebellar symptoms. Archetypal clinical presentations of SCA10, such as seizures and ophthalmological findings, were generally not observed.
Genetic Examination
High-molecular-weight DNA was extracted from peripheral blood leukocytes using conventional methods to test for SCA10 expansion after negative results for SCA1, 2, 3, 6, 7, 12, 17 and DRPLA were received. The proband (IV-2) tested positive for SCA10 expansion on RP-PCR, as did his son (V-1), mother (III-2), maternal aunts (III-7, III-10), full sisters (IV-4, IV-5), and cousins (IV-7, IV-8) (Fig. 2a). Southern blotting further confirmed that the expansion fell within the pathology range (Fig. 2b). PCR, followed by Sanger sequencing, unveiled that the intragenic haplotype of the proband (IV-2) was homozygous for the C (rs5764850)-expansion-G (rs72556348)-G (rs72556349)-C (rs72556350) haplotype (Table 2), which is consistent with the previously described genotype [8, 9, 14, 15].

Genetic examinations of the available affected individuals. a Fluorescent repeat-primed PCR analysis in SCA10 fluorescent triplet-primed PCR analysis of the ATXN10 gene revealed an expanded ATTCT pentanucleotide repeat in the proband (IV-2) and his son (V-1) compared to the unaffected daughter (V-2).as negative control; b southern blot analysis in SCA10 lanes 1 and 12 are size markers, lanes 2–9 are DNA samples from family members, lanes 10 and 11 are negative control. Southern blot analysis shows an SCA10 repeat expansion in the proband (IV-2), his mother (III-2), his sisters (IV-4, IV-5), and cousin (IV-7)
Discussion
SCA is a subset of neurodegenerative diseases with autosomal dominant inheritance that is characterized by progressive cerebellar dysfunction and other extracerebellar manifestations [16]. More than 40 subtypes of SCA have been identified based on clinical characteristics and pathogenic mutations [17], containing over 30 disease-causing genes with overlapping clinical manifestations. Therefore, genetic testing is beneficial for detecting SCA. According to different pathogeneses, SCA can be divided into 3 categories based on different types of mutation as follows: (1) repeat expansion SCA can be further classified as CA with an abnormal trinucleotide (CAG) expansion in the translation region (also known as polyglutamine SCA) or SCA with abnormal expansions in non-coding areas; (2) non-repeat expansion SCA caused by traditional mutations as missense, deletion, and insertion; (3) SCA with an unclear pathogenesis. Previous studies have shown that the incidence of undiagnosed SCA cases in China ranged between 16 and 80% [18], suggesting a low prevalence of rare subtypes in the Chinese cohort, especially when considering the relatively low diagnosis rate in patients excluded for (CAG)n expansion [19, 20]. The dominance of SCA types differs worldwide; Chinese well-known familial SCA subtypes are Machado-Joseph disease/SCA3, SCA2, SCA1 SCA6, and SCA7 [20, 21]. SCA10 is a rare SCA subtype with increasing incidence, particularly in Asia [22], and only two independent SCA10 families have been found to date [8, 9].
SCA10 is phenotypically heterogeneous and has various manifestations in different regions. For example, epilepsy has been identified as a distinguishing feature of SCA10 in patients in Mexico, Venezuela, Argentina [6], Peru [7], China [8], and Japan [9]. However, cases from southern Brazil in a specific area present with pure ataxia [10]. The intention tremor is another feature worth noting. Cases with intention tremor have been identified in Brazil [10] and Peru[7], and all patients with tremors are female. Among the 16 Brazilian families described, pedigree with tremor patients showed the earliest age of onset and the highest proportion of female patients. The clinical characteristics of our cases are generally in line with those of general SCA studies, including cerebellar ataxia, dysarthria, dysphagia, cognitive impairment, and other extracerebellar symptoms. Representative clinical manifestations of SCA10, on the other hand, are not commonly observed, including seizures and ophthalmologic findings. (i) Seizures: as mentioned above, epilepsy is an integral trait observed in SCA10 patients, comprising only two Asian cases [8, 9]. Nevertheless, none of the eight patients in this pedigree had seizures. (ii) Ocular abnormalities: ocular abnormalities, including diplopia, ophthalmoplegia, gaze-evoked nystagmus, fragmented ocular pursuit, and slowed saccades, are universal in SCA10. This lineage lacks ocular manifestations, except in one maternal aunt (III-7). These phenomena are likely attributable to clinical heterogeneity. Repeat interruptions in pentanucleotide expansion may function as a modified motif to affect the clinical phenotype, which is a highly suspicious cause of atypical manifestations. Subsequent studies should be conducted to further explore its genetic pathogenesis.
After SCA10 was first identified in Mexico, reports have surfaced in Brazil and other American continents [7, 14]. Cases have recently been reported in other regions including Europe [7] and Asia [8, 9]. Prior to the discovery of Asian patients, the founder effect was the prevailing hypothesis, suggesting that Amerindians and Native Americans shared a mutual ancestor. Their descendants have spread across North and South America since trekking from East Asia via the Bering Strait. Contrary to cases in Europe that are almost exclusively Peruvian immigrants, patients in China and Japan are native inhabitants with no immigration history. Their haplotype analyses were identical with earlier studies [8, 9, 14, 15], together with the fact that Northern Chinese shared similar genetic structure with Japanese who has East Asian ancestry [23]; we verified the novel opinion that the original SCA10 mutation may have arisen before Amerindians diverged from East Asia [8, 9]. Considering the universality of the haplotypes and geographical migration of populations, the possibility of isolated expansion events or population backflow through Beringia cannot be ruled out.
To the best of our knowledge, we report the third Asian SCA10 family observed to date, which consists of eight patients with available information. Though the two former cases present with typical SCA10 features commonly observed in Amerindians including seizures, these characteristics are not prominent in this pedigree. Combined with haplotype analysis, our findings provide more forceful evidence on the latest hypothesis of SCA10 geographical distribution depicted above. We also offer novel insights into the differential diagnosis of ataxia, suggesting that SCA10 should be considered for people of Asian descent, even in the absence of representative traits. Considering the prevalence of genetic anticipation in SCA that consecutive generations may have earlier ages of onset with increasing severity [24,25,26,27], genetic counseling and prenatal examination are prerequisites for patients with a relevant family history.
References
Ashizawa T. Spinocerebellar ataxia type 10. Handb Clin Neurol. 2012;103:507–19.
Xia G, et al. Purkinje cell loss is the major brain pathology of spinocerebellar ataxia type 10. J Neurol Neurosurg Psychiatry. 2013;84(12):1409–11.
Matsuura T, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. 2000;26(2):191–4.
Lin X, Ashizawa T. Recent progress in spinocerebellar ataxia type-10 (SCA10). Cerebellum. 2005;4(1):37–42.
Schüle B, et al. Parkinson’s disease associated with pure ATXN10 repeat expansion. NPJ Parkinsons Dis. 2017;3:27.
Gatto EM, et al. Ethnic origin and extrapyramidal signs in an Argentinean spinocerebellar ataxia type 10 family. Neurology. 2007;69(2):216–8.
Leonardi L, et al. Spinocerebellar ataxia type 10 in Peru: the missing link in the Amerindian origin of the disease. J Neurol. 2014;261(9):1691–4.
Wang K, et al. Spinocerebellar ataxia type 10 in Chinese Han. Neurol Genet. 2015;1(3):e26.
Naito H, et al. First report of a Japanese family with spinocerebellar ataxia type 10: The second report from Asia after a report from China. PLoS One. 2017;12(5):e0177955.
Domingues BMD, et al. Clinical and genetic evaluation of spinocerebellar ataxia type 10 in 16 Brazilian families. Cerebellum. 2019;18(5):849–54.
McFarland KN, et al. Paradoxical effects of repeat interruptions on spinocerebellar ataxia type 10 expansions and repeat instability. Eur J Hum Genet. 2013;21(11):1272–6.
McFarland KN, et al. Repeat interruptions in spinocerebellar ataxia type 10 expansions are strongly associated with epileptic seizures. Neurogenetics. 2014;15(1):59–64.
Matsuura T, Ashizawa T. Polymerase chain reaction amplification of expanded ATTCT repeat in spinocerebellar ataxia type 10. Ann Neurol. 2002;51(2):271–2.
Bushara K, et al. Expansion of the Spinocerebellar ataxia type 10 (SCA10) repeat in a patient with Sioux Native American ancestry. PLoS One. 2013;8(11):e81342.
Almeida T, et al. Ancestral origin of the ATTCT repeat expansion in spinocerebellar ataxia type 10 (SCA10). PLoS One. 2009;4(2):e4553.
Sullivan R, et al. Spinocerebellar ataxia: an update. J Neurol. 2019;266(2):533–44.
Coarelli G, Brice A, Durr A. Recent advances in understanding dominant spinocerebellar ataxias from clinical and genetic points of view. F1000Res. 2018;7:F1000 Faculty Rev-1781.
Ruano L, et al. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology. 2014;42(3):174–83.
Coutelier M, et al. A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain. 2017;140(6):1579–94.
Chen Z, et al. Updated frequency analysis of spinocerebellar ataxia in China. Brain. 2018;141(4):e22.
Bettencourt C, Lima M. Machado-Joseph Disease: from first descriptions to new perspectives. Orphanet J Rare Dis. 2011;6:35.
Depondt C, et al. MME mutation in dominant spinocerebellar ataxia with neuropathy (SCA43). Neurol Genet. 2016;2(5):e94.
Cao Y, et al. The ChinaMAP analytics of deep whole genome sequences in 10,588 individuals. Cell Res. 2020;30(9):717–31.
Pulst SM. Degenerative ataxias, from genes to therapies: the 2015 Cotzias Lecture. Neurology. 2016;86(24):2284–90.
Chen Z, et al. (CAG)n loci as genetic modifiers of age-at-onset in patients with Machado-Joseph disease from mainland China. Brain. 2016;139(Pt 8):e41.
Matsuura T, et al. Mapping of the gene for a novel spinocerebellar ataxia with pure cerebellar signs and epilepsy. Ann Neurol. 1999;45(3):407–11.
Zu L, et al. Mapping of a new autosomal dominant spinocerebellar ataxia to chromosome 22. Am J Hum Genet. 1999;64(2):594–9.
Funding
This study was funded by the National Natural Science Foundation of China (Grant U1904207, 91849115 and 81530037 to Dr. Yuming Xu, Grant 81771290 and 81974211 to Dr. Changhe Shi, Grant 81901300 to Dr. Chengyuan Mao), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (Grant 2020-PT310-01 to Dr. Yuming Xu), the National Key Research and Development Program of China (Grant 2017YFA0105003 to Dr. Yuming Xu), and the Scientific and Technological Project of Henan Province (Grant SBGJ202003020 to Dr. Chengyuan Mao).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Chengyuan Mao, Xinwei Li, and Yun Su. The first draft of the manuscript was written by Chengyuan Mao and Xinwei Li. Analysis or interpretation of data was performed by Yun Su, Haiyang Luo, Liyuan Fan, Huimin Zheng, Yu Fan, Zhihua Yang, Shuo Zhang, Zhengwei Hu, and Xiaoyan Hao. This study was supervised by Yuming Xu and Changhe Shi. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University.
Consent to Participate
Informed consent was obtained from all individual participants included in this study.
Consent for Publication
The participants have consented to the submission of the article to the journal.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mao, C., Li, X., Su, Y. et al. Spinocerebellar Ataxia Type 10 with Atypical Clinical Manifestation in Han Chinese. Cerebellum 22, 355–362 (2023). https://doi.org/10.1007/s12311-022-01405-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-022-01405-4