
Abstract
The plant-specific NAC transcription factor (TFs) plays crucial role in plant growth as well as in stress resistance. In the present study, 87 Zea mays NAC TFs were obtained from the transcriptome analysis using drought-resistant maize inbred line Y882 as experimental material under PEG stress and rewatering treatment. Comprehensive analyses were conducted including genes structure, chromosomal localization, phylogenetic tree and motif prediction, cis-elements and expression patterns. The results showed that the 87 ZmNAC genes distributed on 10 chromosomes and were categorized into 15 groups based on their conserved gene structure and motifs. Phylogenetic tree analysis was also constructed referencing to the counterparts of Arabidopsis and rice, and the stress-related cis-elements in the promoter region were also analyzed. 87 ZmNAC genes exhibited different expression levels at 3 treatment points, indicating different response to drought stress. This genome-wide analysis of 87 ZmNAC genes will provide basis for further gene function detection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Maize is one of the most important food crop in China with strong adaptability, high economic value and widely utilization. In China, especially in the north and north–west region, the water resources are very limited and drought stresses with different degrees occur every year. Under drought stress, plants undergo various biochemical changes in gene or protein levels to adapt against adverse environmental conditions (Fang et al. 2015). If water stress does not exceed a certain threshold, then plants can actively produce a compensation mechanism to cope with water shortage (Shan 2003). Studies show that the recovery ability of plants after rewatering and drought resistance is more important than drought tolerance under drought stress (Kamoshita et al. 2004). The response of maize plants to rewatering after drought stress is the rapid growth (Bu et al. 2009), and many genes are involved in the process.
In regulating processes of plants, the expression of stress-related genes are largely governed by specific transcription factors. Research findings show that numerous transcription factors (TFs) play an essential roles in improving plant tolerance to abiotic stress (Puranik et al. 2012; Wang and Dane 2013). NAC (NAM, ATAF1,2 and CUC2) transcription factors belong to the plant-specific transcription factor family and functional studies demonstrate that NAC TFs involve in responses to drought, salinity, and cold stresses (Borrill et al. 2017; Kadier et al. 2017; Kou et al. 2014; Saidi et al. 2017; Sun et al. 2018). In Arabidopsis, ATAF1 gene was induced by drought stress and ABA treatment. Overexpression of ATAF1 in transgenic Arabidopsis plants resulted in enhanced drought tolerance (Wu et al. 2009). Overexpression of TaNAC69 in transgenic wheat improved dehydration tolerance and enhanced the expression levels of genes up-regulated under stress (Xue et al. 2011). Another wheat NAC transcription factor TaNAC29 was involved in response to drought, salt and ABA treatments (Xu et al. 2015). Overexpression of SNAC3 in rice showed an enhanced plant tolerance to high temperature and drought, whereas suppression of SNAC3 by RNAi exhibited increased sensitivity to these stresses (Fang et al. 2015).
NAC transcription factors have been well studied in various species such as Arabidopsis (Capella et al. 2014), rice (Nuruzzaman et al. 2010), soybean (Dung Tien et al. 2011) and wheat (Borrill et al. 2017), but only a few NAC members have been analyzed in maize. ZmSNAC1 and ZmNAC55 are strongly induced by drought, cold, and ABA treatments, overexpression of ZmSNAC1 and ZmNAC55 in Arabidopsis induce enhanced drought tolerance, respectively (Lu et al. 2012; Mao et al. 2016). Based on the maize genome sequences from relative database, 148 and 128 maize NAC members were identified, respectively (Ge et al. 2015; Peng et al. 2015), Shiriga identified 152 NAC TFs in maize and selected 11 NAC genes for expressing analysis between a drought-tolerant genotype and a susceptible genotype during drought stress (Shiriga et al. 2014).
In the present study, we used 20% PEG6000 to simulate drought stress and carried out a transcriptome analysis using a drought-resistant maize inbreed line Y882 under PEG stress and rewatering treatment. The genes structure, chromosomal localization, gene ontology, phylogenetic tree and motif prediction, cis-elements and expression patterns were investigated. This study provides the basis for further function detection of NAC genes.
Materials and methods
Plant growth and stress treatments
Maize inbred line Y882 was used in this study. The initial experiment indicated that Y882 was a drought-resistant line (data unpublished). Seeds were surface sterilized and germinated in an incubator for 24 h at 28 °C. Seedlings were grown in a greenhouse with 14 h/10 h light/dark photoperiod, 60% relative humidity, and light intensity of 120 μmol m−2 s−1. Seedlings were grown in half-strength modified Hoagland’s nutrient solution (pH 5.8), which was refreshed every 3 days. Seedlings at the 3-fully expanded leaf stage were transferred to nutrient solution containing 20% polyethylene glycol PEG-6000 for stress treatment. Leaves were harvested at 60 and 96 h of the drought treatment and rewatered at 3 d (denoted as T60, T96 and TR3d, respectively). Control seedlings were grown under the same conditions. Three plants from three different containers of each treatment were used as biological replicates. All samples were immediately frozen in liquid nitrogen and stored at −80 °C.
RNA extraction, cDNA library construction and transcriptome sequencing
Total RNA was extracted using TRIzol Reagent (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. The concentration and quality of the RNA were verified using an Agilent 2100 Bioanalyzer (Agilent Technologies, USA) and NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, USA). The construction of cDNA library and transcriptome sequencing was performed by Genedenovo Technology Company in Guangzhou, China. Equal amounts of total RNA extracted from the three replicate plants at each treatment point to construct the cDNA library using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® by protocols. Double-stranded cDNAs were synthesized using the reverse-transcriptase and random hexamer primers. The cDNA fragments were purified using QIA quick PCR kit and washed with EB buffer. Following the reparation of poly (A) addition and ligated to sequencing adapters, the fragments with the expected sizes were purified by agarose gel electrophoresis and enriched by PCR to construct the cDNA library. The cDNA library was sequenced on the Illumina sequencing platform (Illumina HiSeq™2500) using the paired-end technology.
Reads obtained from the sequencing machines were filtered and mapped to ribosome RNA database using short reads alignment tool Bowtie2 (Langmead and Salzberg 2012). The tools used for read alignment and expression quantification were TopHat2 (Kim et al. 2013) and Cufflinks (Trapnell et al. 2012), respectively. The gene expression level was normalized by using fragments per kilobase of transcript per million mapped reads (FPKM).
Identification of ZmNAC genes and differential gene expression (DEG) analysis
ZmNAC genes were identified from the transcriptome data and the whole-genomic sequence and gene localization information were downloaded from the Ensembl Plants database (http://plants.ensembl.org/index.html). The NAC domains were screened using Plant Transcription Factor Database v5.0 (http://planttfdb.cbi.pku.edu.cn/download.php). The chromosome location of NAC transcription factors were analyzed using MG2C software (http://mg2c.iask.in/mg2c_v2.0/?tdsourcetag=s_pcqq_aiomsg). The level of NAC genes expression was normalized by calculating the Fragments Per Kilobase of transcript per Million mapped reads (FPKM). The differentially expressed genes (DEGs) were identified with |log2 (fold change)| ≥ 1 and FDR value < 0.05. Gene Ontology (Go) analysis was also conducted for gene functional classification.
Phylogenetic analysis and motif prediction
The phylogenetic tree was constructed in MEGA 6.0 using the neighbor-joining method (Bootstrap method: 1000), according to the classification method of NAC TFs in Arabidopsis and rice (Xu et al. 2015). The protein sequences of ZmNAC genes were downloaded from the Ensembl Plants database (http://plants.ensembl.org/index.html). The online MEME software was used to analyze the conserved motifs (http://meme-suite.org/) according to the default program, the maximum number of motif was 20.
cis-Elements analysis in the promoter regions of ZmNAC genes
Upstream regions (− 2000 bp) of ZmNAC genes were selected for cis-elements analysis using Plant CARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/). Five cis-elements were selected in this study including ABRE, MBS, TC-rich, CGTCA and DRE.
qRT-PCR analysis
qRT-PCR was performed on CFX96 real-time PCR (Bio-Rad). The specific primers were designed according to the gene sequence and listed in Table S1. Action 18S (GenBank accession number: AF168884.1) was used as an internal control. Three technical replicates were analyzed for each gene. The relative expression levels of the candidate genes were calculated using the 2−ΔΔCt method (Livak and Schmittgen 2001).
Results
Identification of NAC family members in maize under PEG stress and rewatering
We screened the drought-rewatering transcriptome data and totally obtained 147 ZmNAC genes; then we eliminated 40 NAC genes from the total 147 ZmNAC genes, because the FPKM value of the 40 ZmNAC genes was zero, respectively, indicating that they were not responsive to drought stress and rewatering at any of three treatment point, and finally a total of 87 ZmNAC genes were identified. Comparing with the Plant Transcription Factor Database (V5.0), the coverage was 66%. The relevant biological information parameters such as protein sequences, Molecular weight, PI value, the conserved domain of 87 ZmNAC genes were downloaded from the Ensembl Plants and Plant TFDB v5.0 website and listed in Table 1. The proteins encoded by 87 ZmNAC genes ranged from 171 to 1400 amino acid (aa) residues in length, with the average 395 aa, the molecular weight (MW) varied from 19.9012 to 155.9226 kDa, the Isoelectric point (PI) ranged from 4.18 to 12.01.
The Gene Ontology (GO) analysis was conducted to predict the functions of proteins encoded by ZmNAC genes. The gene products were grouped into molecular function, biological process and cell component categories (Fig. 1). Comparing with the molecular function and cell component category, most of the 87 ZmNAC genes were involved in the regulation of biological process, such as the regulation of gene expression (GO:0010468), biosynthetic process (GO 0009058) and regulation of metabolic process (GO: 0050789).

Gene ontology (GO) analysis of ZmNAC genes. The green, red, and blue columns represent the molecular function, biological process and cellular component, respectively (colour figure online)
Chromosomal locations and phylogenetic analysis
87 ZmNAC genes were distributed unevenly on the ten chromosomes of maize. There were eleven ZmNAC genes mapped on Chromosome 2 and 4, respectively, which was the largest number, followed by ten genes on chromosome 3, nine genes on chromosome 6 and 1, and eight genes on chromosome 7, 8, and 9, respectively; seven genes on chromosome 5, only six genes on chromosome 10. These genes were present in different regions of the chromosomes, most of them were located on both telomeric ends (Fig. 2). Most of the genes on chromosome 2 were positioned on the short arm, while ZmNAC genes on chromosome 3 and 10 were located on the long arms.

Distributions of 87 ZmNAC genes on the 10 chromosomes of maize
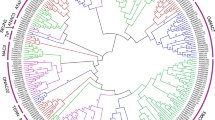
Using MEGA6.0 and the NJ method (Bootstrap = 1000), a phylogenetic tree was constructed, some of NAC genes selected from Arabidopsis and rice as reference. 87 NAC proteins were divided into 12 sub-groups, except ZmNAC63, ZmNAC79, ZmNAC99, ZmNAC134 and ZmNAC94, such as NAM, OSNAC7, TIP, ONAC022, ATAF, SEN5, ONAC003, NAC1, ANAC011, NAC2, ANAC104 and AtNAC3 (Fig. 3), the sequence alignment of each sub-group was listed in Figure S1. Each group had different members, the biggest sub-group was ONAC022, including 16 ZmNAC genes, followed by ATAF, NAC1 and ONAC003. ANAC011 sub-family contained five members, they were ZmNAC57, ZmNAC78, ZmNAC18, ZmNAC28 and ZmNAC43; the sub-group NAC2 had ZmNAC112, ZmNAC38, ZmNAC120 and ZmNAC26 four members. ZmNAC63 and ZmNAC79 was divided into one group, but we could not find the homologous corresponded to Arabidopsis and rice, the same with ZmNAC99, ZmNAC134 and ZmNAC94.

Evolutionary relationship of 87 ZmNAC genes and some homologous proteins in Arabidopsis thaliana and rice
Gene structure and motifs identification of ZmNAC genes
The number of introns and exons of 87 ZmNAC genes revealed significant diversity (Fig. 4). The same sub-group members had the same or similar gene structures. The members in NAC1 group had three exons; most of members had more than 4 exons in ANAC011 and ONAC003 sub-group. Most of NAC genes had 5′ or 3′ UTR region, except ZmNAC79, ZmNAC103, ZmNAC46, ZmNAC41, ZmNAC31, ZmNAC134, ZmNAC94, ZmNAC8 and ZmNAC56. ZmNAC41, ZmNAC134 and ZmNAC 94 had no introns, accounting for 3.44%, 14 ZmNAC genes only had one introns, accounting for 16.10%, 45 ZmNAC genes had two introns, accounting for 51.72%, 7 ZmNAC genes contained three introns, and 16 ZmNAC genes contained 4-12 introns.

Exon-intron structure analyses of 87 ZmNAC genes (colour figure online)
To further identify the diversity of 87 ZmNAC genes, putative motifs were predicted using MEME software (http://meme-suite.org/tools/meme). There was no motifs predicted in ZmNAC79 and ZmNAC63, and the motifs of 85 ZmNAC genes were exhibited in Fig. 5. We could find that the motif pattern was clustered in the same way as the sub-families pattern, and the same clusters had the similar motifs compositions, indicating that the phylogenetic analysis results were accurate. Motif 1-6, and 11 were the common element in most of sub-families, except ZmNAC99 and ONAC003 sub-group. Motif 17 only appeared in ZmNAC57, ZmNAC112, ZmNAC38 and ZmNAC3; Motif 20 appeared in ZmNAC117, ZmNAC77 and ZmNAC61; Motif 16 appeared in ZmNAC60, ZmNAC49, ZmNAC45 and ZmNAC125. Motif 7, 8, 9, 10, 12, 16 and 18 were appeared in ONAC003 sub-group members, and ZmNAC99 was specific and only had six motifs.

Motif distributions of 87 ZmNAC genes (colour figure online)
Stress-related Cis-elements analysis in promoters of ZmNAC genes
In order to better understand the potential regulatory mechanisms of ZmNAC genes under PEG treatment and rewatering, we selected five cis-element ABRE, MBS, TC-rich, CGTCA, DRE and scanned the promoter regions (− 2000 bp upstream of the translation start site) of 87 ZmNAC genes, finally 39 ZmNAC genes were listed and the result was shown in Fig. 6. ABRE is the element with the most frequency and was found in 37 selected promoter regions among 39 ZmNAC genes; sixteen ABRE elements were found in ZmNAC49, eight ABRE were found in ZmNAC43 and ZmNAC54, respectively. TC-rich cis- acting element was involved in defense and stress responsiveness, and was found in 14 selected ZmNAC genes, three were found in ZmNAC78. MBS was MYB binding site involved in drought-induced ability, and was found in 21 selected ZmNAC genes, five were found in ZmNAC16 and ZmNAC112, respectively. The DRE element was specific induced under drought and osmotic stress and only was found in ZmNAC77. The CGTCA element was found in 34 selected gene promoter regions such as ZmNAC42, ZmNAC17 and ZmNAC21.

cis-elements predication in the promoter regions of ZmNAC genes (colour figure online)
Digital expression patterns of ZmNAC genes under PEG stress and rewatering
The expression patterns of 87 ZmNAC genes were different at 60 h, 96 h and 3 d treatment point (Fig. 7). At 60 h treatment point, the number of differentially expressed genes (DEGs) was 18, 12 genes were up-regulated and 6 genes were down-regulated; at 96 h point, the number of DEGs was 11, 5 genes were up-regulated and 6 genes were down-regulated; there were 3 up-regulated genes and 28 down-regulated genes at 3 d point. We also found that the number of up-regulated genes was decreasing, and the number of down-regulated genes was increasing, especially at 3 d point, indicating that the gene expression were more stimulated by rewatering after drought stress. There were 4, 5 and 6 same differentially expressed genes between CK60-vs-T60, CK96-vs-T96 and CK3d-vs-T3d, respectively, and the expression level reaching the significant level at the three treatment point were ZmNAC36 and ZmNAC93.

Statistical analyses of differentially expressed ZmNAC genes under PEG treatment and rewatering
Heatmap of 87 ZmNAC genes was also conducted according to the transcriptome result (Fig. 8). Under PEG treatment and rewatering, the expression patterns of 87 ZmNAC genes exhibited different trend. The expression of some of genes were up-regulated under drought stress, down-regulated after rewatering, such as ZmNAC40, ZmNAC51 and ZmNAC20; some were down-regulated expression under drought stress while up-regulated expression after rewatering, such as ZmNAC45, ZmNAC95 and ZmNAC103; the expression of some genes exhibited random pattern. Under drought stress, plants accordingly make much effort to adapt at molecular, cellular, physiological and metabolic levels in order to survive or avoid adverse effects, and many genes were involved in the process. The expression of drought-resistant genes would be up-regulated under drought stress, while their transcript level would be reduced when the plants were rewatered and returned to normal growth state, and vice versa. Our research goal is to find highly expressed under PEG treatment and down-expressed after rewatering, so ZmNAC40, ZmNAC51, ZmNAC36, ZmNAC104, ZmNAC82, ZmNAC20, ZmNAC49 were selected as candidate genes.

Expression patterns of 87 ZmNAC genes under PEG stress and rewatering (colour figure online)
Expression profiles of six ZmNAC genes under PEG stress and rewatering by qRT-PCR analysis
To further validate the accuracy of transcriptome result and explore the expression patterns of ZmNAC genes, six randomly selected ZmNAC genes were investigated under drought stress and rewatering using qRT-PCR analysis. As shown in Fig. 9, six ZmNAC genes were expressed at different times during PEG stress and rewatering. The expression of ZmNAC118, ZmNAC122, ZmNAC49, ZmNAC25, ZmNAC20 and ZmNAC74 was up-regulated at 60 h and 96 h under PEG stress, then the transcript level was sharply reduced after rewatering at 3 d, which was consistent with the transcriptome result.

Expression patterns of ZmNAC genes in response to drought treatment. Different lowercase letters indicate significant differences at p < 0.05 (Duncan’s test)
Discussion
Drought, like other environment stresses, has adverse effect on maize yield. As water resources becoming more limiting, discovering drought-resistant genes and cultivating drought-resistant lines become more urgent (Bruce et al. 2002). NAC is one of the largest plant- specific transcription factor families and play an essential role in plants’ responses to abiotic stress (Nakashima et al. 2012; Shang et al. 2016; Yang et al. 2015). Genome-wide analysis of NAC family genes have been identified in Arabidopsis, rice, soybean, Medicago truncatula and white pear (Capella et al. 2014; Dung Tien et al. 2011; Gong et al. 2019; So and Lee 2019). Peng (Peng et al. 2015) and Ge (Ge et al. 2015) performed the genome-wide analysis of NAC gene family in maize, respectively, while their studies were based on the bioinformatics analysis. In this study, we focused on exploring ZmNAC genes with drought-resistance and conducted a high-throughput transcriptome, and obtained 87 ZmNAC genes that were responsive to drought stress and rewatering. GO analysis showed that 87 ZmNAC genes were mainly involved the regulation of biological process, such as the regulation of biological process, biosynthetic process and gene expression. The 87 ZmNAC genes were unevenly distributed on 10 maize chromosomes. It had been reported that the N-terminus of NAC proteins was a highly homologous region containing the DNA-binding NAC domain, which was approximately 150 amino acids in length and contained five conserved regions (A to E). Phylogenetic analysis showed that 87 ZmNAC genes were classified 12 sub-groups, with the exception of ZmNAC63, ZmNAC79, ZmNAC99, ZmNAC134 and ZmNAC94. Ten ZmNAC genes belonged to ATAF subfamily and each sub-group had the homologous with Arabidopsis or rice, illustrating not only the accuracy of the phylogenetic tree, but also indicating that genes in the same sub-group have similar functions. The analysis of gene structure was also conducted, and the arrangement and number of introns and exons shed light on the evolution and origin of a given gene (Schwartz et al. 2009). Previous study showed that the expression of ATAF1 was obviously induced by drought, high-salinity and abscisic acid (ABA) and the overexpression of ATAF1 in Arabidopsis increased plant sensitivity to drought, ABA and salt (Liu et al. 2016; Wu et al. 2009); the overexpression of ONAC003 in rice resulted in enhanced tolerance to high temperature, drought (Fang et al. 2015), which indicated that genes in the ATAF1 and ONAC003 sub-family might be involved in abiotic stress responses.
Cis-element analysis is an effective method to study potential transcriptional regulation of genes (Tran et al. 2004). Among them, ARBE was involved in the abscisic acid (ABA) and drought responsiveness, DRE especially indicated the drought and osmotic stress induction in maize, and MBS was MYB binding site involved in drought-induce ability. According to our result, DRE cis-element only appeared in ZmNAC77, 21 candidated genes contained MBS cis-element and 37 candidate genes contained ARBE cis-element. ZmNAC40, ZmNAC95, and ZmNAC20 contained 4 kinds of cis-element, respectively. Through cis-element analysis, we concluded that ZmNAC genes were likely related to drought stress.
Under drought stress and rewatering, different genes exhibited different expression patterns. Under drought stress, the expressions of some genes were up-regulated and then decreased after rewatering, such as ZmNAC49, ZmNAC94, and ZmNAC20; some genes gave the opposite trend, the gene expression was down-regulated under drought stress, and then up-regulated after rehydration, such as ZmNAC8, ZmNAC59, and ZmNAC50. But the expressions of some genes does not follow a regular pattern, which is inconsistent with our research purpose. From the result, we also found that number of DEGs at 3 d was higher than that at 60 h and 96 h, and the number of up-regulated genes was less than that of down-regulated genes. Under drought stress, there were complicated reactions occurred in plants to adapt to the stress, and the expression of drought-resistant genes were motivated during drought stress. When plants were rewatered after drought stress, greater changes took placed reflecting in growth restored, and much genes were involved in the changes. To further explore the expression of ZmNAC genes, the expression analysis of six ZmNAC genes was performed using qRT-PCR, respectively, and the result revealed that under drought stress, ZmNAC118, ZmNAC25, ZmNAC49, ZmNAC74, ZmNAC122 and ZmNAC20 were up-regulated during PEG treatment, and the transcript levels were dropped sharply after rewatering at 3 d, further revealing the accuracy of the transcriptome result and cis-element analysis. This study provides solid basis for further genes function identification and resistant molecular breeding.
References
Borrill P, Harrington SA, Uauy C (2017) Genome-wide sequence and expression analysis of the NAC transcription factor family in polyploid wheat. G3 Genes Genomes Genet 7:3019–3029. https://doi.org/10.1534/g3.117.043679
Bruce WB, Edmeades GO, Barker TC (2002) Molecular and physiological approaches to maize improvement for drought tolerance. J Exp Bot 53:13–25. https://doi.org/10.1093/jexbot/53.366.13
Bu LD, Zhang RH, Han MM, Xue JQ, Chang Y (2009) The physiological mechanism of compensation effect in maize leaf by rewatering after draught stress. Acta Agric Boreal Occident Sin 18:88–92. https://doi.org/10.3969/j.issn.1004-1389.2009.02.020
Capella M, Re DA, Arce AL, Chan RL (2014) Plant homeodomain-leucine zipper I transcription factors exhibit different functional AHA motifs that selectively interact with TBP or/and TFIIB. Plant Cell Rep 33:955–967. https://doi.org/10.1007/s00299-014-1576-9
Dung Tien L, Nishiyama R, Watanabe Y, Mochida K, Yamaguchi-Shinozaki K, Shinozaki K, Lam-Son Phan T (2011) Genome-wide survey and expression analysis of the plant-specific NAC transcription factor family in soybean during development and dehydration stress. DNA Res 18:263–276. https://doi.org/10.1093/dnares/dsr015
Fang Y, Liao K, Du H, Xu Y, Song H, Li X, Xiong L (2015) A stress-responsive NAC transcription factor SNAC3 confers heat and drought tolerance through modulation of reactive oxygen species in rice. J Exp Bot 66:6803–6817. https://doi.org/10.1093/jxb/erv386
Ge SS, Tang GY, Bi YP, Liu ZJ (2015) Genome-wide identification and analysis of NAC gene family in maize. Shandong Agric Sci 47:1–6. https://doi.org/10.14083/j.issn.1001-4942.2015.02.001
Gong X, Zhao L, Song X, Lin Z, Gu B, Yan J, Zhang S, Tao S, Huang X (2019) Genome-wide analyses and expression patterns under abiotic stress of NAC transcription factors in white pear (Pyrus bretschneideri). BMC Plant Biol. https://doi.org/10.1186/s12870-019-1760-8
Kadier Y, Zu Y-y, Dai Q-m, Song G, Lin S-w, Sun Q-p, Pan J-b, Lu M (2017) Genome-wide identification, classification and expression analysis of NAC family of genes in sorghum Sorghum bicolor (L.) Moench. Plant Growth Regul 83:301–312. https://doi.org/10.1007/s10725-017-0295-y
Kamoshita A, Rodriguez R, Yamauchi A, Wade LJ (2004) Genotypic variation in response of rainfed lowland rice to prolonged drought and rewatering. Plant Prod Sci 7:406–420. https://doi.org/10.1626/pps.7.406
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. https://doi.org/10.1186/gb-2013-14-4-r36
Kou X, Wang S, Wu M, Guo R, Xue Z, Meng N, Tao X, Chen M, Zhang Y (2014) Molecular characterization and expression analysis of NAC family transcription factors in tomato. Plant Mol Biol Rep 32:501–516. https://doi.org/10.1007/s11105-013-0655-3
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. https://doi.org/10.1038/nmeth.1923
Liu Y, Sun J, Wu Y (2016) Arabidopsis ATAF1 enhances the tolerance to salt stress and ABA in transgenic rice. J Plant Res 129:955–962. https://doi.org/10.1007/s10265-016-0833-0
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods (San Diego, Calif) 25:402–408. https://doi.org/10.1006/meth.2001.1262
Lu M, Ying S, Zhang D-F, Shi Y-S, Song Y-C, Wang T-Y, Li Y (2012) A maize stress-responsive NAC transcription factor, ZmSNAC1, confers enhanced tolerance to dehydration in transgenic Arabidopsis. Plant Cell Rep 31:1701–1711. https://doi.org/10.1007/s00299-012-1284-2
Mao H, Yu L, Han R, Li Z, Liu H (2016) ZmNAC55, a maize stress-responsive NAC transcription factor, confers drought resistance in transgenic Arabidopsis. Plant Physiol Biochem 105:55–66. https://doi.org/10.1016/j.plaphy.2016.04.018
Nakashima K, Takasaki H, Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K (2012) NAC transcription factors in plant abiotic stress responses. Biochim Biophys Acta Gene Regul Mech 1819:97–103. https://doi.org/10.1016/j.bbagrm.2011.10.005
Nuruzzaman M, Manimekalai R, Sharoni AM, Satoh K, Kondoh H, Ooka H, Kikuchi S (2010) Genome-wide analysis of NAC transcription factor family in rice. Gene 465:30–44. https://doi.org/10.1016/j.gene.2010.06.008
Peng X, Zhao Y, Li X, Wu M, Chai W, Sheng L, Wang Y, Dong Q, Jiang H, Cheng B (2015) Genomewide identification, classification and analysis of NAC type gene family in maize. J Genet 94:377–390. https://doi.org/10.1007/s12041-015-0526-9
Puranik S, Sahu PP, Srivastava PS, Prasad M (2012) NAC proteins: regulation and role in stress tolerance. Trends Plant Sci 17:369–381. https://doi.org/10.1016/j.tplants.2012.02.004
Saidi MN, Mergby D, Brini F (2017) Identification and expression analysis of the NAC transcription factor family in durum wheat (Triticum turgidum L. ssp. durum). Plant Physiol Biochem 112:117–128. https://doi.org/10.1016/j.plaphy.2016.12.028
Schwartz S, Meshorer E, Ast G (2009) Chromatin organization marks exon-intron structure. Nat Struct Mol Biol 16:990–995. https://doi.org/10.1038/nsmb.1659
Shan L (2003) Issues of science and technology on water saving agricultural development in China. Agric Res Arid Areas. https://doi.org/10.3321/j.issn:1000-7601.2003.01.001
Shang H, Wang Z, Zou C, Zhang Z, Li W, Li J, Shi Y, Gong W, Chen T, Liu A, Gong J, Ge Q, Yuan Y (2016) Comprehensive analysis of NAC transcription factors in diploid Gossypium: sequence conservation and expression analysis uncover their roles during fiber development. Sci China Life Sci 59:142–153. https://doi.org/10.1007/s11427-016-5001-1
Shiriga K, Sharma R, Kumar K, Yadav SK, Hossain F, Thirunavukkarasu N (2014) Genome-wide identification and expression pattern of drought-responsive members of the NAC family in maize. Meta Gene 2:407–417. https://doi.org/10.1016/j.mgene.2014.05.001
So H-A, Lee J-H (2019) NAC transcription factors from soybean (Glycine max L.) differentially regulated by abiotic stress. J Plant Biol 62:147–160. https://doi.org/10.1007/s12374-018-0285-2
Sun H, Hu M, Li J, Chen L, Li M, Zhang S, Zhang X, Yang X (2018) Comprehensive analysis of NAC transcription factors uncovers their roles during fiber development and stress response in cotton. BMC Plant Biol. https://doi.org/10.1186/s12870-018-1367-5
Tran LSP, Nakashima K, Sakuma Y, Simpson SD, Fujita Y, Maruyama K, Fujita M, Seki M, Shinozaki K, Yamaguchi-Shinozaki K (2004) Isolation and functional analysis of Arabidopsis stress-inducible NAC transcription factors that bind to a drought-responsive cis-element in the early responsive to dehydration stress 1 promoter. Plant Cell 16:2481–2498. https://doi.org/10.1105/tpc.104.022699
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. https://doi.org/10.1038/nprot.2012.016
Wang Z, Dane F (2013) NAC (NAM/ATAF/CUC) transcription factors in different stresses and their signaling pathway. Acta Physiol Plant 35:1397–1408. https://doi.org/10.1007/s11738-012-1195-4
Wu Y, Deng Z, Lai J, Zhang Y, Yang C, Yin B, Zhao Q, Zhang L, Li Y, Yang C, Xie Q (2009) Dual function of Arabidopsis ATAF1 in abiotic and biotic stress responses. Cell Res 19:1279–1290. https://doi.org/10.1038/cr.2009.108
Xu Z, Gongbuzhaxi Wang C, Xue F, Zhang H, Ji W (2015) Wheat NAC transcription factor TaNAC29 is involved in response to salt stress. Plant Physiol Biochem 96:356–363. https://doi.org/10.1016/j.plaphy.2015.08.013
Xue G-P, Way HM, Richardson T, Drenth J, Joyce PA, McIntyre CL (2011) Overexpression of TaNAC69 leads to enhanced transcript levels of stress up-regulated genes and dehydration tolerance in bread wheat. Mol Plant 4:697–712. https://doi.org/10.1093/mp/ssr013
Yang X, Wang X, Ji L, Yi Z, Fu C, Ran J, Hu R, Zhou G (2015) Overexpression of a Miscanthus lutarioriparius NAC gene MlNAC5 confers enhanced drought and cold tolerance in Arabidopsis. Plant Cell Rep 34:943–958. https://doi.org/10.1007/s00299-015-1756-2
Acknowledgements
The manuscript “Genome-Wide Analysis of NAC Transcription Factor Family in Maize under Drought Stress and Rewatering” was supported by the National Natural Science Foundation of China (No. 31471452), and the National Key Research and Development Program of China (No. 2017YFD0301106).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

{kind=link}
Cite this article
Wang, G., Yuan, Z., Zhang, P. et al. Genome-wide analysis of NAC transcription factor family in maize under drought stress and rewatering. Physiol Mol Biol Plants 26, 705–717 (2020). https://doi.org/10.1007/s12298-020-00770-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12298-020-00770-w