
Abstract
Mutations in STUB1 have been identified to cause autosomal recessive spinocerebellar ataxia type 16 (SCAR16), also named as Gordon Holmes syndrome, which is characterized by cerebellar ataxia, cognitive decline, and hypogonadism. Additionally, several heterozygous mutations in STUB1 have recently been described as a cause of autosomal dominant spinocerebellar ataxia type 48. STUB1 encodes C-terminus of HSC70-interacting protein (CHIP), which functions as an E3 ubiquitin ligase and co-chaperone and has been implicated in several neurodegenerative diseases. In this study, we identified two SCAR16 pedigrees from 512 Taiwanese families with cerebellar ataxia. Two compound heterozygous mutations in STUB1, c.[433A>C];[721C>T] (p.[K145Q];[R241W]) and c.[433A>C];[694T>G] (p.[K145Q];[C232G]), were found in each SCAR16 family by Sanger sequencing, respectively. Among them, STUB1 p.R241W and p.C232G were novel mutations. SCAR16 seems to be an uncommon ataxic syndrome, accounting for 0.4% (2/512) of our cohort with cerebellar ataxia. Clinically, the three patients from the two SCAR16 families presented with cerebellar ataxia alone or in combination with cognitive impairment. The brain MRIs showed a marked cerebellar atrophy of the patients. In conclusion, SCAR16 is an important but often neglected diagnosis of cerebellar ataxia of unknown cause, and the isolated cerebellar ataxia without involvement of other systems cannot be a basis to exclude the possibility of STUB1-related disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hereditary cerebellar ataxias are a clinically and genetically heterogeneous group of cerebellar disorders characterized by slowly progressive gait unsteadiness usually associated with poor coordination of hands, speech, and eye movements. Mutations in the STIP1 homology and U-Box containing protein 1 (STUB1) gene have been originally identified to cause autosomal recessive spinocerebellar ataxia type 16 (SCAR16), also named as Gordon Holmes syndrome, which usually manifests cerebellar ataxia, hypogonadotropic hypogonadism, and cognitive dysfunction [1, 2]. However, a wider clinical spectrum of recessive STUB1 mutations was demonstrated later, presenting as cerebellar ataxia in combination with a variable degree of dementia, spastic tetraparesis, epilepsy, autonomic dysfunction, extrapyramidal symptoms, and hypogonadism [3,4,5,6]. Furthermore, heterozygous mutations in STUB1 have recently been described as a cause of autosomal dominant spinocerebellar ataxia (SCA) type 48 (SCA48) with a later disease onset and similar clinical manifestations to SCAR16 [7, 8].
The STUB1 gene encodes the C-terminus of HSC70-interacting protein (CHIP), which is a 35-kDa protein and contains three domains, including an N-terminal three tetratricopeptide repeat (TPR) domain, a highly charged middle coiled coil domain, and a carboxyl-terminal U-box domain [2, 9,10,11]. CHIP functions as both an E3 ubiquitin ligase and a molecular cochaperone [9,10,11]. Its TPR domain mediates interactions with heat shock proteins, the coiled coil domain influencing dimerization of CHIP, while the U-box domain of CHIP serves as an ubiquitin ligase [9,10,11]. CHIP facilitates ubiquitylation and maintains protein homeostasis by controlling chaperone levels during stress and recovery [12]. Loss of ubiquitin ligase activity of CHIP has been demonstrated as the molecular mechanism for SCAR16 [2].
Currently, studies about STUB1 mutations in Han Chinese populations remain sparse, and STUB1 mutations have rarely been screened in large inherited cerebellar ataxia cohorts before. The aim of this study is to investigate the frequency, clinical manifestations, and spectrum of STUB1 mutations in a Taiwanese cohort of 512 pedigrees with cerebellar ataxia.
Methods
Patients
All participants were recruited from the Neurology service of Taipei Veterans General Hospital. Mutations in STUB1 were screened in 108 molecularly unassigned index patients from 512 pedigrees with cerebellar ataxia, after mutations responsible for SCA1, 2, 3, 6, 7, 8, 10, 12, 17, 22, 23, 26, 27, 28, 35, and 36, and DRPLA had been excluded. All participants were of Han Chinese descent. Genetic diagnosis of SCAR16 was made in two index patients by identifying biallelic STUB1 mutations. Nine individuals from the two Taiwanese SCAR16 families were enrolled into the study, including 3 patients, 5 individuals carrying a single mutant STUB1 allele, and one with wild type STUB1 alleles only (Fig. 1a). The patients were thoroughly evaluated by neurological examinations. Age at onset (AO) was defined as the age when the earliest symptoms of truncal or appendicular ataxia occurred according to the statements given by the patients. The clinical severity of ataxia was evaluated using the 40-point (0 being normal) validated Scale for the Assessment and Rating of Ataxia (SARA) [13, 14]. The presence and severity of non-ataxia signs was determined by the Inventory of Non-Ataxia Signs (INAS) [15]. The cognitive function was assessed with mini-mental screening exam (MMSE) [16] and cognitive ability screening inventory (CASI), Chinese version [17]. Brain magnetic resonance imaging (MRI) and magnetic resonance spectroscopy (MRS) were performed with a 1.5-T system (Signa EXCITE, GE Medical Systems, Milwaukee, WI). Nerve conduction studies (NCS) and electromyography (EMG) were also performed. Written informed consent was obtained from the participants and the study was approved by the Institutional Review Board of Taipei Veterans General Hospital.

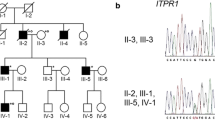
Pedigrees and electropherograms of the STUB1 mutations, a Pedigree structures of the two families with autosomal recessive spinocerebellar ataxia type 16 (SCAR16). The probands are denoted by an arrow. The squares and circles denote males and females. Filled symbols represent affected members with cerebellar ataxia, and open symbols indicate unaffected individuals. The STUB1 genotypes are labeled for each individual and “WT” stands for the wildtype allele. b The electropherograms of the compound heterozygous STUB1 mutations identified in the three affected individuals; c.[433A>C];[721C>T] (p.[K145Q];[R241W]) for A-II-2 and c.[433A>C];[694T>G] (p.[K145Q];[C232G]) for B-II-1 and B-II-2
Mutation Analysis
Genomic DNA was extracted from peripheral blood white cells. The exons and their flanking regions of STUB1 were analyzed by PCR amplification and Sanger sequencing with the intronic primers. The Sanger sequencing was performed by using the Big Dye 3.1 dideoxy terminator method (Applied Biosystems, Foster City, CA, USA) on an ABI Prism 3700 Genetic Analyzer (Applied Biosystems). Amplicon sequences were compared with the reference STUB1 coding sequence (NM_005861.4). The sequence variations were confirmed by sequencing both sense and antisense strands of the amplicons. The pathogenicity of the novel STUB1 variants were evaluated by in silico prediction of the functional effects by three bioinformatics tools, Mutation Taster (http://www.mutationtaster.org) [18], Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) [19], and Combined Annotation Dependent Depletion (CADD) (https://cadd.gs.washington.edu/) [20]. Given that STUB1-related cerebellar ataxia is a rare disease, the pathogenic variants in STUB1 should be absent or very rare in the general population. Hence, we surveyed the allele frequencies of the novel STUB1 variants in the Genome Aggregation Database (gnomAD, v2.1.1) (https://gnomad.broadinstitute.org) and Taiwan biobank (https://taiwanview.twbiobank.org.tw/index) which contains genome data of 1517 healthy Taiwanese individuals. Evolutionary conservation of the mutation sites was analyzed by aligning amino acid sequences of human CHIP and its orthologues utilizing the UniProt website (http://www.uniprot.org) [21].
Results
Genetic Analysis
Mutational analysis of STUB1 in the 108 index patients with cerebellar ataxia revealed two compound heterozygous variants in STUB1, of which c.[433A>C];[721C>T] (p.[K145Q];[R241W]) was identified in patient A-II-2 and c.[433A>C];[694T>G] (p.[K145Q];[C232G]) was found in patients B-II-2 (Fig. 1b). The parents of the patient A-II-2 and patients B-II-2 all carried only one single heterozygous STUB1 mutation. The STUB1 p.C232G and p.R241W were novel.variants, while the p.K145Q had been well demonstrated as a pathogenic mutation in previous studies [5, 22, 23].
To evaluating the pathogenicity of STUB1 p.C232G and p.R241W, we utilized three bioinformatics programs. Mutation Taster and polyphen-2 predicted the two STUB1 variants as disease causing and probably damaging. The CADD v1.4 PHRED scores were 26.7 and 26 for the p.C232G and p.R241W variants, suggesting that they were in the top 0.25% of most deleterious variants in the genome [20]. Both STUB1 p.C232G and p.R241W were not found in the 1517 ethnically matched control genomes in the Taiwan Biobank database. The p.C232G was absent, and the p.R241W was present with an allele frequency of 0.000799% (2/250190) in the gnomAD, which contained exome or genome data of more than 140,000 individuals. Both mutations alter the amino acid residues of CHIP which are evolutionarily conserved at least from human to fish (Fig. 2).

Schematic representation of CHIP, the protein encoded by STUB1, with an N-terminal tetratricopeptide repeat (TPR) domain, a highly charged middle coiled coil domain, and a carboxyl-terminal U-box domain. The STUB1 p.K145Q, p.C232G, and R241W mutations all alter an evolutionarily conserved amino acid residue, as shown by aligning the amino acid sequences of CHIP orthologues from various species
Clinical Features
Three patients with ataxia from the two SCAR16 families were investigated (Fig. 1a). The first patient (A-II-2) began to have an insidious onset of gait unsteadiness and dysarthria at age of 29 years (Fig. 1, family A). Neurological examination at age 30 revealed a moderate degree of dysarthria, mild gait ataxia, and cogwheel pursuits, without focal weakness or sensory deficits. The score of SARA was 10.5, and the INAS count was 0. His parents were unremarkable clinically and on the brain MRI. The second patient (B-II-2) started to have progressive gait unsteadiness since age of 22 years. He had had one motorcycle accident at age 18 resulting in a head concussion and multiple fractures, with no subsequent sequela or functional impairment. On neurological exam at age 25, he was found to have gait ataxia, dysarthria and cogwheel pursuits. His score of SARA was 21, and the INAS count was 1 for mild cognitive dysfunction. His elder brother (B-II-1) had also experienced dysarthria and gait unsteadiness since age of 37 years (Fig. 1, family B). Their parents had been doing well with normal imaging features on the brain CT. Their younger brother, allegedly also being afflicted with some gait difficulties, was unwilling to be evaluated.
Neuroimaging and Other Laboratory Evaluations
The brain MRI demonstrated a marked cerebellar atrophy without other visible abnormalities (Fig. 3). The MR spectroscopy of brain with the volume of interest located in the cerebellar hemispheres and cerebellar vermis revealed decreased ratios of NAA/Cr in all three patients. Cognitive studies revealed that the cognition was normal in patient A-II-2 (MMSE 30/30; CASI 93/100) but was impaired in patient B-II-2 (MMSE 26/30; CASI 71/100) with lower scores in short-term memory (7/12), abstract thinking (8/12), drawing (6/10), attention (5/8), and verbal fluency (5/10) in CASI. No evidence of hypogonadism was found in the three patients with normal levels of serum follicle–stimulating hormone (FSH), luteinizing hormone (LH), and testosterone. The NCS and EMG showed normal pattern. The tilting table test evaluating the autonomic nervous system function in the three subjects revealed no postural hypotension or postural tachycardia.

Features of T1-weighted brain MRI in patients with autosomal recessive spinocerebellar ataxia type 16 (SCAR16), At examination, patients A-II-2 was a 30-year-old man with disease duration of 1 year, patient B-II-1 was a 39 year-old man with disease duration of 2 years, and patient B-II-2 was a 25-year-old man with disease duration of 3 years
Discussion
To understand the features and contribution of STUB1 mutations in hereditary cerebellar ataxia in our population, we screened 108 unrelated Taiwanese patients with cerebellar ataxia for mutations in the STUB1 gene and identified two compound heterozygous mutations, p.[K145Q];[R241W] and p.[K145Q];[C232G], in two patients, respectively. No simple heterozygous mutation in STUB1 was identified. Since the 108 patients were selected from 512 unrelated patients with hereditary cerebellar ataxia after excluding known causative mutations, SCAR16 accounted for 0.4% (2/512) of the Taiwanese cohort with cerebellar ataxia and SCA48 should be absent or very rare in our population. Among the STUB1 mutations, the p.C232G and p.R241W were novel. Their pathogenicity was supported by the following findings. Firstly, the two mutations alter the evolutionarily conserved amino acid of the U-box domain of CHIP and may compromise its ubiquitin ligase function. Secondly, the STUB1 p.R241G mutation, which affects the same amino acid as the p.R241W mutation, has been reported in a patient with SCAR16 before [22]. Then, they were absent in the 1517 ethnically matched control genomes in the Taiwan biobank database and absent or present with an extremely rare allele frequency (2/250190) in the gnomAD. Furthermore, either of the two mutations are present in trans with the known pathogenic mutation, p.K154Q, in the patients. Moreover, the p.C232G and p.R241W mutations are both predicted to be pathogenic by Mutation Taster, Polyphen-2, and CADD programs. Based on these findings, the STUB1 p.c232G and p.R241W mutations may be classified as likely pathogenic variants, according to the guidelines by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [24].
The clinical presentations and age of disease onset may vary among patients with SCAR16. Autosomal recessive cerebellar ataxia is usually associated with involvement of multiple neurological systems, and SCAR16 has been reported to manifest cerebellar ataxia with a variable combination of hypogonadotropic hypogonadism, cognitive dysfunction, spastic quadriparesis, epilepsy, autonomic dysfunction, and extrapyramidal symptoms [3,4,5,6]. However, in our study, the SCAR16 patients presented isolated cerebellar ataxia or in combination with cognitive impairment only. The age of disease onset of SCAR16 in the previous studies is usually before 20 years [1,2,3,4,5], while the onset ages of the patients in the present study range from 22 to 37 years. Hence, SCAR16 may manifest adult-onset isolated cerebellar ataxia without involvement of other systems. In addition to the SCAs belonged to the group of autosomal dominant cerebellar ataxia (ADCA) type III [25], such as SCA6, the differential list of isolated cerebellar ataxia should be expanded to incorporate SCAR16.
Since mutations sharing similar features may have similar effects on the encoded protein, it is interesting to correlate the phenotype and genotype of STUB1 mutations in SCAR16. One study analyzed the clinical features, STUB1 mutations, and changes of CHIP function in 24 SCAR16 patients and found that STUB1 mutations affecting the U-box domain of CHIP were linked to cognitive dysfunction [26]. In the present study, only one of the two SCAR16 patients with clear clinical evaluation had cognitive impairment; however, both had one mutant allele altering the U-box domain and the same second mutation, p.K145Q, located in the middle coiled coil domain of CHIP. The discrepancy of the phenotypic and mutational features between these two patients may come from many factors. CHIP does not exert its function alone, and any genetic or environmental factor influencing the functional pathways which CHIP participates in may modify the SCAR16 phenotype. Actually, intra-familial phenotypic variation may exist in SCAR16 and present with variation in age of ataxia onset as well as presence or absence of cognitive dysfunction [1, 5]. In the other hand, SCAR16 is a rare disease, and only a small number of SCAR16 cases with variable homozygous or compound heterozygous STUB1 mutations had been reported, so it may be premature to conclude whether there is a clear genotype–phenotype relationship in SCAR16.
One limitation of the present study is that we might underdiagnose the SCAR16 cases in our ataxia cohort because of limitation of the methodology. We did not investigate large segment deletions of total or a part of the STUB1 gene in this cohort. Since the molecular mechanism of SCAR16 has been demonstrated as loss of ubiquitin ligase activity, large segment deletion affecting partial or entire STUB1 may result in loss of CHIP function and cause SCAR16. However, we did not find any ataxia patient carrying a single STUB1 mutation by Sanger sequencing in our cohort, and biallelic large segment deletion of STUB1 should be extremely rare. Therefore, this issue might not influence our study substantially.
Conclusion
This study expands the clinical and mutational spectrum of SCAR16. We identified three SCAR16 patients carrying compound heterozygous mutations in STUB1 and manifesting an adult-onset cerebellar ataxia alone or in combination with cognitive impairment from a Taiwanese cohort with cerebellar ataxias. STUB1 p.R241W and p.C232G are novel pathogenic mutations. Mutations in STUB1 are not a common cause of cerebellar ataxia; however, SCAR16 is still an important but often neglected diagnosis of cerebellar ataxia of unknown cause, and the isolated cerebellar ataxia without involvement of other systems cannot be a basis to exclude the possibility of STUB1-associated ataxia.
References
Shi Y, Wang J, Li JD, Ren H, Guan W, He M, et al. Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS One. 2013;8(12):e81884.
Shi CH, Schisler JC, Rubel CE, Tan S, Song B, McDonough H, et al. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet. 2014;23(4):1013–24.
Cordoba M, Rodriguez-Quiroga S, Gatto EM, Alurralde A, Kauffman MA. Ataxia plus myoclonus in a 23-year-old patient due to STUB1 mutations. Neurology. 2014;83(3):287–8.
Synofzik M, Schüle R, Schulze M, Gburek-Augustat J, Schweizer R, Schirmacher A, et al. Phenotype and frequency of STUB1 mutations: next-generation screenings in Caucasian ataxia and spastic paraplegia cohorts. Orphanet J Rare Dis. 2014;9:57.
Hayer SN, Deconinck T, Bender B, Smets K, Züchner S, Reich S, et al. STUB1/CHIP mutations cause Gordon Holmes syndrome as part of a widespread multisystemic neurodegeneration: evidence from four novel mutations. Orphanet J Rare Dis. 2017;12(1):31.
Gazulla J, Izquierdo-Alvarez S, Sierra-Martínez E, Marta-Moreno ME, Alvarez S. Inaugural cognitive decline, late disease onset and novel STUB1 variants in SCAR16. Neurol Sci. 2018;39(12):2231–3.
Genis D, Ortega-Cubero S, San Nicolás H, Corral J, Gardenyes J, de Jorge L, et al. Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). Neurology. 2018;91(21):1988–98.
De Michele G, Lieto M, Galatolo D, Salvatore E, Cocozza S, Barghigiani M, et al. Spinocerebellar ataxia 48 presenting with ataxia associated with cognitive, psychiatric, and extrapyramidal features: a report of two Italian families. Parkinsonism Relat Disord. 2019;65:91–6.
Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, et al. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19(6):4535–45.
Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Höhfeld J, et al. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 2001;3(1):93–6.
Jiang J, Ballinger CA, Wu Y, Dai Q, Cyr DM, Höhfeld J, et al. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J Biol Chem. 2001;276(46):42938–44.
Qian SB, McDonough H, Boellmann F, Cyr DM, Patterson C. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature. 2006;440(7083):551–5.
Schmitz-Hubsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66:1717–20.
Lee YC, Liao YC, Wang PS, Lee IH, Lin KP, Soong BW. Comparison of cerebellar ataxias: a three-year prospective longitudinal assessment. Mov Disord. 2011;26:2081–7.
Jacobi H, Rakowicz M, Rola R, Fancellu R, Mariotti C, Charles P, et al. Inventory of Non-Ataxia Signs (INAS): validation of a new clinical assessment instrument. Cerebellum. 2013;12(3):418–28.
Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98.
Lin KN, Wang PN, Liu CY, Chen WT, Lee YC, Liu HC. Cutoff scores of the cognitive abilities screening instrument, Chinese version in screening of dementia. Dement Geriatr Cogn Disord. 2002;14(4):176–82.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
The Uniprot Consortium. Activities at the universal protein resource (UniProt). Nucleic Acids Res. 2014;42:D191–8.
Depondt C, Donatello S, Simonis N, Rai M, van Heurck R, Abramowicz M, et al. Autosomal recessive cerebellar ataxia of adult onset due to STUB1 mutations. Neurology. 2014;82(19):1749–50.
Sun M, Johnson AK, Nelakuditi V, Guidugli L, Fischer D, Arndt K, et al. Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet Med. 2019;21(1):195–206.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Harding AE. Clinical features and classification of inherited ataxias. Adv Neurol. 1993;61:1–14.
Madrigal SC, McNeil Z, Sanchez-Hodge R, Shi CH, Patterson C, Scaglione KM, et al. Changes in protein function underlie the disease spectrum in patients with CHIP mutations. J Biol Chem. 2019;294:19236–45.
Funding
This work was supported by research grants from the Ministry of Science and Technology (MOST), Taiwan, to B.W.S. (103-2314-B-010-049-MY3, 104-2745-B-010-004, 106-2321-B-010-010, 107-2314-B-010-017). This work was also financially supported by the Brain Research Center, National Yang-Ming University from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.
Author information
Authors and Affiliations
Contributions
Y.C. Lee and B.W.S. conceived and designed the study. H.H.C., C.T.H., and B.W.S. collected patient information and executed clinical analyses. Y.S.T. performed the mutational and bioinformatics analysis. H.H.C. and Y.C. Lee drafted the manuscript. C.T.H, Y.C. Liao, Y.C. Lee, and B.W.S. revised the manuscript. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chiu, HH., Hsaio, CT., Tsai, YS. et al. Clinical and Genetic Characterization of Autosomal Recessive Spinocerebellar Ataxia Type 16 (SCAR16) in Taiwan. Cerebellum 19, 544–549 (2020). https://doi.org/10.1007/s12311-020-01136-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-020-01136-4