
Abstract
Wheat spot blotch, caused by Bipolaris sorokiniana, is a serious constraint to wheat production, reducing grain yield and consequently having severe economic impact. Several plant miRNAs have recently been discovered as regulators of gene expression involved in cellular and metabolic functions. So far reports on the roles of miRNAs in B. sorokiniana infection response of wheat are scanty. To further understand the defence mechanism of miRNAs- regulated cellular functions, we examined the expression patterns of 17 miRNAs and their targets involved in the interaction between wheat and B. sorokiniana in two contrasting wheat genotypes, Chiriya-1 and WH-147. All of the miRNAs and target genes were shown to be expressed differentially in both genotypes after B. sorokiniana infection. Seven and nine miRNAs were observed as up-regulated in the resistant genotype Chiriya-1 and the susceptible genotype WH147, respectively. Among the up-regulated miRNAs, ptc-miR901 (~ 10.21 times) accumulated the most in Chiriya-1 followed by ptc-miR1450 (~ 7.6 times) in WH-147. Furthermore, only two miRNAs, tae-miR156 and ptc-miR482c showed a complete inverse relation with their target genes, SPL and NBS-LRR, respectively. This research sheds light on the temporal differential regulation of miRNAs and their targets, which may play a role in wheat adaptation to B. sorokiniana infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Wheat (Triticum aestivum L.) is one of the important cereal crops grown worldwide that serves as major source of carbohydrates and protein in economically deprived continents. Recent climate changes have aggravated the disease scenario in India which leads to increase in the incidence of spot blotch of wheat caused by Bipolaris sorokiniana (Sacc.) Shoemaker. It is a seed-borne disease, in which primary infection initiated from inoculum surviving on conidia on soil, crop residues or collateral hosts. B. sorokiniana causing spot blotch, root-rot, leaf-spot disease, black point, seedling and head blight in cereal crops, first colonizes the older leaves at the base of wheat plant and then progresses to the upper part of the canopy (Nizam et al. 2012). Yield losses caused by B. sorokiniana were estimated to be 15 to 25% (Dublin and van Ginkel 1991), which can be devastating for farmers in the Eastern Gangetic Plains. On the other hand, to cope up with fungal infection, plants have evolved cascades of molecular and biochemical networks to elicit the defence response. Changes in expression patterns for genes implicated in a wide range of cellular, physiological and metabolic processes are one of the most visible aspects of fungus tolerance adaptation (Vinocur and Altman 2005). Elucidation of the molecular events underlying the biotic and abiotic stresses is important for increased survivability of the crop (McIntosh and Pretorius 2011).
The regulatory role of sRNA (small non-coding RNAs) that is 20–24 nucleotides in length has become the leading area of research. MicroRNAs (miRNAs) have been implicated to play critical roles in gene expression regulation at the post-translational level (Bartel 2004; Jones-Rhoades 2006; Singroha et al. 2021). The genes coding for miRNAs are transcribed by RNA polymerase II as pri-miRNAs which are processed by a dicer-like enzyme (DCLs) to form Precursor miRNAs (pre-miRNAs), and further cleaved by DICER LIKE 1 (DCL1) into a miRNA/miRNA* duplex followed by the transfer of duplex from the nucleus to cytoplasm. These molecules are methylated by the HUA ENHANCER 1 (HEN1). Finally, the mature miRNA is incorporated into a RNA-INDUCED SILENCING COMPLEX (RISC) (Eldem et al. 2013) and reaches its target mRNA transcript by binding with AGRONAUTE 1 (AGO1) protein. After RISC loading, the AGO1 protein unwinds the miRNA:miRNA* duplexes, and one strand of the duplex miRNA is directed to the exosome for degradation by a small-RNA degrading nuclease, while the mature miRNA is inserted into the RISC, which contains AGO proteins (Arribas-Hernández et al. 2016). Finally, the mature miRNA directs the RISC to target mRNAs that are complementary to it.
MiRNAs with high homology to the target mRNA cause site-specific cleavage of the mRNA, whereas miRNAs with poor base pairing to the target mRNA cause translational repression and/or mRNA degradation (Sun et al. 2012). In plants, miRNAs act as negative regulators, mainly inhibiting gene expression at the level of post-transcription. The latest release of miRBase (Release 22.1, October 2018, http://www.mirbase.org) contains 38,589 hairpin precursors and 48,860 mature miRNAs, from 271 organisms are publicly available. Amongst them, 125 of them belong to bread wheat, while 738 and 71 miRNAs are found for rice and barley, respectively.
With the availability of the wheat genome sequence, several groups have discovered wheat miRNAs which are conserved across plant species (Achakzai et al. 2018). However, owing to the complexity of the wheat genome and the spatial/temporal nature of miRNA expression, a large number of miRNAs still remain to be discovered. Recent findings support that miRNAs are involved in plant responses to a wide array of biotic stresses such as powdery mildews (Wu et al. 2014) and rusts in wheat (Feng et al. 2015; Gupta et al. 2012; Kumar et al. 2017). Seeing the huge genome size of the bread wheat, more number of miRNAs is expected to be identified (Brenchely et al. 2012). Recently, identification of miRNAs using high-throughput sequencing techniques has become low cost and easily available. Meng et al. (2013) identified 605 miRNAs representing 540 families at grain filling stage in wheat using deep sequencing approach. Though, computational predictions using new bioinformatics tools can also be used for miRNA and its target prediction (Pandey et al. 2013; Jaiswal et al. 2019). Transcriptional regulation of many miRNAs and genes in response to biotic stress has been widely reported in various crops (Gupta et al. 2012; Kumar et al. 2017; Inal et al. 2014; Feng et al. 2015). However, the role of miRNAs in wheat infected by B. sorokiniana is very poorly known. In this work, we investigated the expression profiling of miRNAs associated in cellular and biochemical functions related to host defence response in two contrasting wheat genotypes, Chiriya-1 and WH-147 which has been infected with B. sorokiniana. In addition, we also used qPCR to validate target transcripts of responsive miRNAs.
Material and methods
Plant material
The plant material included 64 wheat genotypes representing registered genetic stocks for different traits, advanced lines, known resistant sources of spot blotch and few released varieties (Supplementary Table 1). Planting was conducted in the first week of December so that the post anthesis stage coincided with the warm and humid conditions required for proper disease development. The genotypes were planted in the poly-house in a single row plot of 1 m length keeping row to row and plant to plant distances of 25 cm and 5 cm, respectively at ICAR-Indian Institute Wheat Barley Research, Karnal, India. To enhance disease development and spread, a mixture of susceptible genotypes was planted as spreader rows at borders and every 10 rows. To grow a good crop, recommended agronomic practices were used.
Creation of epiphytotic conditions and phenotyping
Spot blotch disease was created artificially in the poly-house to minimise environmental effects. A mixture of B. sorokiniana isolates collected from Faizabad, Pantnagar, Coochbehar and Kalyani, all of which have been identified as hot spots for spot blotch incidence in India. The pure cultures of individual B. sorokiniana isolates were maintained on potato dextrose agar media (Chand et al. 2003; Pandey et al. 2008) and sorghum grains were used for large-scale multiplication of B. sorokiniana spores (Chand et al. 2013) was multiplied on autoclaved sorghum grains. The inoculated sorghum grains were incubated for about 6 weeks at room temperature with constant shaking to promote the fungal development. The infected sorghum grains were subsequently sprinkled in between the planted rows as well as in the spreader rows. Apart from that the spores were extracted from the infected sorghum grains in water. The spore suspension was adjusted to about 104 spores/ml of water which was evenly sprayed at three different Zadoks growth stages viz., tillering (GS-20), flag leaf emergence (GS-37) and anthesis (GS-65) in the evening hours. To maintain sufficient humidity, misting as well as irrigation was given to the plants after inoculations. Disease assessment was done for each plot visually at early dough stage (GS-83) as per Zadoks scale (Zadoks et al. 1974). Recording of spot blotch incidence was done following a double-digit scale (00–99) as a modification of Saari and Prescott’s severity scale (Saari and Prescott, 1975). The upper most leaves, the flag leaf (digit D1) and the penultimate leaf (digit D2) were considered for scoring the disease. The disease severity (DS) was calculated as given below:
Genotypes were classified into six groups based on the reactions to spot blotch as highly resistant (0–10), resistant (11–20), moderately resistant (21–40), moderately susceptible (41–60), susceptible (61–80) and highly susceptible (81–100) based on disease severity.
Selection of miRNAs and target genes, Primer designing
Based on a review of the literature, we identified 17 miRNAs and five target genes involved in defence of different fungal pathogens (Table 2). The mature miRNA sequences were retrieved and downloaded from miRNA Registry database (http://miRNA.sanger.ac.uk). MiRNA-specific forward primers and a universal reverse primer along with internal control were designed using BioEdit software version 5.09.04 (Supplementary Table 2). Similarly, primers for target genes of miRNAs were designed by using PrimerBlast (http://www.ncbi.nlm.nih.gov/tools/primer-blast) (Supplementary Table 3). β-Actin (Forward Primer:5’- CAA ATC ATG TTT GAG ACC TTC AAT G -3’ and Reverse primer 5’-ACC AGA ATC CAA CAC GAT ACC TG-3’) was used as a reference gene for data normalization of target genes as described earlier (Kaur et al. 2017). These primers were then used for analysis of hairpin structure, homodimer and heterodimer formation by using OligoAnalyser (http://eu.idtdna.com/calc/analyzer), and primers were made final if they were having ∆G less than − 9 kcal/mol. All the primer sequences were validated by using gel electrophoresis of PCR amplicons and the presence of just one peak on the thermal dissociation (Tm) curve generated by the thermal denaturation protocol, which was performed after each real-time PCR run.
Isolation of miRNAs and total RNAs
For the miRNA investigations, two contrasting wheat genotypes viz., Chiriya-1 (resistant) and WH-147 (sensitive) for B. sorokiniana were used. After 48 h, samples were collected from the leaves of plants that had been sprayed with sterile water (mock) and infected with disease and kept at − 80 °C until use.
To analyse miRNA expression behaviour, total miRNAs were isolated form 100 mg of leaf tissue using mirVana™ isolation Kit (Ambion) according to the manufacturer’s instructions. Total RNA was extracted for target transcripts using TRIzol® Reagent (Ambion, USA) following the manufacturer’s protocol. The quality and quantity of miRNAs were determined using a Nano-Drop spectrophotometer, ND-1000 (NanoDrop Technologies, USA).
srcDNA library construction
For the miRNAs expression profiles, a small RNA (srcDNA) library was generated as described previously (Seungil et al. 2006). miRNAs were poly A tailed at 37 °C for 45 min in 50 μl reaction volume comprising 0.3 μg RNA and 5U E. Coli poly(A) polymerase (NEB). The poly (A) tailed small RNA was isolated from sample using a purification cartridge included in mirVana Probe and Marker Kit (Ambion) and following the manufacturer’s instructions and also reported by Gupta et al. (2012).
Expression analysis of miRNA targeted transcripts
To quantify the expression profile of selected miRNAs target transcripts, we synthesized the cDNA from total RNA using Novagen® first-strand cDNA synthesis kit (Merck, Germany) according to the user’s manual for the expression analysis of various target genes of selected miRNAs and as previously described (Kaur et al. 2017). The concentration and purity of srcDNA and cDNA were quantified by using Nano-Drop spectrophotometer, ND-1000 (NanoDrop Technologies, USA).
Validation of miRNAs and target genes using qPCR
Before conducting the expression study, we tested the primer efficiency by PCR-amplifying all of the miRNAs and their corresponding target genes. To validate, a srcDNA library was used to amplify 17 miRNAs using a specific forward primer and an RTQ universal reverse primer (Supplementary Table 2). About ~ 93 bp amplicon was recovered using 3% agarose gel. After PCR confirmation as described (Gupta et al. 2012), quantitative real-time PCR was performed on CFX real time system (BioRad, USA) as follow: the total volume of the PCR reaction was 10 μl of 29 Light CyclerR 480 SYBR Green 1 master mix (Roche), 0.8 μl RTQ-UNIr primer (10 μM), 0.8 μl forward primer (10 μM) and 2 μl (10 ng/μl) of srcDNA to a final reaction volume of 20 μl. The threshold cycle (Ct) value of the technical triplicates and standard deviation was calculated. The relative expression level of all the miRNAs was calculated by using the comparative 2−ΔΔCt method. For internal control in qRT-PCR and data normalization, β-actin and U6 snRNA were used.
Results
Screening of wheat genotype for B. sorokiniana infection under field conditions
Spot blotch disease of wheat, caused by B. sorokiniana, is a serious disease of wheat in the Warm and humid environment of South East Asia. In poly-house conditions, the genotypes were tested against mixture of B. sorokiniana isolates. Disease score was recorded and genotypes were classified into six groups viz., highly resistant (0–10), resistant (11–20), moderately resistant (21–40), moderately susceptible (41–60), susceptible (61–80) and highly susceptible (81–100) based on disease severity (Fig. 1). Out of 64 lines, 11 genotypes were recorded as highly resistant, 7 resistant, 5 moderately resistant, 17 moderately susceptible, 14 susceptible and 10 highly susceptible (Table 1). The genotypes Chirya-1, GS/2019–20/5048, GS/2019–20/5048, GS/2019–20/9003 and GS/2019–20/9003 were recorded highly resistant to spot blotch whereas genotypes DBW 46, FLW 13, FLW 31, PAU 16,058, Sonalika, WH 147, GS/2019–20/4036, GS/2019–20/4045, GS/2019–20/4046 and GS/2019–20/4057 were recorded highly susceptible.

Histogram of spot blotch disease severity in selected advance wheat lines and released wheat genotypes
MicroRNA profiling in resistant cultivar Chiriya-1 during B. sorokiniana infection
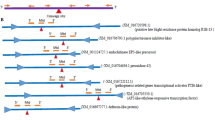
Based on the previous research describing the function of miRNAs in crop-fungus interactions, 17 miRNAs were chosen to explore their crucial role during B. sorokiniana infection in two contrasting wheat genotypes, Chiriya-1 (resistant) and WH-147 (sensitive). These 17 miRNAs derived from different plant species, includes Triticum aestivum (6), Physcotrella trichocarp (6), Oryza sativa (2), Physcomitrella patens (1), Chlamydomonas reinhardtii (1), and Solanum lycopersicum (1). These miRNAs have a role in various cellular and molecular responses, including disease resistance (Table 2). qPCR study of these miRNAs revealed varying degree of accumulation in Chiriya-1 genotypes infected with B. sorokiniana (Fig. 2). We observed that 7 miRNAs were involved in up-regulation, including ptc-miR169-3p, ptc-miR166a, ptc-miR1450, osa-miR2118a, osa-miR1869, ppt-miR901 and cre-miR1169-3p while 10 were down-regulated, including ptc-miR482c, ptc-miR1447, ptc-miR1448, tae-miR156, tae-miR160, tae-miR167, tae-miR396-5p, tae-miR444a, tae-miR1138, sly-miR1917 in resistant wheat genotype Chiriya-1 (Figs. 2, 3 and 4). Furthermore, ppt-miR901 was observed as significantly elevated (~ 10.21 times), followed by cre-miR1169-3p (~ 8.2 times), and ptc-miR169-3p (~ eight times). Similarly, amongst down regulated miRNAs, tae-miR156 (~ 0.12 times) and tae-miR396-5p (~ 0.12 times) were substantially down-regulated (Figs. 3 and 4).

qRT-PCR validation of selected miRNAs from Populus trichocarpa. The histograms display the relative values for the quantified expressions of the miRNAs. The analyses were performed as triplicates, and the error bars were indicated on each column

qRT-PCR validation of selected miRNAs from Triticum aestivum. The histograms indicate the relative values for the quantified expressions of the miRNAs. The analyses were performed as triplicates, and the error bars were indicated on each column

qRT-PCR validation of selected miRNAs from Oryza, Physcomitrella patens, Chlamydomonas reinhardtii and Solanum lycopersicum. The histograms show the relative values for the quantified expressions of the miRNAs. The analyses were performed as triplicates, and the error bars were indicated on each column
MicroRNA profiling in susceptible cultivar WH-147 during B. sorokiniana infection
The comparative expression study of these 17 miRNAs selected from different plant species in susceptible wheat genotype WH-147 revealed the same trend of differential expression behaviour as Chiriya-1. We observed that, as compared to Chiriya-1, 9 miRNAs were up-regulated: ptc-miR169-3p, ptc-miR482c, ptc-miR1448, ptc-miR1450, tae-miR156, tae-miR167, osa-miR2118a, ppt-miR901, cre-miR1169-3p while 8 miRNAs viz. ptc-miR166a, ptc-miR1447, tae-miR160, tae-miR396-5p, tae-miR444a, tae-miR1138, osa-miR1869 and sly-miR1917 were down-regulated in wheat genotype WH-147 genotypes (Figs. 2, 3 and 4). Among the 9 up-regulated miRNAs, five were found to exhibit a consistent pattern of expression in both genotypes: ptc-miR169-3p, ptc-miR1450, osa-miR2118a, ppt-miR901 and cre-miR1169-3p (Figs. 2 and 4). Nonetheless, six miRNAs, namely ptc-miR1447, tae-miR160, tae-miR396-5p, tae-miR444a, tae-miR1138, and sly-miR1917 showed common trend of accumulation in both the genotypes (Figs. 2, 3 and 4). Amongst up-regulated miRNAs, ptc-miR1450 (~ 7.6 times), osa-miR2118a (~ 7.06 times) and tae-miR167 (~ 5.5 times) was observed to be the most strongly up-regulated (Figs. 2, 3 and 4). Similarly, tae-miR160 (~ 0.04 times), tae-miR444a (~ 0.16 times), and osa-miR1869 (~ 0.19 times) were shown to be down-regulated miRNAs (Figs. 3 and 4).
Validation of target genes of selected miRNAs using qPCR and regulatory network analysis
In order to see the effect of different miRNAs on the expression behaviour of their corresponding genes in resistant and susceptible wheat genotypes, we performed the expression analysis of five target genes namely SPL, MADS box, ARF, HDZ and NBS-LRR targeted by tae-miR156, tae-miR444a, tae-miR160, ptc-miR166a, and ptc-miR482c, respectively (Fig. 5). We observed a difference in the expression patterns of all the target genes when compared to the associated miRNAs (Fig. 5). Only two miRNAs, tae-miR156 and ptc-miR482c, shows a perfect inverse relationship with their target genes (Fig. 5). Furthermore, the target gene (NBS-LRR) of ptc-miR482c exhibited the greatest up-regulation (3.05 times), whereas HDZ protein expression (0.07 times) was the least (ptc-miR166a) (Fig. 5).

qRT-PCR validation of selected miRNAs and their target genes. Histogram show the relative values for the quantified expressions of the miRNAs. The analyses were performed as triplicates, and the error bars were indicated on each column
To explore the possible regulatory association between miRNA and their targets, the putative miRNA-target relationship was investigated by searching against the analysed miRNAs using the psRNAtarget tool (https://www.zhaolab.org/psRNATarget/). The results showed that 17 miRNAs target a total of 179 wheat transcripts (Supplementary Table 4) and a possible regulatory network as shown in Fig. 6 was constructed using cytoscape (http://cytoscape.org/), among which osa-miR2118a target TRIAECS424BL1044950.1(Mitogen-activated protein kinase 6) while tae-miR167 targets TRIAECS422BL0383990.1, TRIAECS420286040.2, TRIAECS422DL0501640.1 and TRIAECS423B0732040.2 (Auxin responsive factor 8) further validate the role of miRNA imparting role in disease resistance.

A schematic representation of the regulatory network relationships between the putative miRNAs and their targets in wheat. The 17 miRNAs that regulate target genes of wheat are shown as circles
Discussion
MicroRNAs control gene expression in response to abiotic and biotic stress. The onset of the disease triggers early host defence responses. These defensive responses comprise of hormone signalling pathway regulated by lipoxygenase activity, a cell to cell signalling mechanism involving lignin biosynthesis and a translational initiation factor-regulated protein synthesis.
So far, a large number of miRNAs from model plant species have been identified but only a few of these miRNAs has been functionally characterized. In the current investigation some of the chosen miRNAs had not previously been shown to be stress responsive, whereas others were recognised to be implicated in fungal stress (Zhao et al. 2012). miR156 has been shown to have a role in the response of fungal stress in pine, poplar, and wheat (Xin et al. 2010).
MiR156 has been shown to have a role in the response to fungal stress in pine, poplar, and wheat (Xin et al. 2010). miR169 expression was shown to be down-regulated in soybean in response to pathogen stress (Subramanian et al. 2008), but up-regulated in poplar in response to fungal attack (Gupta et al. 2014). miR169 has the ability to target a number of RNAs, including Pto kinase 1, a well-known resistance gene (Inal et al. 2014). As a result, its expression level in infected samples is predicted to be greater compared to control. Our result of increased accumulation of ptc-miR169-3p in resistant cultivar Chiriya-1 upon B. sorokiniana infection suggests its active role in defence mechanism in wheat compared to WH147. It has been reported that a nucleotide-binding site leucine-rich repeat (NBS-LRR) protein, targeted by miR482, was suppressed in tomato when inoculated with Pseudomonas syringe (DC3000) (Gupta et al. 2014). Since a resistant cultivar needs to synthesize more defence proteins than a susceptible cultivar does, higher expression levels of such target genes in resistant cultivar was expected. Our results showed that compared to susceptible genotype WH-147, down regulation of ptc-miR482c-5p in resistant genotype Chiriya-1 might be crucial to accumulation more defence related target gene in Chiriya-1. Similarly, miR166 has been reported to target a fungal virulent gene, Ca2+-dependent cysteine protease (Clp-1), which decreases disease resistance against fungal pathogen in cotton and the expression of miR166 was observed to be higher in resistant cotton cultivar upon fungal infection suggesting decreased virulence compared to susceptible cultivars (Zhang et al. 2016). Our result of increased expression of ptc-miR166a in Chiriya-1 might suggest same defence mechanism operating in cotton. miR1447, miR1448 which were reported to target disease resistance proteins (Lu et al. 2005, 2008) were observed to be differentially regulated upon D. gregaria infection with miR1447 being increased and miR1448 is being decreased. In contrast, we observed reverse relation of expression of miR1447 and miR1448 in both the resistant and susceptible genotype (Fig. 3 and 4) suggesting a different mechanism of defence response in wheat. Zhao et al. (2012) showed up-regulation of miRNA1450 targeting LRR-transmembrane protein kinase gene involved in defence mechanism in Populus trichocarpa against Botryosphaeria dothidea (Zhao et al. 2012). We also observed increased expression of this miRNA in both the wheat cultivars suggesting similar defence mechanism.
The six miRNAs namely tae-miR156, tae-miR160, tae-miR167, tae-miR396-5p, tae-miR444a, tae-miR1138 have been validated for their different role in different plants exposed to various fungal pathogen (Gupta et al. 2014). miR156 has been reported to be down-regulated in T. aestivum and P. taeda exposed to Erysiphe graminis f. sp. tritici and Cronartium quercuum, respectively while up-regulated in Populus beijingensis and P. trichocarpa exposed to Dothiorella gregaria and B. dothidea respectively. Similarly, miR160 showed up-regulation in Populus trichocarpa while down-regulation in P. taeda upon exposure with B. dothidea and C. quercuum respectively (Lu et al. 2007; Xin et al. 2010; Chen et al. 2012, 2018; Zhao et al. 2012). Our result showed down regulation of these two miRNA in resistant cultivar Chiriya-1 while inverse relation in WH-147 upon B. sorokiniana infection suggesting the different function of same miRNA to different function in different species under different set of pathogens. Likewise, expression of miR164, miR444 and miR1138 have been shown to be up-regulated by targeting their corresponding target genes ARF, MADS box and eIF−4b under Puccinia graminis f. sp. tritici infection (Gupta et al. 2012) while miR396 targeting 50S ribosomal protein L20 was reported to be up-regulated in P. beijingensis under D. gregaria fungal infection respectively (Chen et al. 2012). Our result of differential expression of these four miRNAs in resistant and susceptible cultivars signifies their role during hormonal imbalances and translation machinery during B. sorokiniana infection. Upon infection with B. sorokiniana, wheat plants activates miRNA mediated defense pathway involing disease signal transduction based on different kinases, hormone signaling, and production of reactive oxygen species (ROS). These pathways are significantly modulated by interaction of miRNAs and target genes, usually transcription factors (TFs) in response to B. sorokiniana infection. These regulatory interaction of miRNAs and TFs could reorganize the molecular machinery to impart resistance.Thus, we propose the possible regulatory interaction model of miRNA-target pairs during B. sorokiniana infection in wheat (Figs. 6 and 7).

Possible disease resistance regulatory mechanism during Bipolaris sorokiniana infection in wheat. ROS: reactive oxygen species
MiR2118 and miR1917 by targeting TIR-NBS-LRR and triple response 4 protein respectively, have been characterized for their role during disease resistance in cotton under V. dahlia fungal invasion where they were observed as down regulated (Yin et al. 2012). Up-regulation of miR2118 and down-regulation of miR1917 in both the cultivars, Chiriya-1 and WH-147 suggesting the interference in accumulation of their corresponding target genes modulating the disease response under B. sorokiniana infection. Leucine-rich repeat receptor, and copine family proteins which are targeted by miR1169-3p, and miR1869 are induced upon pathogen attack (Jambunathan et al. 2001; Kulcheski et al. 2011; Eldem et al. 2013). Inal et al. (2014) showed that the expression of miR901 is down-regulated in wheat upon fungal-inoculation with F. culmorum, B. sorokiniana in the both resistant and susceptible cultivars. The contrast result of inverse expression in our study in both the cultivars might be linked to different genetic background leading to differential response. Recognizing mRNAs regulated by miRNAs will help us better understand the biological functions of miRNAs (Tan et al. 2012). We constructed interaction networks of the differentially expressed miRNA-targets. osa-miR2118a target was predicted to regulate cognate target gene (MAP kinases) while tae-miR167 predicted to regulate three genes coding for Auxin responsive factor 8 regulators. The overall analyses indicated that fungal-responsive wheat miRNAs were differentially regulated in a pathogen and cultivar specific manner. This suggested that such genetic variation among cultivars could be amenable to selection for disease resistance in terms of miRNA expression upon fungal stresses.
References
Achakzai HK, Barozai MYK, Din M, Baloch IA, Achakzai AKK (2018) Identification and annotation of newly conserved microRNAs and their targets in wheat (Triticum aestivum L.). PLoS ONE 13(7):e0200033
Arribas-Hernández L, Kielpinski LJ, Brodersen P (2016) mRNA decay of most Arabidopsis miRNA targets requires slicer activity of AGO1. Plant Physiol 171:2620–2632. https://doi.org/10.1104/pp.16.00231
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Brenchley R, Spannagl M, Pfeifer M, Barker GL, D’Amore R, Allen AM et al (2012) Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491:705–710
Chand R, Pandey S, Singh H, Kumar S, Joshi A (2003) Variability and its probable cause in natural populations of spot blotch pathogen Bipolaris sorokiniana of wheat (T. aestivum L.) in India. J Plant Dis Prot 110:27–35
Chand R, Yadav O, Bashyal B, Prasad L, Joshi A (2013) Technique for the maintenance of heterokaryotic isolates of Bipolaris sorokiniana. Indian Phytopathol 66:61–65
Chen L, Ren Y, Zhang Y, Xu J, Zhang Z, Wang Y (2012) Genome-wide profiling of novel and conserved Populus microRNAs involved in pathogen stress response by deep sequencing. Planta 235(5):873–883
Chen X, Liu Z, Shi G et al (2018) MIR167a transcriptionally regulates ARF6 and ARF8 and mediates drastically plant Pi-starvation response via modulation of various biological processes. Plant Cell Tissue Organ Cult 133:177–191
Dubin HJ, Van Ginkel M (1991) The status of wheat diseases and disease research in the warmer areas. In: Saunder DA, Hettel GP (eds) Wheat for non-traditional warm areas in heat stressed environments: Irrigated Dry Areas. UNDP/CIMMYT, Mexico, pp 125–145
Eldem V, Okay S, Unver T (2013) Plant microRNAs: new players in functional genomics. Turk J Agric for 37:1–21
Feng H, Wang B, Zhang Q, Fu Y, Huang L, Wang X, Kang Z (2015) Exploration of microRNAs and their targets engaging in the resistance interaction between wheat and stripe rust. Front Plant Sci 6:469
Gupta O, Permar V, Koundal V, Singh U, Praveen S (2012) MicroRNA regulated defense responses in Triticum aestivum L. during Puccinia graminis f. sp. tritici infection. Mol Biol Rep 39(2):817–824
Gupta OP, Sharma P, Gupta RK, Sharma I (2014) Current status on role of miRNAs during plant–fungus interaction. Physiol Mol Plant Pathol 85:1–7
Inal B, Turktas M¸ Eren H, Ilhan E, Okay S, Atak M, et al (2014) Genome-wide fungal stress responsive miRNA expression in wheat. Planta 240:1287–1298
Jaiswal S, Iquebal MA, Arora V et al (2019) Development of species specific putative miRNA and its target prediction tool in wheat (Triticum aestivum L.). Sci Rep 9:3790
Jambunathan N, Siani JM, McNellis TW (2001) A humidity-sensitive Arabidopsis copine mutant exhibits precocious cell death and increased disease resistance. Plant Cell 13:2225–2240
Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAs and their regulatory roles in plants. Ann Rev Plant Biol 57:19–53
Kaur A, Gupta OP, Meena NL, Grewal A, Sharma P (2017) Comparative temporal expression analysis of microRNAs and their target genes in contrasting wheat genotypes during osmotic stress. Appl Biochem Biotechnol 181:613–626
Kulcheski F, de Oliveira L, Molina L, Almerao M, Rodrigues F et al (2011) Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genomics 12:307
Kumar D, Dutta S, Singh D, Prabhu K, Kumar M, Mukhopadhyay K (2017) Uncovering leaf rust responsive miRNAs in wheat (Triticum aestivum L.) using high-throughput sequencing and prediction of their targets through degradome analysis. Planta 245(1):161–182
Lu S, Sun YH, Shi R, Clark C, Li L, Chiang VL (2005) Novel and mechanical stress responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell 17:2186e203
Lu S, Sun YH, Amerson H, Chiang VL (2007) MicroRNAs in loblolly pine (Pinus taeda L.) and their association with fusiform rust gall development. Plant J 51:1077–1098
Lu S, Sun YH, Chiang VL (2008) Stress-responsive microRNAs in Populus. Plant J 55:131–151
McIntosh RA, Pretorius ZA (2011) Borlaug Global Rust Initiative provides momentum for wheat rust research. Euphytica 179:1–2
Meng F, Liu H, Wang K, Liu L, Wang S, Zhao Y, Yin J, Li Y (2013) Development-associated microRNAs in grains of wheat (Triticum aestivum L.). BMC Plant Biol 13:140
Nizam S, Verma S, Singh K, Aggarwal R, Srivastava KD, Verma PK (2012) High reliability transformation of the wheat pathogen Bipolaris sorokiniana using Agrobacterium tumefaciens. J Microbiol Meth 88:386–392
Pandey SP, Sharma S, Chand R, Shahi P, Joshi A (2008) Clonal variability and its relevance in generation of new pathotypes in the spot blotch pathogen, Bipolaris sorokiniana. Curr Microbiol 56:33–41
Pandey B, Gupta OP, Pandey DM, Sharma I, Sharma P (2013) Identification of new stress-induced microRNA and their targets in wheat using computational approach. Plant Signal Behav 8:e23932
Saari EE, Prescott JM (1975) A scale for appraising the foliar intensity of wheat disease. Plant Dis Rep 59:377–380
Seungil R, Park C, Jin J, Sanders KM, Yan W (2006) A PCR-based method for detection and quantification of small RNAs. Biochem Biophys Res Commun 351(3):756–763
Singroha G, Sharma P, Sunkur R (2021) Current status of miRNA-mediated regulation of drought stress responses in cereals. Physiol Plan 172:1808–1821
Subramanian S, Fu Y, Sunkar R, Barbazuk WB, Zhu J-K, Yu O (2008) Novel and nodulation-regulated microRNAs in soybean roots. BMC Genomics 9:160
Sun G (2012) MicroRNAs and their diverse functions in plants. Plant Mol Biol 80:17–36
Sun G, Stewart CN, Xiao P, Zhang B (2012) MicroRNA expression analysis in the cellulosic biofuel crop Switchgrass (Panicum virgatum) under abiotic stress. PLoS ONE 7:e32017
Tan GN, Victoriano AF, Okamoto T (2012) Evaluation of online miRNA resources for biomedical applications. Genes Cells 17:11–27
Vinocur B, Altman A (2005) Recent advances in engineering plant tolerance to abiotic stress: achievements and limitations. Curr Opin Biotechnol 16(2):123–132
Wu F, Guo O, Zhang W, Jin W (2014) Identification and analysis of powdery mildew-responsive miRNAs in wheat. J Phytopathol 163:264–270
Xin M, Wang Y, Yao Y, Xie C, Peng H, Ni Z et al (2010) Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol 10:123
Yin Z, Li Y, Han X, Shen F (2012) Genome-wide profiling of miRNAs and othersmall non-coding RNAs in the Verticillium dahliaee inoculated cotton roots. PLoS ONE 7(4):e35765
Zadoks JC, Chang TT, Konzak CF (1974) A decimal code for the growth stages of cereals. Weeds Res 14:415–421
Zhang T, Zhao YL, Zhao JH, Wang S, Jin Y, Chen ZQ et al (2016) Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nat Plants 2(10):16153
Zhao JP, Jiang XL, Zhang BY, Su XH (2012) Involvement of microRNA-mediated gene expression regulation in the pathological development of stem canker disease in Populus trichocarpa. PLoS ONE 7(9):e44968
Acknowledgements
This work was supported by the Indian Council of Agricultural Research of New Delhi, India (Grant-in-Aid 1008018 LBSYOA to PS). We also thank Director, ICAR-IIWBR for providing the facility for conductance of this experiment and Dr R Selvakumar, Dr Sonia and lab members for kind support for during the experimentation.
Author information
Authors and Affiliations
Contributions
PS conceptualized and performed the experiments. VG did phenotyping and generated data, PS and OPG wrote the first draft, PS, GS, and GPS edited the final draft of the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Consent to participate
NA.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sharma, P., Gupta, O.P., Gupta, V. et al. Differential expression profiling of microRNAs and their target genes during wheat-Bipolaris sorokiniana pathosystem. Physiol Mol Biol Plants 27, 2567–2577 (2021). https://doi.org/10.1007/s12298-021-01092-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12298-021-01092-1