
Abstract
In wheat, meta-QTLs (MQTLs), ortho-MQTLs, and candidate genes (CGs) were identified for nitrogen use efficiency and root system architecture. For this purpose, 1788 QTLs were available from 24 studies published during 2006–2020. Of these, 1098 QTLs were projected onto the consensus map resulting in 118 MQTLs. The average confidence interval (CI) of MQTLs was reduced up to 8.56 folds in comparison to the average CI of QTLs. Of the 118 MQTLs, 112 were anchored to the physical map of the wheat reference genome. The physical interval of MQTLs ranged from 0.02 to 666.18 Mb with a mean of 94.36 Mb. Eighty-eight of these 112 MQTLs were verified by marker-trait associations (MTAs) identified in published genome-wide association studies (GWAS); the MQTLs that were verified using GWAS also included 9 most robust MQTLs, which are particularly useful for breeders; we call them ‘Breeder’s QTLs’. Some selected wheat MQTLs were further utilized for the identification of ortho-MQTLs for wheat and maize; 9 such ortho-MQTLs were available. As many as 1991 candidate genes (CGs) were also detected, which included 930 CGs with an expression level of > 2 transcripts per million in relevant organs/tissues. Among the CGs, 97 CGs with functions previously reported as important for the traits under study were selected. Based on homology analysis and expression patterns, 49 orthologues of 35 rice genes were also identified in MQTL regions. The results of the present study may prove useful for the improvement of selection strategy for yield potential, stability, and performance under N-limiting conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nitrogen (N) is a major limiting factor for plant growth and development (Raun and Johnson 1999) so that during the four decades of the green revolution, a seven-fold rise in the use of nitrogen fertilizer was needed to achieve the doubling of agricultural food production worldwide (Hirel et al. 2007). A side-effect of this excess use of N has also been witnessed in the form of a negative impact on the ecosystem (Li et al. 2015). In order to limit the excess use of N without any adverse effect on yield, the use of cultivars with improved efficiency for N uptake and utilization has been recommended, particularly under low N input management (Good and Beatty 2011). In wheat, the availability of genetic variation in the germplasm suggests that the development of cultivars with high nitrogen use efficiency (NUE) is possible and can be achieved through breeding (Balyan et al. 2016). However, due to the complex genetic architecture of this trait and strong genotype-environment interactions (G × E) involved, efforts for breeding wheat cultivars for high NUE met with only limited success.
NUE is commonly defined as grain yield per unit of N supplied (Moll et al. 1982); it largely depends on two major components: Nitrogen Uptake Efficiency (NUpE) and Nitrogen Utilization Efficiency (NUtE). NUpE is the quantity of N taken up by the plant per unit of N provided, whereas, NUtE is the grain yield per unit of N taken up by the plant. As a result, NUE is only the sum of NUpE and NUtE (Moll et al. 1982; Good et al. 2004). Other traits that are most relevant for measuring NUE in wheat include the following, besides others: (i) chlorophyll content, (ii) plant height, (iii) biomass, (iv) fresh and dry weight of plants, (v) N accumulation partitioning indices, (vi) leaf area index, (vii) grain N, (viii) grain protein content, (ix) enzymatic activities in different plant parts/tissues, (x) agronomic traits such as grain number, grain weight, grain filling duration, days to heading/flowering/maturity, tiller number, and spike related traits (Greef 1994; Cirilo et al. 2009; Sun et al. 2013; Balyan et al. 2016; Brasier et al. 2020). Root system architecture (RSA) is also critical while breeding for high NUpE (Lopez-Bucio et al. 2003; Garnett et al. 2009). A desirable root RSA for improved NUpE, particularly under low-N soils, includes increase in root dry weight, root length, root density, root number, root diameter, and length of lateral roots/root hairs (Meister et al. 2014; Li et al. 2016; Forde 2014), Meister et al. 2014; Li et al. 2015).
In wheat, due to lack of complete knowledge about the genetic basis of NUE, and associated G × E interactions, selection of cultivars with RSA-based NUE has been suggested (Garnett et al. 2009). QTL studies involving all RSA traits (mentioned above) are also available in wheat. Numerous wheat QTLs for NUE traits at physiological and agronomic levels have been reported (Fan et al. 2019; Zhang et al. 2019; Brasier et al. 2020). Similarly, QTLs that regulate RSA have also been identified (Salarpour et al. 2020; Yang et al. 2020). Despite these significant advances in information about the genetics of NUE and RSA, only a very small fraction of these QTLs and the associated markers could be utilized in actual breeding. The reasons for this failure have been widely discussed (Misztal, 2006; Collard and Mackill, 2008; Cobb et al. 2019).
In order to overcome the above limitations in using QTLs for the development of wheat cultivars with higher NUE, a meta-QTL (MQTL) analysis seems to be an appropriate approach, which allows identification of the most robust and stable MQTLs (Goffinet and Gerber, 2000; Veyrieras et al. 2007). These MQTLs have shown to be more effective than QTLs for their use in marker-assisted selection (MAS) because the MQTLs generally have reduced confidence intervals (CIs) and improved phenotypic variation explained (PVE%). MQTLs have also been shown to be useful for the identification of promising candidate genes (CGs) associated with the trait in question. Improved software and algorithms have become available and are being increasingly utilized for meta-QTL analysis (Sosnowski et al. 2012; de Oliveira et al. 2014). The approach has already been used to identify MQTLs for a variety of traits in all major crops, including wheat (Online Resource 1), rice (Kumar and Nadarajah 2020; Khahani et al. 2020, 2021), barley (Zhang et al. 2017) and maize (Zhao et al. 2018).
Meta-QTL analysis in wheat has already been conducted for a number of traits, which are listed in Online Resource 1. A solitary study on MQTL for traits relevant to NUE was also conducted, almost ten years ago (Quraishi et al. 2011), where 11 MQTLs based on only three studies were reported. Similarly, reports on MQTLs for RSA using QTLs reported till 2017 are also available (Darzi-Ramandi et al. 2017; Soriano and Alvaro, 2019). A large number of studies involving interval mapping for NUE have also been conducted in different crops including wheat during the last 10 years (Online Resource 2). Since hundreds of QTLs for NUE in wheat have been reported during the last decade (after the only report of MQTL for NUE published in 2011), a fresh meta-QTL analysis is warranted. The MQTLs for the same trait have also been used for identification of the so-called ortho-MetaQTLs, which seem to be conserved over cereals and therefore will be more reliable and can be used in more than one cereals. This identification of ortho-MQTLs has been facilitated by a high level of synteny and collinearity between wheat and other cereals.
Keeping in view the above, the present study was planned, which involved meta-analysis for NUE and RSA in wheat and ortho-MQTL analysis involving wheat and maize. For NUE, QTLs reported during 2006–2020, and for RSA, QTLs reported during 2018–2020 were utilized for the present study. Since MQTLs for NUE are also known in maize (Ahn et al. 1993; Bennetzen and Chen, 2008), ortho-MQTLs for wheat and maize were also worked out with the hope that the results of the present study may be used not only for wheat and maize but also for other cereals (due to conserved nature of ortho-MQTLs). MQTLs for NUE/RSA were not available in other cereals like rice, barley, etc. for identification of ortho-MQTLs involving all the cereals in the present study. Results of the meta-analysis were also compared with the available results from GWAS, and integrated with transcriptomics, and rice–wheat orthology to identify the promising genomic regions and important CGs, which affect NUE/RSA in wheat. The findings of the present study should prove useful for the improvement of wheat cultivars for yield potential, stability, and performance under N-limiting conditions.
Materials and methods
Bibliographic search and data collection
A comprehensive bibliographic search was carried out on wheat QTLs related to NUE published during 2006–2020; QTL information associated with RSA traits under different levels of N contents, published during 2018–2020 was also collected. Details on 15 studies for QTLs associated with NUE and 9 studies for QTLs associated with RSA are summarized in Online Resource 2. Each study was utilized for collecting the following information: (i) data on associated traits, (ii) type and size of the mapping population, (iii) position of QTLs (peak position and genetic CI), (iv) LOD (logarithm of the odds) score for each QTL, (v) R2 or PVE (phenotypic variation explained) for each QTL, (vi) closely linked or flanking markers. Studies with incomplete information were not included.
Construction of consensus map and QTL projection
Following three high-quality linkage maps containing different types of markers, extensively used in QTL mapping studies, were utilized to prepare a consensus map: (i) Wheat_Composite_2004’ available at GrainGenes database (http://wheat.pw.usda.gov), (ii) ‘Wheat, Consensus SSR, 2004’ (Somers et al. 2004), (iii) the integrated map constructed by Marone et al. (2013). Markers flanking individual QTLs, identified in all 24 QTL mapping studies, were also included on this consensus map. The R package “LP merge” (Endelman and Plomion 2014) was used to construct a consensus map for meta-QTL analysis. Construction of consensus map using LPmerge involves two steps. First, it computes the number of consensus bins, markers, and ordinal conflicts (if available). If there are inconsistencies in the order of markers in the linkage maps, it resolves them by removing ordinal constraints using statistical manipulations. Second, it generates 1 to 4 consensus maps (K = 1 to 4, where K is the maximum interval size); from which one or more maps can be chosen using the associated statistics developed for this purpose; this is accomplished by estimating the root mean square error (RMSE, based on mean and standard deviation) between each initial map and the consensus map. The final map for further analysis was the consensus map with a length close to the mean length of the component linkage maps associated with the lowest standard deviation.
The QTLs having the following information were selected for projection on the consensus map: (i) PVE, (ii) LOD scores, (iii) peak positions, and (iv) CIs. For QTLs, lacking original CI, 95% CI was calculated based on population-specific equations derived by Darvasi and Soller, (1997) and Guo et al. (2006). The QTL positions were then projected on the consensus chromosome map utilizing a scaling rule between the marker interval of the QTLs and the corresponding CI on the consensus map (Arcade et al. 2004; Veyrieras et al. 2007). BioMercator v.4.2 (https://urgi.versailles.inra.fr/Tools/BioMercator-V4) was used for this purpose. The new CI of each QTL on the consensus map was computed using Gaussian distribution (Veyrieras et al. 2007).
Meta-QTL analysis and verification using GWAS
After QTL projection, meta-analysis was performed for each chromosome, separately, using BioMercator v.4.2. Since the number of QTLs per chromosome was > 10, an algorithm, involving two-step analysis, developed by Veyrieras et al. (2007) was used for the analysis. In the first step, five different models (based on the presence of 1, 2, 3, 4, or N real QTLs) were used, and the best model was defined using Akaike (AIC) statistics. The second step involved setting up suitable parameters for further analysis; these parameters included the actual number of MQTLs or real QTLs to be mapped on the concerned chromosome. The details of statistical approaches and algorithms that are available in the Biomercator software are described in several earlier studies (e.g. de Oliveira et al. 2014). The LOD and PVE values of MQTLs were calculated as averages of LOD and PVE values of the QTLs involved.
The nucleotide sequences of the markers flanking the MQTLs were retrieved either from the published literature or the databases such as GrainGenes (https://wheat.pw.usda.gov/GG3/) and CerealDB (https://www.cerealsdb.uk.net/cerealgenomics/CerealsDB/indexNEW.php). The retrieved sequences were used for BLAST against the wheat reference genome (RefSeq v1.0) available in EnsemblPlants to identify the physical coordinates of the MQTLs. Physical positions of some SNPs were directly obtained from the JBrowse wheat genome browser (https://wheat-urgi.versailles.inra.fr/Tools/Jbrowse).
Information on marker-trait associations (MTAs) for NUE and RSA identified in recent GWA studies was collected and used for a comparison with MQTLs identified during the present study. Similarly, the physical positions of markers (significantly associated with the trait) were obtained either from databases or through BLAST searches. GWAS-MTAs that occurred within 5 Mb physical regions around the MQTLs were considered to be co-located with the MQTLs (Yang et al. 2021).
Identification and characterization of ortho-MQTLs using wheat and maize MQTLs
Ortho-MQTLs were identified using synteny and collinearity between wheat and maize MQTLs. For this purpose, only robust wheat MQTLs each involving at least 15 initial QTLs were utilized along with available information on maize MQTLs (Guo et al. 2018). In each case, a set of CGs underlying the wheat MQTLs was used for the identification of syntenic regions in maize MQTLs using the EnsemblPlants database.
Mining of candidate genes and expression analysis
Physical coordinates of MQTLs were utilized for the identification of CGs using the BioMart tool of EnsemblPlants. Only MQTLs with ≤ 2 Mb CI were initially considered for identification of available CGs; for other MQTLs, where the physical interval was > 2 Mb, the physical position of the MQTL peak was calculated according to Jan et al. (2021) and then a 2 Mb region around the MQTL peak was used for identification of CGs.
In-silico expression of CGs identified as above was conducted using the Expression Visualization and Integration Platform (expVIP) (http://www.wheat-expression.com/); the available relevant transcriptomics datasets were utilized for this purpose (e.g., Ramírez-González et al. 2018). CGs with expression levels > 2 transcripts per million (TPM) in relevant plant parts/tissues (e.g., roots, shoots, leaves, and grains) were considered for further analysis following the criteria suggested by Wagner et al. (2013). For better visualization of the patterns of gene expression, heat maps were generated using an online platform ‘Morpheus’ (https://software.broadinstitute.org/morpheus/). CGs with known functions were accepted as high-confidence CGs for individual MQTLs.
Wheat orthologues for NUE-related rice genes
Basic information on NUE-related rice genes was collected from two recently published research papers (Han et al. 2016; Zhang et al. 2020a, 2020b); protein sequences of these rice genes were extracted from the NCBI database (https://www.ncbi.nlm.nih.gov/). Wheat orthologues of these rice genes were identified from the wheat reference genome using extracted proteins sequences for BLASTP searches. The genes found within MQTL regions were accepted as wheat orthologues for NUE traits. These wheat orthologues were subjected to expression analysis utilizing the relevant transcriptomics datasets available at the expVIP. Levels of gene expression were assessed using TPM values, which were then used for developing heat maps.
Results and discussion
Salient features of the QTLs
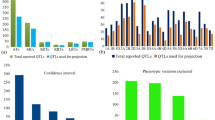
A total of 1,788 QTLs were available from 24 earlier studies, carried out during 2006–2020 (Online Resource 2, 3). These studies involved 20 DH/RIL populations ranging in size from 91 to 227 lines. Some of the essential features of this data include the following: (i) The 1,788 initial QTLs were distributed on all the 21 wheat chromosomes (Fig. 1a). The sub-genome B carried the maximum number of QTLs (721 QTLs) and the D sub-genome carried the minimum (457 QTLs) which is in agreement with previous meta-QTL studies conducted for NUE and RSA in wheat (Quraishi et al. 2011; Darzi-Ramandi et al. 2017; Soriano and Alvaro, 2019). (ii) The number of QTLs per component trait ranged from 77 for enzymatic activities to 602 for agronomic traits (ATs) (Fig. 1b). All the traits associated with NUE and RSA were grouped into 6 main categories (Online Resource 4). (iii) CIs for QTLs ranged from zero to 96 cM, with an average of 10.01 cM (Fig. 1c). (iv) The PVE of individual QTLs ranged from 1.55 to 55.81%, with an average of 10.65% (Fig. 1d). The QTL data collected in this study has been made publicly available through a recently developed WheatQTL database (http://wheatqtldb.net/) (Singh et al. 2021).

Frequencies of QTLs used for identification of MQTLs in wheat, based on four different criteria: a number of QTLs for 21 chromosomes arranged according to 7 homoeologous groups (1–7), each group with three sub-genomes (A, B, D), b frequencies of QTLs for each of the six traits, c frequencies of QTLs with different size of CI (in cM), and d frequencies of QTLs with different levels of phenotypic variation explained (PVE%)
Wheat consensus map
The consensus genetic map included a total of 1,40,166 markers (Online Resource 5, 6) spread over a length of 6853.27 cM, thus giving a density of 20.45 markers/cM for the whole genome. For individual chromosomes, the size of the genetic map ranged from 127.70 (6B) to 661.02 cM (2D) (Fig. 2), the number of markers ranged from 1177 on 4D to 15,716 on 3B and the density of markers ranged from 4.07 markers/cM for 1B to 83.14 markers/cM for 6B (Fig. 2, Online Resource 6). The sub-genome A covered a distance of 2538.18 cM with 61,949 markers (24.41 markers/cM), the sub-genome B covered a distance of 2289.26 cM with 58,846 markers (25.70 markers/cM) and the sub-genome D covered a distance of 2025.82 cM with 19,371 markers (9.56 markers/cM). These features are not very different from those on chromosome maps used in earlier meta-QTL studies (Venske et al. 2019; Jan et al. 2021). The map developed in the present study is much better, relative to those prepared and used in three earlier studies for meta-QTL studies for the traits under study (Quraishi et al. 2011; Darzi-Ramandi et al. 2017; Soriano and Alvaro, 2019).

Details of the consensus map, showing frequencies of mapped markers on each of the 21 chromosomes; on top of each bar representing individual chromosomes are given length of each chromosome (in cM) and marker density (markers/cM)
MQTLs for NUE and RSA
Only 1703 QTLs of the available 1788 QTLs could be used for projection onto the consensus map; the remaining 85 QTLs did not have complete information required for the projection; 118 MQTLs involving 1098 QTLs were identified using meta-QTL analysis (Online Resource 7) (Fig. 3). The remaining 605 QTLs could not be assigned to any MQTL, since they either lacked common markers between consensus and initial maps, or the QTLs had relatively low R2 values and/or large CI. The distribution of MQTLs on 21 individual chromosomes was not proportionate to the number of QTLs carried by them. For instance, chromosomes 4B, 5A, 6A, and 7B carried fewer MQTLs and were not proportionate to the density of QTLs on these chromosomes.

Distribution of 118 MQTLs on 21 wheat chromosomes. The boxes on the right of each chromosome represent the positions of MQTLs (with name of MQTL in each case) for three different features, (red boxes: GWAS-verified MQTLs, green boxes: breeder’s QTLs, black boxes: un-verified MQTLs). On the left of each bar representing a chromosome, are given distances in cM, and on the right are given pairs of flanking markers for each MQTL
The number of QTLs involved in an individual MQTL varied from 2 to 52 (Fig. 4a); however, the following five MQTLs were each based on > 25 QTLs: MQTL4A.2 (26), MQTL5D.6 (26), MQTL5A.1 (29), MQTL4B.4 (38), and MQTL4B.1 (52). This is in sharp contrast to several earlier studies, where none of the individual MQTLs carried > 18 QTLs (Quraishi et al. 2011; Darzi-Ramandi et al. 2017; Soriano and Alvaro, 2019). Twenty (20) of the 118 MQTLs identified in the present study had genomic locations that overlapped with MQTLs found in at least one of the three previous studies (Darzi-Ramandi et al. 2017; Soriano and Alvaro, 2019; Quraishi et al. 2011) (Online Resource 8); 14 of these 20 MQTLs were also verified using GWAS results. All these verified MQTLs may be used with high confidence for MAS and for further basic studies including positional cloning and functional studies of underlying CGs.

Basic information of MQTLs identified, a frequencies of MQTLs harbouring different number of QTLs, b comparison of CIs of QTLs and those of meta-QTLs, showing fold level of reduction in the size of CI
The CI of MQTLs, on an average, was reduced 8.56 fold relative to the CIs in QTLs used (the range of CI in QTLs was 0 to 96 cM with a mean of 10.27 cM), although the average reduction in the size of CI for individual MQTLs widely differed in individual chromosomes and could reach 24.87 fold reduction on chromosome 5D and 40.52 fold reduction on chromosome 4B followed by 19.34 fold in 4D and 15.78 fold in 5A (Fig. 4b). These reductions in the size of CIs are in sharp contrast to those reported in several earlier studies on NUE/RSA, where CI was reduced by merely 2.98 times (14.8/4.96) in one study (Soriano and Alvaro, 2019) and 3.25 (20.8/6.4) in another study (Darzi-Ramandi et al. (2017).
The MQTLs were anchored to the physical map of the wheat reference genome; for only 6 MQTLs, physical coordinates could not be worked out, since these MQTLs were flanked by markers like AFLP, which did not have any sequence data required for BLAST searches (Online Resource 7). The physical lengths of 112 mapped MQTLs differed with a range of 0.02 Mb for MQTL3A.3 to 666.18 Mb for MQTL1B.7, with a mean of 94.36 Mb (29 of these MQTLs each covered a physical distance of < 2 Mb).
Several MQTLs exhibited a clustered distribution with some overlapping MQTLs within a cluster. This clustering of MQTLs was particularly apparent in chromosome 1A (3 MQTLs within 527.18 to 591.64 Mb region), 1B, (8 MQTLs within 17.32–683.81 Mb), and 2A (3 MQTLs clustered within 25.6–43.3 Mb). Other clusters with some overlapping regions were observed on the following wheat chromosomes: 2B (4.48–41.47 Mb), 4A (601.04–695.53 Mb), 6B (30.25–694.99 Mb), and 7A (2.72–210.63 Mb), etc. (Online Resource 7). Such clustering of MQTLs was also reported in some earlier studies (Jan et al. 2021; Saini et al. 2021; Yang et al. 2021) and is a desirable feature for the transfer of more than one MQTL during MAS. These clusters may prove useful for marker-assisted breeding (MAB).
Individual MQTLs also differed for the number of traits each controlled; the number of traits controlled by an individual MQTL ranged from a solitary trait controlled by MQTL3B.2 to as many as 26 traits controlled by MQTL4B.1. As many as 70 MQTLs out of 118 each carried QTLs for several traits including both NUE and RSA. According to one study, RSA shows a significant positive correlation with NUpE but no correlation with NUE, implying that only N acquisition, not N utilisation, is most probably related to the function of roots (Li et al. 2015; Garnett et al. 2009). The MQTLs were used for a further selection of some promising MQTLs using the following criteria: (a) CI < 2 cM, (b) average PVE > 10%, (c) average LOD > 5, and (d) involvement of at least 10 initial QTLs with the MQTL. This exercise resulted in the selection of 9 promising MQTLs, which we like to call breeder’s QTLs; these breeders’ QTLs with their essential features are listed in Table 1. All these breeder’s QTLs (except MQTL2A.2) involved QTLs for both NUE and RSA, hence, these can be used by breeders for the improvement of both NUE and RSA.
Comparison of MQTLs with results of GWAS
The physical positions of MQTLs were also used for comparison with MTAs for NUE/RSA reported in earlier GWAS conducted on durum (tetraploid) and bread wheat (hexaploid). Information on MTAs for NUE and RSA identified in 17 GWA studies published during 2017–2021 is available in Online Resource 9. For this purpose, only 112 physically mapped MQTLs could be considered; 88 of these MQTLs each matched one or more known MTAs from among 412 MTAs (Online Resource 10; Table 1) that were available for comparison. Among them, 45 MQTLs were verified using GWAS for durum wheat, and 81 were verified using GWAS for hexaploid wheat (38 MQTLs were verified with both durum and bread wheat).
The number of matching MTAs for each MQTL also differed, so that as many as 21 MQTLs each matched with at least 10 MTAs identified in 17 GWA studies; of these MQTL5B.2 matched with 60 MTAs, followed by MQTL6B.4, 1B.7, and 1B.4 with 55, 49, and 40 MTAs, respectively. Some of the MQTLs (e.g., MQTL1B.2, 1B.3, and 4B.4) each involving 18 or more initial QTLs matched with more than 20 MTAs (Online Resource 10). These MQTLs overlapping MTAs may be used for mining CGs affecting the NUE and RSA traits. The frequency distribution of some GWAS-validated MQTLs is depicted in Fig. 5.

Frequencies of known GWAS-based MTAs which matched each of 21 MQTLs; for either of the two different traits (NUE, RSA); MQTLs with at least 10 MTAs are included in this figure. MTA results from GWAS using 17 different natural populations were used for this purpose
A comparison MQTLs with GWAS-based MTAs, as above, has been undertaken only sparingly in wheat, where two such studies are available (Aduragbemi and Soriano, 2021; Yang et al. 2021). In these two studies, only 38.66% and 61.37% of MQTLs, respectively, could be verified with the GWAS results. These values of MQTLs verified by GWAS in earlier studies seem to be in agreement with our results. One can perhaps speculate about the reasons for the lack of verification of the remaining MQTLs with MTAs from GWAS. The simplest explanation is that none of the studies (meta-QTL analysis and GWAS) covered the entire genetic variation and that the material used for two sets of studies differed widely.
Ortho-MQTLs in maize
In order to identify ortho-MQTLs for wheat and maize, the syntenic regions of each of the 19 selected wheat MQTLs were compared with previously published maize MQTLs for a variety of traits (Guo et al. 2018). Another solitary study of MQTLs for NUE in maize (Liu et al. 2012) could not be utilized due to the non-availability of complete data required for such analysis. Nine ortho-MQTLs were identified for RSA, which were located on the following six maize chromosomes: 3, 4, 5, 8, 9, and 10; the number of MQTLs for individual ortho-MQTL ranged 1 to 3. The detailed information on ortho-MQTLs is available in Online Resources 11, and 12.
Ortho-MQTLs in wheat were also studied in two earlier studies (Quraishi et al. 2011; Saini et al. 2021). In one of these studies, an ortho-MQTL associated with NUE was identified for four different related crops including wheat, maize, sorghum, and rice; the study also included the identification of a structurally and functionally conserved gene called glutamate synthase (GoGAT) at orthologous locations in these closely related species. Two conserved flanking markers were also developed for the GoGAT gene (Quraishi et al. 2011). Such orthologous genes for NUE/RSA can also be identified using the results of the present study for further detailed study. A conserved orthologous set (COS) of markers may also be developed for their use in MAS for cereal breeding programs (Quraishi et al. 2009).
Candidate genes and their expression analysis
In the genomic regions defined by 112 of the 118 MQTLs, a total of 1991 CGs (Online Resource 13) were found. Several genes/gene families with related functions were detected frequently in different MQTL regions (Fig. 6). These genes included the following: 98 genes encoding proteins containing kinase domain, 79 genes for proteins containing F-box-like domains, 40 genes for cytochrome P450 proteins, 32 genes for glycoside hydrolases, 22 genes for UDP-glucosyltransferases, 16 genes for NAC TFs, 14 genes for expansins, 13 genes for early nodulin-93 proteins, 11 genes for GRAS TFs and 10 genes for ABC transporter-like proteins. (Fig. 6). Association of these genes/gene families with NUE and/or RSA has been reported in several earlier studies (Zhang et al. 2020a, 2020b; Meijón et al. 2014; Jun et al. 2015; Minic 2008; Dong et al. 2020; He et al. 2015; Marowa et al. 2016; Bi et al. 2009; Do et al. 2018). In the present study, some genes that are known to be associated with NUE and RSA appeared in clusters. These clusters may prove useful for marker-assisted breeding (MAB). These genes belonged to the following classes: (i) protein kinase-like domain superfamily (Yang et al. 2020), (ii) F-box-like domain superfamily (Meijón et al. 2014), (iii) expansins (Marowa et al. 2016), (iv) glutathione S-transferase (Dalton et al. 2009), (vi) zinc finger C2H2-type (Han et al. 2020), and (vii) cytochrome P450 (Xu et al. 2015), etc. (Online Resource 13).

Frequencies of candidate genes encoding each of 15 different proteins associated with NUE/RSA
The in-silico expression analysis of the CGs permitted identification of 930 CGs (belonging to 89 MQTLs), with at least 2 TPM, and 307 CGs with more than 5 TPM expressions in different plant tissues; these CGs could be classified in the following two classes based on their expression patterns: (i) Class I genes, mostly expressed in roots and shoots/leaves tissues at the vegetative stages, suggesting their roles in the regulation of NUpE (Zhang et al. 2020a, 2020b; Han et al. 2016); (ii) Class II genes, mainly expressed in the spike (including rachis, stamen, pistil, stigma, and ovary, etc.) and grains (including endosperm, embryo, seed coat, and aleurone layer, etc.). The genes of class II are generally involved in regulating NUE and in turn affecting grain yield and related traits (Mosleth et al. 2015; Karunarathne et al. 2020) (e.g., Online Resource 14). Some CGs encoding uncharacterized or unpredicted proteins also exhibited significant expression in different plant tissues. These genes should be investigated further to see if they play a role in the regulation of NUE in wheat. The expression pattern of some CGs expressed in roots, and shoots/leaves are shown in Fig. 7.

Heat map exhibiting levels of differential expressions of important proteins encoded by candidate genes underlying the following 7 MQTLs: MQTL1A.6, MQTL1B.2, MQTL1B.7, MQTL2A.2, MQTL4A.2, MQTL4A.5, and MQTL5D.6; names of proteins encoded by genes are given on the top X axis of the heat map and the names of 31 classes of organs, where expression of each protein was studied are shown on the left on Y-axis. The scale drawn on the upper right of the heat map represents the range of expression shown by different intensities of blue and red colours
As many as 97 high confidence CGs were detected using the following criteria (i) significant expression in relevant tissues, and (ii) functions previously reported to be important for traits related to either NUE or RSA (Table 2). The molecular and biological functions of these CGs can be studied by cloning/characterization and transgenic approaches. A meta-analysis conducted for three major crops (wheat, rice, and maize) using the data collected from 130 independent genetic transformation studies indicated a promising potential of genetic transformation strategies for improving different NUE parameters (Li et al. 2020).
Orthologues of rice genes in wheat MQTL regions
Efforts were also made to find if the wheat MQTLs identified during the present study carry genes that are orthologues of known NUE/RSA genes from other cereals, since synteny and collinearity between wheat genome and genomes of other cereals are widely known now (Gaut, 2002; Kumar et al. 2009). This became possible due to the availability of results from the following studies: (i) In several crops including rice, the molecular basis of NUE or its associated phenotype has been examined using several forward and reverse genetics approaches (Hu et al. 2016; Sun et al. 2021; Xiang et al. 2021; Yu et al. 2021; Zhang et al. 2020a, 2020b). (ii) In rice, some of the important classes of genes involved or implicated in NUE include kinases, transporters, and transcription factors (TFs) (Vidal et al. 2020; Zhang et al. 2020a, 2020b). (iii) Recently, Kumari and Raghuram (2020) and Kumari et al. (2021) compiled a comprehensive list of phosphatases involved in the N-response and/or NUE in crops, and the genes encoding for transporters, TFs, microRNAs, kinases, and phosphatases. They also confirmed differential expression of some promising NUE-candidate genes in a tissue/stage-specific manner through in silico expression analysis (Kumari et al. 2021). The functional studies of many more genes particularly in rice have made it possible to identify orthologues of these genes in complex genomes of related species, including wheat (Han et al. 2016; Zhang et al. 2020a, 2020b). Several crucial genes affecting the NUE of rice have already been reported to have similar functions in wheat, such as TaNFYA-B1, AlaAT, TaASN2-1A, TaGS1.1-4A, TaASN2-1B, TaGDH1-5A, TaNRT2.1, and TaNRT2.4-6A (Qu et al. 2015; Tiong et al. 2021; Karunarathne et al. 2020), which suggests that analysis of homology between related species can be used to select relevant Cgs.
For the present study, 60 rice genes were available (Online Resource 15); 35 of these genes could be utilized for the identification of 49 wheat orthologues in MQTL regions identified during the present study. Of these 35 genes, 18 genes showed more than one wheat orthologue, whereas 17 genes showed only one orthologue in wheat MQTL regions (Table 3). In some cases, orthologues of more than one gene were detected within the same MQTL region. For instance, the three MQTLs, namely MQTL1A.5, 1B.3, and 6D.5 carried orthologues of 8, 5, and 4 different rice genes, respectively. The wheat orthologues of rice genes could be divided into five major categories (Table 3); these are briefly described below:
-
(i)
Genes involved in signaling: Three wheat orthologues (viz., TaSAPK6-1A, 1B, and TaSAPK7-1D) of two rice genes encoding for sucrose non-fermenting-1 related kinases (SnRKs) were identified in MQTL1A.5, 1B.3, and 1D.3 regions, respectively (Table 3). Overexpression of SnRK1 increased carbon absorption and nitrogen uptake in rice (Han et al. 2016). The SnRKs are known to play a major role at the interface of metabolic and stress signaling, implying that they could be manipulated to boost crop performance under diverse conditions (Coello et al. 2011).
-
(ii)
Genes involved in N assimilation: Two wheat orthologues viz., TaNiR1-6B, 6D of a rice nitrite reductase gene were detected in MQTL6B.2 and 6D.5 regions, respectively (Table 3). Overexpression of a ferredoxin-dependent nitrite reductase gene in Arabidopsis elevated NO2 assimilation (Takahashi et al. 2001). In the second phase of the nitrate assimilation pathway, it catalyses the six-electron reduction of nitrite to ammonium. In addition to rice, this gene has also been cloned in various other crop plants (Han et al. 2016), including maize (Lahners et al., 1988) and Arabidopsis (Tanaka et al., 1994). Several genes involved in primary N uptake and assimilation have also been identified as potential bioengineering targets to increase the NUE of important crops (Li et al. 2020).
-
(iii)
Genes involved in amino acid biosynthesis: Twenty-two (22) wheat genes were identified, which are orthologous to 17 rice genes involved in the biosynthesis of different amino acids (Table 3). Of these 22 genes, two genes, TaASN2-1A, and TaASN2-1B have already been functionally characterized in wheat (reviewed in Karunarathne et al. 2020). The orthologues of these genes are key players in driving NUE in important crops and have been studied in some detail in major crops including rice and barley (Beatty et al. 2013; Han et al. 2016; Zhang et al. 2020a, 2020b). Among these genes, the GOGAT gene is found in higher plants in two antigenically distinct forms, namely, NAD(P)H-GOGAT and Fd-GOGAT. The wheat MQTL3B.3 was found to carry TaGOGAT1-3B and MQTL3D.3 was found to carry TaGOGAT3D.3 gene. These enzymes differ in size of the proteins encoded, tissue localization, and physiological roles (Esposito et al. 2005). NAD(P)H-GOGAT is expected to play a major role in primary N assimilation since it is regulated by N-status in the plant in response to available N (Vanoni and Curti, 1999). The deletion of the OsNADH-GOGAT2 gene resulted in a significant reduction in yield and plant biomass in rice (Yamaya, 2011).
-
(iv)
Genes encoding TFs: Wheat MQTLs were also found to carry 9 genes (Table 3) that were orthologous to 6 rice genes encoding TFs. Among these genes, only one gene, TaNAC9-6B (or GPC-B1/NAM-B1) has been functionally characterized in wheat which is known to improve the nutrient remobilization and grain protein content (Uauy et al. 2006). The remaining genes are also important genes involved in N assimilation and their roles in N remobilization and NUE has been demonstrated in several crops (Han et al. 2016; Chiasson et al. 2014).
-
(v)
Genes encoding transporters: A total of 13 wheat orthologues of 9 rice genes encoding different transporters were also identified in different wheat MQTL regions (Table 3). These 13 wheat genes included two ammonium transporter genes (TaAMT1.2/1.3-6B, 6D), 3 wheat nitrate transporter genes (viz., TaNRT2.3-3B, 3D; TaNRT2.4-7B) of the NRT2 family (high-affinity transporters), four wheat genes (TaNPF7.7-1A, 1B, 1D and TaNRT1-4B) of NRT1/NPF family (low affinity transporters) and three NRT2 partner proteins (TaNAR2.1-4A; TaNAR2.2-6B, 6D) (Table 3). In addition, a lysine histidine transporter (i.e., TaLHT1-7A) was detected in MQTL7A.6 region.
In plants, N can be taken up either as ammonium or nitrate directly from the soil through roots. The ammonium transporter genes facilitate the movement of ammonium ions to intracellular compartments, where it is then converted into a variety of organic compounds, including amino acids, via the GS/GOGAT pathway (Oaks, 1994). Nitrate, on the other hand, is primarily carried into the cell by nitrate transporters and then converted to nitrite by nitrate reductase before being reduced to ammonium salts by nitrite reductase (Han et al. 2016; Zhang et al. 2020a, 2020b). Increasing the efficiency of N-related transporters is another biotechnological approach for improving N uptake (Zhang et al. 2020a, 2020b). Since N uptake by different transporters relies on proper carbon skeletons to allow for the synthesis of the various transported molecules, simply increasing the expression of these N-related transporters without proper carbon skeletons would not always result in an increase in NUE in plants (Hawkesford, 2012). This information is useful for the utilization of transporter genes for improving NUE in wheat.
Among the above 49 wheat orthologues occurring in wheat MQTLs, 44 showed significant expressions (>2 TPM) in different tissues. A summary of some wheat genes that were found to be orthologous to rice genes is presented in Fig. 8. The genes encoding transporter proteins, TFs, and signaling molecules involved in signaling were found to exhibit expression in almost all tissues examined, although the highest expression was noticed in roots tissues. This suggests that these genes are involved in regulating both NUpE and NUtE, thus affecting uptake and utilization of N, affecting yield (Han et al. 2016). N assimilation genes exhibited high expression in leaves/shoots tissues, thus matching their role in N assimilation (Masclaux-Daubresse et al. 2010). Genes involved in amino acid biosynthesis showed expression in all the tissues including roots, leaves/shoots, spike organs, and grains suggesting their roles in multiple physiological functions in plants (Masclaux-Daubresse et al. 2010).

Expression patterns (shown in the form of heat maps) of some promising wheat orthologues of known genes in rice (belonging to five major categories each shown outside an arrow) in 11 different tissues including the following: Ra radicle, R root, RAM root apical meristem, AR axillary roots, FL flag leaf, S spike, SP spikelets, G grain, EP embryo proper, L leaf, SH shoots
Conclusions
The meta-QTL analysis provides stable and robust QTLs with relatively narrower CIs, thus improving their utility in MAB. In the present study, we identified 118 MQTLs for NUE and RSA; most of the MQTLs involved QTLs for both NUE and RSA, suggesting a relationship between these two traits. As many as 88 MQTLs including 9 robust major QTLs (described as breeders’ QTLs) were also validated using the results from GWAS. Although, being a complex trait, NUE can not be easily improved using an individual major MQTL, clustered MQTLs identified during the present study should prove useful for MAB. After due validation, ortho-MQTL identified in the present study can also be used across cereals.
As many as 1991 CGs underlying the 112 MQTLs were also identified during the present study; >300 of these CGs showed >5 TPM expressions in relevant plant parts/tissues. Ninety-seven (97) high confidence CGs with functions previously reported as important for studied traits were also listed for 53 robust and GWAS-verified MQTLs. These CGs may be further validated using approaches like overexpression, gene editing, and gene knockout, etc. In addition, 49 orthologues for 35 known rice NUE genes were also identified in MQTL regions, suggesting that the knowledge from rice, the model cereal, can also be utilized for the identification of useful genes in wheat. Overall, the breeder’s MQTLs identified in the present study have the potential for MAB for NUE traits and the CGs are the potential targets that can be exploited using biotechnological techniques like transgenesis and gene editing for improving NUE in wheat.
Availability of data and material
Data generated or analysed during this study are included in this published article (and its Supplementary Material).
References
Aduragbemi A, Soriano JM (2021) Unravelling consensus genomic regions conferring leaf rust resistance in wheat via meta-QTL analysis. bioRxiv. doi:https://doi.org/10.1101/2021.05.11.443557
Ahn S, Anderson JA, Sorrells ME, Tanksley SD (1993) Homoeologous relationships of rice, wheat and maize chromosomes. Mol Gen Genet 241:483–490. https://doi.org/10.1007/BF00279889
Arcade A, Labourdette A, Falque M, Mangin B, Chardon F, Charcosset A, Joets J (2004) BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics 20:2324–2326. https://doi.org/10.1093/bioinformatics/bth230
Balyan HS, Gahlaut V, Kumar A, Jaiswal V, Dhariwal R, Tyagi S, Agarwal P, Kumari S, Gupta PK (2016) Nitrogen and phosphorus use efficiencies in wheat: physiology, phenotyping, genetics, and breeding. Plant Breed Rev 40:167–234. https://doi.org/10.1002/9781119279723.ch4
Beatty PH, Carroll RT, Shrawat AK, Guevara D, Good AG (2013) Physiological analysis of nitrogen-efficient rice overexpressing alanine aminotransferase under different N regimes. Botany 91:866–883
Bennetzen JL, Chen M (2008) Grass genomic synteny illuminates plant genome function and evolution. Rice 1:109–118. https://doi.org/10.1007/s12284-008-9015-6
Bi YM, Kant S, Clark J, Gidda S, Ming F, Xu J, Rochon A, Shelp BJ, Hao L, Zhao R, Mullen RT (2009) Increased nitrogen-use efficiency in transgenic rice plants over-expressing a nitrogen-responsive early nodulin gene identified from rice expression profiling. Plant Cell Environ 32:1749–1760
Brasier K, Ward B, Smith J, Seago J, Oakes J, Balota M, Davis P, Fountain M, Brown-Guedira G, Sneller C, Thomason W (2020) Identification of quantitative trait loci associated with nitrogen use efficiency in winter wheat. PLoS ONE 15:e0228775. https://doi.org/10.1371/journal.pone.0228775
Chiasson DM, Loughlin PC, Mazurkiewicz D, Mohammadidehcheshmeh M, Fedorova EE, Okamoto M, McLean E, Glass AD, Smith SE, Bisseling T, Tyerman SD (2014) Soybean SAT1 (Symbiotic Ammonium Transporter 1) encodes a bHLH transcription factor involved in nodule growth and NH4+ transport. Proc Natl Acad Sci 111:4814–4819. https://doi.org/10.1073/pnas.1312801111
Cirilo AG, Dardanelli J, Balzarini M, Andrade FH, Cantarero M, Luque S, Pedrol HM (2009) Morpho-physiological traits associated with maize crop adaptations to environments differing in nitrogen availability. Field Crops Res 113:116–124. https://doi.org/10.1016/j.fcr.2009.04.011
Cobb JN, Biswas PS, Platten JD (2019) Back to the future: revisiting MAS as a tool for modern plant breeding. Theor Appl Genet 132:647–667. https://doi.org/10.1007/s00122-018-3266-4
Coello P, Hey SJ, Halford NG (2011) The sucrose non-fermenting-1-related (SnRK) family of protein kinases: potential for manipulation to improve stress tolerance and increase yield. J Exp Bot 62:883–893. https://doi.org/10.1093/jxb/erq331
Collard BCY, Mackill DJ (2008) Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos Trans R Soc Lond B Biol Sci 363:557–572
Dalton DA, Boniface C, Turner Z, Lindahl A, Kim HJ, Jelinek L, Govindarajulu M, Finger RE, Taylor CG (2009) Physiological roles of glutathione S-transferases in soybean root nodules. Plant Physiol 150:521–530
Darvasi A, Soller M (1997) A simple method to calculate resolving power and confidence interval of QTL map location. Behav Genet 27:125–132. https://doi.org/10.1023/a:1025685324830
Darzi-Ramandi H, Shariati J V, Tavakol E, Najafi-Zarini H, Bilgrami SS, Razavi K (2017) Detection of consensus genomic regions associated with root architecture of bread wheat on groups 2 and 3 chromosomes using QTL meta–analysis. Aust J Crop Sci 777–785.
Do THT, Martinoia E, LeeY, (2018) Functions of ABC transporters in plant growth and development. Curr Opin Plant Biol 41:32–38
Dong NQ, Sun Y, Guo T, Shi CL, Zhang YM, Kan Y, Xiang YH, Zhang H, Yang YB, Li YC, Zhao HY (2020) UDP-glucosyltransferase regulates grain size and abiotic stress tolerance associated with metabolic flux redirection in rice. Nat Commun 11:1–16
Endelman JB, Plomion C (2014) LPmerge: an R package for merging genetic maps by linear programming. Bioinformatics 30:1623–1624. https://doi.org/10.1093/bioinformatics/btu091
Esposito S, Guerriero G, Vona V, Di Martino RV, Carfagna S, Rigano C (2005) Glutamate synthase activities and protein changes in relation to nitrogen nutrition in barley: the dependence on different plastidic glucose-6P dehydrogenase isoforms. J Exp Bot 56:55–64
Fan X, Cui F, Ji J, Zhang W, Zhao X, Liu J, Meng D, Tong Y, Wang T, Li J (2019) Dissection of pleiotropic QTL regions controlling wheat spike characteristics under different nitrogen treatments using traditional and conditional QTL mapping. Front Plant Sci 10:187. https://doi.org/10.3389/fpls.2019.00187
Forde BG (2014) Nitrogen signaling pathways shaping root system architecture: an update. Curr Opin Plant Biol 21:30–36
Garnett T, Conn V, Kaiser BN (2009) Root based approaches to improving nitrogen use efficiency in plants. Plant Cell Environ 32:1272–1283. https://doi.org/10.1111/j.1365-3040.2009.02011.x
Gaut BS (2002) Evolutionary dynamics of grass genomes. New Phytol 154:15–28
Goffinet B, Gerber S (2000) Quantitative trait loci: a meta-analysis. Genetics 155:463–473
Good AG, Shrawat AK, Muench DG (2004) Can less yield more? Is reducing nutrient input into the environment compatible with maintaining crop production? Trends Plant Sci 9:597–605. https://doi.org/10.1016/j.tplants.2004.10.008
Good AG, Beatty PH (2011) Biotechnological approaches to improving nitrogen use efficiency in plants: alanine aminotransferase as a case study. In: Malcolm JH, Peter B (ed) The molecular and physiological basis of nutrient use efficiency in crops. Wiley, New York, pp 165–191. doi:https://doi.org/10.1002/9780470960707
Greef JM (1994) Productivity of maize (Zea mays L.) in relation to morphological and physiological characteristics under varying amounts of nitrogen supply. J Agron Crop Sci 172:317–326
Guo B, Sleper DA, Lu P, Shannon JG, Nguyen HT, Arelli PR (2006) QTLs associated with resistance to soybean cyst nematode in soybean: meta-analysis of QTL locations. Crop Sci 46:595–602
Guo J, Chen L, Li Y, Shi Y, Song Y, Zhang D, Li Y, Wang T, Yang D, Li C (2018) Meta-QTL analysis and identification of candidate genes related to root traits in maize. Euphytica 214:223. https://doi.org/10.1007/s10681-018-2283-3
Han M, Wong J, Su T, Beatty PH, Good AG (2016) Identification of nitrogen use efficiency genes in barley: searching for QTLs controlling complex physiological traits. Front Plant Sci 7:1587
Han G, Lu C, Guo J, Qiao Z, Sui N, Qiu N, Wang B (2020) C2H2 zinc finger proteins: master regulators of abiotic stress responses in plants. Front Plant Sci 11:115. https://doi.org/10.3389/fpls.2020.00115
Hawkesford MJ (2012) Improving nutrient use efficiency in crops. eLS. doi:https://doi.org/10.1002/9780470015902.a0023734
He X, Qu B, Li W, Zhao X, Teng W, Ma W, Ren Y, Li B, Li Z, Tong Y (2015) The nitrate-inducible NAC transcription factor TaNAC2-5A controls nitrate response and increases wheat yield. Plant Physiol 169:1991–2005
Hirel B, Le Gouis J, Ney B, Gallais A (2007) The challenge of improving nitrogen use efficiency in crop plants: towards a more central role for genetic variability and quantitative genetics within integrated approaches. J Exp Bot 58:2369–2387
Jan I, Saripalli G, Kumar K, Kumar A, Singh R, Batra R, Sharma PK, Balyan HS, Gupta PK (2021) Meta-QTL analysis for stripe rust resistance in wheat. doi:https://doi.org/10.21203/rs.3.rs-380807/v1
Jun XU, Wang XY, Guo WZ (2015) The cytochrome P450 superfamily: key players in plant development and defense. J Integr Agr 14:1673–1686
Karunarathne SD, Han Y, Zhang XQ, Li C (2020) Advances in understanding the molecular mechanisms and potential genetic improvement for nitrogen use efficiency in barley. Agronomy 10:662. https://doi.org/10.3390/agronomy10050662
Khahani B, Tavakol E, Shariati V, Fornara F (2020) Genome wide screening and comparative genome analysis for Meta-QTLs, ortho-MQTLs and candidate genes controlling yield and yield-related traits in rice. BMC Genom 21:294. https://doi.org/10.1186/s12864-020-6702-1
Khahani B, Tavakol E, Shariati V, Rossini L (2021) Meta-QTL and ortho-MQTL analyses identified genomic regions controlling rice yield, yield-related traits and root architecture under water deficit conditions. Sci Rep 11:6942. https://doi.org/10.1038/s41598-021-86259-2
Kumar S, Mohan A, Balyan HS, Gupta PK (2009) Orthology between genomes of Brachypodium, wheat and rice. BMC Res Notes 2:1–9
Kumar IS, Nadarajah K (2020) A meta-analysis of quantitative trait loci associated with multiple disease resistance in rice (Oryza sativa L.). Plants 9:1491. doi:https://doi.org/10.3390/plants9111491
Kumari S, Raghuram N (2020) Protein Phosphatases in N Response and NUE in Crops. In: Pandey G (ed) Protein phosphatases and stress management in plants. Springer, Switzerland, pp 233–244
Kumari S, Sharma N, Raghuram N (2021) Meta-analysis of yield-related and N-responsive genes reveals chromosomal hotspots, key processes and candidate genes for nitrogen-use efficiency in rice. Front Plant Sci 12:627955
Lahners K, Kramer V, Back E, Privalle L, Rothstein S (1988) Molecular cloning of complementary DNA encoding maize nitrite reductase: molecular analysis and nitrate induction. Plant Physiol 88:741–746
Li P, Chen F, Cai H, Liu J, Pan Q, Liu Z, Gu R, Mi G, Zhang F, Yuan L (2015) A genetic relationship between nitrogen use efficiency and seedling root traits in maize as revealed by QTL analysis. J Exp Bot 66:3175–3188. https://doi.org/10.1093/jxb/erv127
Li X, Zeng R, Liao H (2016) Improving crop nutrient efficiency through root architecture modifications. J Integr Plant Biol 58:193–202
Li M, Xu J, Gao Z, Tian H, Gao Y, Kariman K (2020) Genetically modified crops are superior in their nitrogen use efficiency: a meta-analysis of three major cereals. Sci Rep 10:1–9
Liu R, Zhang H, Zhao P, Zhang Z, Liang W, Tian Z, Zheng Y (2012) Mining of candidate maize genes for nitrogen use efficiency by integrating gene expression and QTL data. Plant Mol Biol Rep 30:297–308. https://doi.org/10.1007/s11105-011-0346-x
López-Bucio J, Cruz-Ramırez A, Herrera-Estrella L (2003) The role of nutrient availability in regulating root architecture. Curr Opin Plant Biol 6:280–287
Marone D, Russo MA, Laidò G, De Vita P, Papa R, Blanco A, Gadaleta A, Rubiales D, Mastrangelo AM (2013) Genetic basis of qualitative and quantitative resistance to powdery mildew in wheat: from consensus regions to candidate genes. BMC Genom 14:562. https://doi.org/10.1186/1471-2164-14-562
Marowa P, Ding A, Kong Y (2016) Expansins: roles in plant growth and potential applications in crop improvement. Plant Cell Rep 35:949–965
Masclaux-Daubresse C, Daniel-Vedele F, Dechorgnat J, Chardon F, Gaufichon L, Suzuki A (2010) Nitrogen uptake, assimilation and remobilization in plants: challenges for sustainable and productive agriculture. Ann Bot 105:1141–1157
Meijón M, Satbhai SB, Tsuchimatsu T, Busch W (2014) Genome-wide association study using cellular traits identifies a new regulator of root development in Arabidopsis. Nat Genet 46:77–81. https://doi.org/10.1038/ng.2824
Meister R, Rajani MS, Schachtman RD, DP, (2014) Challenges of modifying root traits in crops for agriculture. Trends Plant Sci 19:779–788
Minic Z (2008) Physiological roles of plant glycoside hydrolases. Planta 227:723–740
Misztal I (2006) Challenges of application of marker assisted selection: a review. Anim Sci Pap Rep 24:5–10
Moll RH, Kamprath EJ, Jackson WA (1982) Analysis and interpretation of factors which contribute to efficiency of nitrogen utilization1. Agron J 74:562. https://doi.org/10.2134/agronj1982.00021962007400030037x
Mosleth EF, Wan Y, Lysenko A, Chope GA, Penson SP, Shewry PR, Hawkesford MJ (2015) A novel approach to identify genes that determine grain protein deviation in cereals. Plant Biotechnol J 13:625–635
Oaks A (1994) Primary nitrogen assimilation in higher plants and its regulation. Can J Bot 72:739–750
de Oliveira Y, Sosnowski O, Charcosset A, Joets J (2014) BioMercator 4: a complete framework to integrate QTL, meta-QTL, and genome annotation. In: European Conference on Computational Biology 2014 Sep 2014, Strasbourg, France
Qu B, He X, Wang J, Zhao Y, Teng W, Shao A, Zhao X, Ma W, Wang J, Li B, Li Z (2015) A wheat CCAAT box-binding transcription factor increases the grain yield of wheat with less fertilizer input. Plant Physiol 167:411–423
Quraishi UM, Abrouk M, Bolot S, Pont C, Throude M, Guilhot N, Confolent C, Bortolini F, Praud S, Murigneux A, Charmet G (2009) Genomics in cereals: from genome-wide conserved orthologous set (COS) sequences to candidate genes for trait dissection. Funct Integr Genom 9:473–484
Quraishi UM, Abrouk M, Murat F, Pont C, Foucrier S, Desmaizieres G, Confolent C, Riviere N, Charmet G, Paux E, Murigneux A (2011) Cross-genome map based dissection of a nitrogen use efficiency ortho-metaQTL in bread wheat unravels concerted cereal genome evolution. Plant J 65:745–756. https://doi.org/10.1111/j.1365-313X.2010.04461.x
Ramírez-González RH, Borrill P, Lang D, Harrington SA, Brinton J, Venturini L, Davey M, Jacobs J, Van Ex F, Pasha A, Khedikar Y (2018) The transcriptional landscape of polyploid wheat. Science. https://doi.org/10.1126/science.aar6089
Raun WR, Johnson GV (1999) Improving nitrogen use efficiency for cereal production. Agronomy 91:357–363
Saini DK, Srivastava P, Pal N, Gupta PK (2021) Meta-QTLs, ortho-metaQTLs and candidate genes for grain yield and associated traits in wheat (Triticum aestivum L.). doi:https://doi.org/10.21203/rs.3.rs-430452/v1
Salarpour M, Pakniyat H, Abdolshahi R, Heidari B, Razi H, Afzali R (2020) Mapping QTL for agronomic and root traits in the Kukri/RAC875 wheat (Triticum aestivum L.) population under drought stress conditions. Euphytica 216:1–19
Singh K, Batra R, Sharma S, Saripalli G, Gautam T, Singh R, Pal S, Malik P, Kumar M, Jan I, Singh S (2021) WheatQTLdb: a QTL database for wheat. Mol Genet Genom. https://doi.org/10.1007/s00438-021-01796-9
Somers DJ, Isaac P, Edwards K (2004) A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.). Theor Appl Genet 109:1105–1114. https://doi.org/10.1007/s00122-004-1740-7
Soriano JM, Alvaro F (2019) Discovering consensus genomic regions in wheat for root-related traits by QTL meta-analysis. Sci Rep 9:10537. https://doi.org/10.1038/s41598-019-47038-2
Sosnowski O, Charcosset A, Joets J (2012) BioMercator V3: an upgrade of genetic map compilation and quantitative trait loci meta-analysis algorithms. Bioinformatics 28:2082–2083. https://doi.org/10.1093/bioinformatics/bts313
Sun JJ, Guo Y, Zhang GZ, Gao MG, Zhang GH, Kong FM, Zhao Y, Li SS (2013) QTL mapping for seedling traits under different nitrogen forms in wheat. Euphytica 191:317–331. https://doi.org/10.1007/s10681-012-0834-6
Sun Y, Hu Z, Wang X, Shen X, Hu S, Yan Y, Kant S, Xu G, Xue Y, Sun S (2021) Overexpression of OsPHR3 improves growth traits and facilitates nitrogen use efficiency under low phosphate condition. Plant Physiol Biochem 166:712–722
Takahashi M, Sasaki Y, Ida S, Morikawa H (2001) Nitrite reductase gene enrichment improves assimilation of NO2 in Arabidopsis. Plant Physiol 126:731–741
Tanaka S, Ida S, Irifune K, Oeda K, Morikawa H (1994) Nucleotide sequence of a gene for nitrite reductase from Arabidopsis thaliana. DNA Seq 5:57–61
Tiong J, Sharma N, Sampath R, MacKenzie N, Watanabe S, Metot C, Lu Z, Skinner W, Lu Y, Kridl J, Baumann U (2021) Improving nitrogen use efficiency through overexpression of alanine aminotransferase in rice, wheat, and barley. Front Plant Sci 12:29
Uauy C, Distelfeld A, Fahima T, Blechl A, Dubcovsky J (2006) A NAC gene regulating senescence improves grain protein, zinc, and iron content in wheat. Science 314:1298–1301
Vanoni MA, Curti B (1999) Glutamate synthase: a complex iron-sulfur flavoprotein. Cell Mol Life Sci 55:617–638
Venske E, Dos Santos RS, Farias DD, Rother V, da Maia LC, Pegoraro C, Costa de Oliveira A (2019) Meta-analysis of the QTLome of fusarium head blight resistance in bread wheat: Refining the current puzzle. Front Plant Sci 10:727. https://doi.org/10.3389/fpls.2019.00727
Veyrieras J-B, Goffinet B, Charcosset A (2007) MetaQTL: a package of new computational methods for the meta-analysis of QTL mapping experiments. BMC Bioinform 8:49. https://doi.org/10.1186/1471-2105-8-49
Vidal EA, Alvarez JM, Araus V, Riveras E, Brooks MD, Krouk G, Ruffel S, Lejay L, Crawford NM, Coruzzi GM, Gutiérrez RA (2020) Nitrate in 2020: thirty years from transport to signaling networks. Plant Cell 32:2094–2119
Wagner GP, Kin K, Lynch VJ (2013) A model based criterion for gene expression calls using RNA-seq data. Theory Biosci 132:159–164. https://doi.org/10.1007/s12064-013-0178-3
Xiang J, Qian K, Zhang Y, Chew J, Liang J, Zhu J, Zhang Y, Fan X (2021) OsLSD1.1 is involved in the photosystem II reaction and affects nitrogen allocation in rice. Plant Physiol Biochem. https://doi.org/10.1016/j.plaphy.2021.06.004
Xu J, Wang X, Guo W (2015) The cytochrome P450 superfamily: Key players in plant development and defense. J Integr Agric 14:1673–1686. https://doi.org/10.1016/S2095-3119(14)60980-1
Yamaya T (2011) Disruption of a novel NADH-glutamate synthase2 gene caused marked reduction in spikelet number of rice. Front Plant Sci 2:57
Yang X, Xia X, Zeng Y, Nong B, Zhang Z, Wu Y, Tian Q, Zeng W, Gao J, Zhou W, Liang H (2020) Genome-wide identification of the peptide transporter family in rice and analysis of the PTR expression modulation in two near-isogenic lines with different nitrogen use efficiency. BMC Plant Biol 20:193. https://doi.org/10.1186/s12870-020-02419-y
Yang Y, Amo A, Wei D, Chai Y, Zheng J, Qiao P, Cui C, Lu S, Chen L, Hu YG (2021) Large-scale integration of meta-QTL and genome-wide association study discovers the genomic regions and candidate genes for yield and yield-related traits in bread wheat. Theor Appl Genet 134:1–27
Yu J, Xuan W, Tian Y, Fan L, Sun J, Tang W, Chen G, Wang B, Liu Y, Wu W, Liu X (2021) Enhanced OsNLP4-OsNiR cascade confers nitrogen use efficiency by promoting tiller number in rice. Plant Biotechnol J 19:167–176
Zhang Y, Yu C, Lin J, Liu J, Liu B, Wang J, Huang A, Li H, Zhao T (2017) OsMPH1 regulates plant height and improves grain yield in rice. PLoS ONE 12:e0180825. https://doi.org/10.1371/journal.pone.0180825
Zhang N, Zhang X, Song L, Su Q, Zhang S, Liu J, Zhang W, Fu X, Zhao M, Sun L, Ji J (2020a) Identification and validation of the superior alleles for wheat kernel traits detected by genome-wide association study under different nitrogen environments. Euphytica 216:1–15
Zhang Z, Gao S, Chu C (2020b) Improvement of nutrient use efficiency in rice: current toolbox and future perspectives. Theor Appl Genet 133:1365–1384
Zhao X, Peng Y, Zhang J, Fang P, Wu B (2018) Identification of QTLs and meta-QTLs for seven agronomic traits in multiple maize populations under well-watered and water-stressed conditions. Crop Sci 58:507–520
Acknowledgements
The Department of Science and Technology (DST) in New Delhi, India, provided DKS with an INSPIRE fellowship, while the Head, Department of Plant Breeding and Genetics, Punjab Agricultural University in Ludhiana, India, provided the essential facilities.
Author information
Authors and Affiliations
Contributions
PKG and PS conceived and planned the study. DKS, YC, NP and AC collected the literature and tabulated the data for meta-QTL analysis. DKS conducted the analysis. DKS and YC interpreted the results and wrote the manuscript. PKG and PS edited and finalized the manuscript with the help of DKS.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Saini, D.K., Chopra, Y., Pal, N. et al. Meta-QTLs, ortho-MQTLs and candidate genes for nitrogen use efficiency and root system architecture in bread wheat (Triticum aestivum L.). Physiol Mol Biol Plants 27, 2245–2267 (2021). https://doi.org/10.1007/s12298-021-01085-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12298-021-01085-0