
Abstract
Schizophrenia is a mental disorder characterized by cognitive impairment resulting in compromised quality of life. Since the regulation of synaptic plasticity has functional implications in various aspects of cognition such as learning, memory, and neural circuit maturation, the dysregulation of synaptic plasticity is considered as a pathobiological feature of schizophrenia. The findings from our recently concluded studies indicate that there is an alteration in levels of synaptic plasticity markers such as neural cell adhesion molecule-1 (NCAM-1), Neurotropin-3 (NT-3) and Matrix-mettaloproteinase-9 (MMP-9) in schizophrenia patients. The objective of the present article is to review the role of markers of synaptic plasticity in schizophrenia. PubMed database (http;//www.ncbi.nlm.nih.gov/pubmed) was used to perform an extensive literature search using the keywords schizophrenia and synaptic plasticity. We conclude that markers of synaptic plasticity are altered in schizophrenia and may lead to complications of schizophrenia including cognitive dysfunction.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Schizophrenia is a chronic psychotic disorder characterized by functional impairment in thoughts, perception, and behavior, often resulting in a compromised quality of life in patients [1]. It is one of the leading contributors of disability globally, and is associated with reduction in life span by 10–20 years [2]. Earlier studies have documented around 10% life time suicidal rate and 18 to 55% suicide attempt in schizophrenic patients [3]. Schizophrenic symptoms are categorized as a constellation of positive symptoms characterized by confusion, hallucinations and delusions, as well as negative symptoms characterized by withdrawal, lack of motivation and emotional blunting [4]. Owing to the heterogeneity in disease presentation, there is no clear consensus in defining the etiopathogenesis of schizophrenia [5]. Neurotransmitter abnormalities, genetic predisposition and environmental factors have all been implicated to play a fundamental role in the etiology of schizophrenia [1, 4] (Table 1).
Synaptic Plasticity
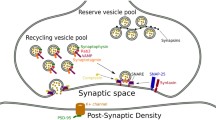
The neurons in the CNS transmit information with one another through neuronal junctions known as synapse. Synaptic plasticity is the ability of synapses to strengthen or weaken over time, in response to increases or decreases in their activity. Plastic change also results from the alteration of the number of neurotransmitter receptors located on a synapse. Neuroplasticity refers to the ability of the brain to modulate its activity in response to the dynamic signals in the central nervous system (CNS) by reorganization of its structure, functions, or connections [6]. Notably, synaptic plasticity is a continuing process that is dependent on activity or experience, and therefore forms the basis for memory and learning [6, 7]. While synaptic plasticity forms the basis of crucial cognitive functions throughout life, it also plays a vital role in neuronal organization and maturation of neural circuits during development [7,8,9].
Regulation of Synaptic Plasticity
Synaptic plasticity is regulated through the proteins released at the synapse, which in turn regulate the synaptic strength, weight or efficacy. The various pattern of expression of these proteins thereby influence the weight of the neural circuit, ultimately influencing key cognitive functions such as short and long-term memory, learning and information processing [6]. These proteins behave as molecular markers of synaptic plasticity and are likely to be dynamically altered during human development and distinctive alteration in such markers may predict the development of psychotic disorders [6, 8].
Dysregulation of Synaptic Plasticity in Schizophrenia
New experimental evidences accumulated over the past few years highlight an unified ‘disconnection hypothesis’ which implicates the dysregulation of synaptic plasticity as key precipitating event in the pathogenesis of schizophrenia syndrome [7,8,9,10]. It has been hypothesized that, healthy human brain in response to an excitatory stimuli, eliminates the weak synapses and strengthen the adaptive synapses through a process termed as synaptic pruning resulting in organization and maturation of neural circuits. However, in schizophrenic brain the synaptic pruning is exacerbated due to impairments in synaptic plasticity resulting in suboptimal maturation of neural circuits, leading to disturbances in cognitive, motor and sensory functions [8].
Impairment in synaptic plasticity may influence neuronal maturation and lead to sensory, motor, cognitive, and psychotic disturbances at various stages of development [8]. Notably, the molecular markers of synaptic plasticity were differentially expressed in specific regions of brain, in response to different stimuli such learning, predator exposure and stress [8]. Earlier investigators have documented alteration in the post-synaptic density (PSD) proteins and the genes encoding for the markers of synaptic plasticity in schizophrenia [8, 12].
In a genome-wide study involving over 36,000 schizophrenic patients, it was reported that variants of 108 genetic loci involved in glutamatergic neurotransmission and synaptic plasticity were associated with schizophrenia [13]. In line with this evidence, proteomic studies have also elucidated the differential expressions of several proteins actively involved in the maintenance of synaptic signaling in schizophrenic patients [14,15,16]. Aberrations of cellular machinery governing synaptic plasticity which results in alterations of synaptic proteins have been demonstrated in schizophrenia [14, 17, 18]. Interestingly, the psychosis-onset in schizophrenia is preceded by subclinical disturbances in sensory, motor and cognitive functions [8]. Considering the key involvement of synaptic plasticity in maintenance and maturity of neural circuits, memory and learning [8] dysregulation of neuronal plasticity could be one of the key underlying features of schizophrenia. Assessment of the molecular markers of synaptic plasticity in the background of schizophrenia could be beneficial in further understanding the pathogenic mechanisms behind this psychotic disorder, as well as assist in its symptomatic diagnosis. This review summarizes the alterations in some of the key molecular markers of synaptic plasticity, and their association with schizophrenia.
Markers of Synaptic Plasticity Associated with Schizophrenia
Neurotrophins
Neurotrophins are vital components in mediating the development as well as the homeostatic maintenance of nervous system [19]. Neurotrophins including Nerve growth factor (NGF), Brain Derived Neurotrophic Factor (BDNF), Neurotrophin-3 (NT-3) and Neurotrophins-4 (NT-4) serve as biomolecules that regulate the growth, survival and plasticity of the dopaminergic, cholinergic, and serotonergic neurons [19]. Considering their key role in synaptogenesis and synaptic plasticity, disruption of neurotrophin signaling and their altered expression could lead to disturbances in neural plasticity [20] and could behave as a precipitating event in the etiopathology of schizophrenia [21].
Neurotrophin-3 (NT-3)
The neurotrophic factor NT-3 is encoded by the NTF3 gene [22]. NT-3 is the third member of NGF family of neurotrophins and is functionally distinct from nerve growth factor (NGF) and Brain derived neurotrophic factor (BDNF) [22]. NT-3 has been implicated in neurogenesis as well as growth, differentiation, and survival of neurons, both in developing as well as mature nervous system [23]. The effects of NT-3 are mediated through its binding to specific low affinity receptor p75 and high affinity tropomyosin receptor kinase (TRK) receptors [22, 23]. Evidences from experimental models indicate the involvement of NT-3 in regulating synaptic formation, reorganization and neuronal plasticity [23,24,25].
Single nucleotide polymorphism (SNP) in the NTF-3 gene has been associated with risk for the onset of schizophrenia. The A3/147 bp allele in a dinucleotide repeat polymorphism of the promoter region of the NT-3 was observed to be more prevalent in male schizophrenic patients when compared to controls in a Japanese population [26, 27]. Moreover, G/-3004/A polymorphism in the promoter region of NTF-3 gene exhibited weak susceptibility for schizophrenia [28].
NT-3 levels were reported to be altered in psychiatric disorders. Experimental studies have revealed alterations in NT-3 expression in specific-regions of brain in response to treatment in patients with major depressive disorder (MDD) [29]. In a recent study from our laboratory, we reported increased serum levels of NT-3 in schizophrenic patients compared to controls and was associated with disease severity [30]. NT-3 levels were found to be elevated in schizophrenic patients who exhibited depressive symptoms [31, 32]. Several factors like chronic stress are known to alter NT-3 levels in blood [33]. NT-3 has been demonstrated to cross the blood brain barrier [34, 35]. Hence it can be hypothesized that elevated NT-3 in blood enters brain and alters the neural plasticity resulting in schizophrenia [30].
Brain Derived Neurotrophic Factor
Brain derived neurotrophic factor (BDNF) is a growth factor known to be involved in the regulation of proliferation and survival of neurons [36]. BDNF signaling has been implicated in the regulation of dopaminergic, serotoninergic and GABAergic neurotransmitter systems, which were found to be disrupted in schizophrenia [36,37,38]. BDNF is released at the synapse and is involved in maintenance of the synaptic plasticity similar to other neurotrophic factors [36]. Impaired BDNF signaling has been associated with alterations in synaptic plasticity and suboptimal neurodevelopment [36,37,38]. BDNF expression was found to be down-regulated in the pre-frontal cortex and hippocampus of suicidal subjects [39]. The genome wide association study (GWAS) reported several SNP’s in the BDNF locus, which were nominally associated with schizophrenia [13]. A functional variation of the BDNF gene, rs6265 (Val → Met substitution), has been correlated with compromised secretion of BDNF in response to activity [36, 40]. Interestingly, when compared to control subjects both Val/Val homozygotes and Met-carriers exhibited reduced hippocampal volumes [41]. These findings reveal that the structural changes in hippocampus may not be dependent primarily on the genotypic variations of BDNF gene [41] and indicate the presence of other mechanisms behind hippocampal volume alterations in schizophrenia. Of interest, in cultured hippocampal neurons, the variants such as Val-BDNF and Met-BDNF were localized differently at the cellular level. While, Val-BDNF was localized in cell body as well as the dendrites, there was absence of Met-BDNF variant in the synapse and distal dendrites [42]. Notably, a study by Skibinska et al., reported that though BDNF levels were reduced in depression but not associated with Val → Met functional polymorphism [43]. These findings are indicative that while genotypic variants of BDNF may not affect their expression, they could be associated with potential alteration in synaptic plasticity.
The serum levels of BDNF were found to be reduced in patients with schizophrenia when compared to control subjects [41, 42]. In a recent study from our laboratory, we reported negative association between BDNF levels with positive symptom score, general psychopathology score and total PANSS score in schizophrenia patients suggesting that low BDNF levels increases severity of schizophrenia [42]. Tang et al. have demonstrated that serum BDNF levels did not vary significantly between deficit and non-deficit schizophrenic patients, indicating that alterations in cognitive performance may not reflected in the circulatory levels of BDNF [44]. BDNF levels were reduced in schizophrenic patients with depression, when compared to patients without depression [32]. Study by Yang et al. observed that reduced BDNF levels in both chronic and first-episode schizophrenic patients was associated with poor memory, speed of processing and visual learning suggesting involvement of disruption of BDNF signaling with cognitive impairment in schizophrenia [45]. Even though the cause of reduction in BDNF in schizophrenia was not known, earlier studies have demonstrated that interleukin-23 was negatively correlated with BDNF levels indicating inflammation may reduce BDNF levels in schizophrenia [42].
Nerve Growth Factor
Nerve growth factor (NGF), the first discovered neurotrophic factor, functions as regulator of nociceptive pain and as mediator of growth, development, and survival of neurons [46]. They are crucial regulators of synaptic plasticity and are reported to be involved in the induction of dendritic outgrowth and the regulation of dendritic length and complexity in cortical neurons [47, 48]. NGF signaling is notably involved in both short- and long-term effects on the neuroanatomical structure. Indeed, evidences link the dysregulation in NGF signaling with structural alterations in brain [49]. Earlier studies have reported alterations in cerebral connectivity and morphology in schizophrenia [9, 50]. Grey matter volume was found to be smaller is schizophrenic patients when compared to control subjects and serum levels of NGF correlated with the gray matter volume in specific brain regions. Serum NGF levels were positively correlated with pons region and negatively correlated with left midcingulate cortex (MCC) and left orbital gyrus in schizophrenic patients [49]. Reduction in left orbital gyrus has been associated with longer duration of illness and poor social functioning in schizophrenia [51].
Circulating levels of NGF in blood correlated well with concentrations of NGF in CNS owing to the capacity of NGF to permeate the blood brain barrier efficiently [47, 50,51,52]. Previous investigators have reported reduced levels of NGF in serum and cerebrospinal fluid in first episode schizophrenic patients when compared to control subjects [52]. Moreover, several polymorphisms variants in the NGF and NGFR gene have been identified and were attributed either as risk factors or as protective factor in schizophrenic patients [53]. Schizophrenia patients treated with atypical antipsychotics were reported to have better efficiency in improving peripheral NGF levels compared to typical antipsychotic drugs [54].
Neural Cell Adhesion Molecule
Neural cell adhesion molecule (NCAM) is an integral membrane glycoprotein plays a key role in the regulation of cell adhesion, neural cell migration and synaptic plasticity [55]. It influences the strength and the morphology of synaptic connections through mediating neurite outgrowth, axonal guidance, synaptic pruning and contact with astrocytes [56, 57]. Abnormalities in NCAM signaling was reported to contribute to the etiopathogenesis of neuropsychiatric disorders such as schizophrenia [58].Experimental studies have demonstrated that intervention with NCAM-1 antibodies attenuate synaptic strength (long term potentiation (LTP)), while NCAM-1 gene knockout leads to deficits in spatial memory in mice [59]. SNP’s in NCAM-1 gene was associated with neurocognitive score in a large sample of schizophrenic patients [60].
In our recent study with 176 chronic schizophrenic patients, serum NCAM-1 was elevated in schizophrenic patients when compared to control subjects. Interestingly we observed that serum NCAM-1 levels were positively associated with attention, language, visuospatial abilities and better cognitive functioning in schizophrenia [61].Notably, reduced NCAM levels were associated with cognitive deficit in schizophrenia, and also independently contributed to changes in the cognitive scores as measured by The MATRICS consensus cognitive battery (MCCB) [62]. Moreover, reduced serum NCAM levels in these patients were associated with hippocampal volume and negatively correlated with disease severity [63]. These findings indicate the association of NCAM with neurodevelopmental alterations, as well as the involvement of disrupted NCAM signaling in early stages of schizophrenic syndrome.
Matrix Metalloproteinase
Matrix Metalloproteinases (MMPs) are extracellularly acting enzymes belonging to the metzincin (zinc dependent and with conserved Met residue) super-family of proteases [64, 65]. Though they were primarily thought to be involved in degradation of extracellular matrix (ECM) components, several evidences implicate their role as regulators of extracellular signaling in central nervous system [64,65,66]. Through the processing of growth factors, cell surface receptors, cell adhesion molecules, and latent biomolecules, MMPs are seen to be involved in initiation and termination of signaling cascades related to synaptic remodeling [64]. In response to a range of stimuli, the synaptic connections are remodeled, with help of modifications of the peridendritic environment, and extracellular matrix. Through the proteolytic disassembly of ECM, MMPs play a key role in remodeling the synaptic plasticity of neurons [67]. In brain injury, exacerbated MMP activity has been associated with the pathological initiation of neuro-inflammation, neuronal degeneration and synaptic loss [68, 69].
MMPs have been implicated to be involved in both structural (dendritic spine modifications) and functional (magnitude of synaptic transmission) modulation of synaptic circuits [64]. Imaging of the hippocampal region using high-resolution microscopy indicated the distribution of MMP-9 throughout the synaptically dense region (neuropil). Notably, MMP9 was seen to co-localize with presynaptic and postsynaptic molecular markers, which could indicate its involvement in the processing of biomolecules involved in regulation of synaptic plasticity [67, 70]. The activity of MMPs is tightly regulated by tissue inhibitors of metalloproteinases (TIMPs). It is hypothesized that TIMPs mediate the formation/re-establishment of synaptic connections through the regulation of MMP activity, and contribute to persistent increase in synaptic strength (LTP) between neurons [71].
In a study by Holland et al., increased plasma MMP-9 levels was associated with negative symptoms in schizophrenic patients. Remarkably, the increase in peripheral MMP-9 was associated with decrease in left and right hippocampal volumes in schizophrenic patients [72]. Considering that structural alteration in hippocampus is closely observed in the pathophysiology of schizophrenia [73, 74] MMP-9 aberration maybe a key mediating mechanism.
In our recent study with schizophrenic patients, our findings indicated elevated serum MMP-9 levels in patients when compared to control subjects. MMP-9 levels negatively correlated with fluency, language and cognitive score in schizophrenic patients. Importantly, we observed through regression analysis that increase in MMP-9 is associated with risk of cognitive impairment in schizophrenia. In our study we did not observe any difference in MMP-9 levels between drug-naïve and chronic schizophrenic patients [30]. Other studies also support this evidence where medication did not alter MMP-9 levels in schizophrenic patients [30, 72, 75] indicating the involvement of alternative mechanisms [76,77,78].
In our study with drug-naïve male schizophrenic patients, we reported increased serum levels of MMP-9 in schizophrenic patients when compared to control subjects (77). Notably, the increase is MMP-9 was associated with an increase in the lipid peroxidation marker, Malondialdehyde (MDA). Moreover, increase in peripheral MMP-9 was negatively associated with total antioxidant status in the serum of schizophrenic patients [77]. In line with previous evidence which suggest that reactive oxygen species is a key stimulus in the activation of latent MMP-9 [79], oxidative stress could be a key factor in initiating MMP-9 over activation in schizophrenia. MMP-9 over activation in turn is seen to mediate neuroinflammation and redox dysregulation, that could result in a central inhibitory/excitatory imbalance found in schizophrenia [76].
Notably, in a phenotype-based genetic association study with over 1000 schizophrenic patients, a C/T SNP (rs20544) in the MMP-9 gene was associated with more severe chronic delusions in schizophrenic patients (78). Of note, the genetic variation in MMP-9 (rs20544) was attributed to modified mRNA structure and activity of MMP-9 at the synapses, which resulted in changes in the dendritic spine morphology (78). The genetic variation in MMP-9 could thereby indicate an increased susceptibility to structural plasticity in response to synaptic stimulation [79].
Neurexin
Neurexins (NRXN) are presynaptic transmembrane proteins that play a fundamental role in synaptic transmission The functionality of NRXNs is primarily at the neuronal surface where they govern the synaptic stability and neuronal adhesion [80]. Several mutations in NRXN genes have been identified in patients with schizophrenia [80, 81]. Moreover, NRXN loss of function rodent model (NRXN KO models) have demonstrated dysregulation of excitatory and inhibitory neurotransmission [82] as well as a reduction in pre-pulse inhibition [83] which is commonly observed in schizophrenic patients [80]. The NRXN synaptic complex has been observed to regulate Disrupted in schizophrenia 1 (DISC1), which is closely implicated in schizophrenic pathology [84]. Studies by Kirov et al., have identified an involvement of copy number variations in NRXN1 gene in the pathogenesis of schizophrenia [81, 85, 86]. A large study including seven European populations highlighted that NXRN1 deletion mutation in exons conferred increased susceptibility for schizophrenia [87]. Functional characterization of rare NRXN gene variants may precipitate the impaired neurodevelopmental phenotype in schizophrenia and warrant further study [88].
Possible Mechanisms for Dysregulation of Synaptic Plasticity Markers
Among the factors related to dysregulation of markers of synaptic plasticity, stress and inflammation plays a crucial role. Stress is a characteristic environmental factor in mood disorders and may represent a key pathogenic mechanism that influences the level of synaptic plasticity markers [89]. Both acute and chronic stress induced reduction in BDNF synthesis in hippocampus/hypothalamus of rodents [90, 91]. On contrary, only chronic stress induced an increase in mRNA levels of NT-3 in the dentate gyrus and hippocampus region in in immobilization induced rodent stress model [92]. Interestingly, the expression of BDNF in dorsal hippocampal region CA1 varied based on the presence or absence of stress stimuli in rodents [11]. Both 2.5 and 24 h restraint stress triggered an alteration in subcellular distribution and activity of MMP-9 in rat hippocampus [93]. The expression of molecular markers of synaptic plasticity was found to be affected by treatment with drugs. When compared to untreated MDD, patients treated with anti-depressants exhibited up-regulation of BDNF expression in parietal cortex and NT-3 in cortical regions, thalamus, putamen, cingulate gyrus and nucleus caudatus regions [29]. Similarly, chronic antidepressant treatment attenuated the NT-3 expression in locus coeruleus of immobilized stress induced rodents [94]. Stress and anti-depressants may exert their clinical outcomes through the alteration of expression of synaptic plasticity markers [94].
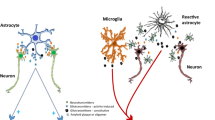
Inflammation has been identified as a key pathogenic factor in altering synaptic plasticity [95,96,97]. Microglial cell activation and immunological aberrant immune response may underlie the alterations in synaptic plasticity in schizophrenia [97]. Microglial cells activated by both neuroinflammation and peripheral inflammation were observed to trigger alterations in synaptic plasticity in hippocampus [96]. Our studies in this regard investigated the relationship between markers of synaptic plasticity with cytokines in psychiatric disorders [98,99,100]. Our data indicated that peripheral BDNF levels negatively correlated with the cytokine IL-23 and disease severity in schizophrenic patients [99]. We also observed that the peripheral IL-17 and IL-10 levels were markedly elevated and associated with disease severity in schizophrenia [100]. In line with this evidence IL-6 has also been identified to regulate synaptic plasticity [101], and alteration in such cytokines could behave as precipitating event for synapse pathology in schizophrenia.
Conclusions
Synaptic plasticity is known to be impaired in schizophrenia and alteration in the markers of synaptic plasticity are reported to be associated with the complications of schizophrenia including cognitive dysfunction. Assessment of molecular markers of synaptic plasticity that are implicated in schizophrenia would help in identification of novel markers for diagnosis of the disease and development of therapeutic strategies to combat this devastating psychiatric disorder. Owing to the relationship between inflammation and alterations in synaptic plasticity, the role of anti-inflammatory drugs in treatment of schizophrenia requires further investigations.
Abbreviations
- BDNF:
-
Brain derived neurotrophic factor
- GWAS:
-
Genome wide association study
- ECM:
-
Extracellular matrix
- LTP:
-
Long term potentiation
- MCC:
-
Midcingulate cortex
- MCCB:
-
The MATRICS consensus cognitive battery
- MDD:
-
Major depressive disorder
- MMP-9:
-
Matrix metalloproteinase-9
- NCAM:
-
Neural adhesion molecule
- NGF:
-
Nerve Growth Factor
- NRXN:
-
Neurexin
- NT3:
-
Neurotrophin-3
- PSA:
-
Polysilic acid
- PSD:
-
Post-synaptic density
- SNP:
-
Single nucleotide polymorphism
- TIMP:
-
Tissue inhibitor for matrix metalloproteinase
- TRK:
-
Tropomyosin receptor kinase
References
Kahn RS, Sommer IE, Murray RM, Meyer-Lindenberg A, Weinberger DR, Cannon TD, et al. Schizophrenia. Nature Reviews Disease Primers. 2015;1:15067.
Moradi H, Harvey PD, Helldin L. Correlates of risk factors for reduced life expectancy in schizophrenia: is it possible to develop a predictor profile? Schizophr Res. 2018;201:388–92.
Sher L, Kahn RS. Suicide in schizophrenia: an educational overview. Medicina (Kaunas). 2019;55(7):361.
Hany M, Rehman B, Azhar Y, Chapman J. Schizophrenia. In StatPearls. Treasure Island, FL: StatPearls; 2021. http://www.ncbi.nlm.nih.gov/books/NBK539864/ (Accessed on 25/10/2021).
Patel KR, Cherian J, Gohil K, Atkinson D. Schizophrenia: overview and treatment options. Pharm Ther. 2014;39(9):638–45.
Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacol. 2008;33(1):18–41.
Crabtree GW, Gogos JA. Synaptic plasticity, neural circuits, and the emerging role of altered short-term information processing in schizophrenia. Front Synaptic Neurosci. 2014;25(6):28.
Forsyth JK, Lewis DA. Mapping the consequences of impaired synaptic plasticity in schizophrenia through development: an integrative model for diverse clinical features. Trends Cogn Sci. 2017;21(10):760–78.
Stephan KE, Baldeweg T, Friston KJ. Synaptic plasticity and dysconnection in schizophrenia. Biol Psychiatry. 2006;59(10):929–39.
Berdenis van Berlekom A, Muflihah CH, Snijders GJ, MacGillavry HD, Middeldorp J, Hol EM, et al. Synapse pathology in schizophrenia: a meta-analysis of postsynaptic elements in postmortem brain studies. Schizophr Bull. 2020;46(2):374–86.
Zoladz PR, Park CR, Halonen JD, Salim S, Alzoubi KH, Srivareerat M, et al. Differential expression of molecular markers of synaptic plasticity in the hippocampus, prefrontal cortex, and amygdala in response to spatial learning, predator exposure, and stress-induced amnesia. Hippocampus. 2012;22(3):577–89.
Föcking M, Lopez LM, English JA, Dicker P, Wolff A, Brindley E, et al. Proteomic and genomic evidence implicates the postsynaptic density in schizophrenia. Mol Psychiatry. 2015;20(4):424–32.
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–7.
MacDonald ML, Ding Y, Newman J, Hemby S, Penzes P, Lewis DA, et al. Altered glutamate protein co-expression network topology linked to spine loss in the auditory cortex of schizophrenia. Biol Psychiatry. 2015;77(11):959–68.
Pennington K, Beasley CL, Dicker P, Fagan A, English J, Pariante CM, et al. Prominent synaptic and metabolic abnormalities revealed by proteomic analysis of the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder. Mol Psychiatry. 2008;13(12):1102–17.
Hill JJ, Hashimoto T, Lewis DA. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2006;11(6):557–66.
Glausier JR, Lewis DA. Dendritic spine pathology in schizophrenia. Neuroscience. 2013;22(251):90–107.
Garey L. When cortical development goes wrong: schizophrenia as a neurodevelopmental disease of microcircuits. J Anat. 2010;217(4):324–33.
Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736.
Durany N, Michel T, Zöchling R, Boissl KW, Cruz-Sánchez FF, Riederer P, et al. Brain-derived neurotrophic factor and neurotrophin 3 in schizophrenic psychoses. Schizophr Res. 2001;52(1–2):79–86.
Buckley PF, Mahadik S, Pillai A, Terry A. Neurotrophins and schizophrenia. Schizophr Res. 2007;94(1):1–11.
Maisonpierre PC, Belluscio L, Squinto S, Ip NY, Furth ME, Lindsay RM, et al. Neurotrophin-3: a neurotrophic factor related to NGF and BDNF. Science. 1990;247(4949 Pt 1):1446–51.
Hernández-Echeagaray E. Neurotrophin-3 modulates synaptic transmission. Vitam Horm. 2020;114:71–89.
Ammendrup-Johnsen I, Naito Y, Craig AM, Takahashi H. Neurotrophin-3 enhances the synaptic organizing function of TrkC–protein tyrosine phosphatase σ in rat hippocampal neurons. J Neurosci. 2015;35(36):12425–31.
Gómez-Pineda VG, Torres-Cruz FM, Vivar-Cortés CI, Hernández-Echeagaray E. Neurotrophin-3 restores synaptic plasticity in the striatum of a mouse model of Huntington’s disease. CNS Neurosci Ther. 2018;24(4):353–63.
Nanko S, Hattori M, Kuwata S, Sasaki T, Fukuda R, Dai XY, Yamaguchi K, Shibata Y, Kazamatsuri H. Neurotrophin-3 gene polymorphism associated with schizophrenia. Acta Psychiatr Scand. 1994;89(6):390–2. https://doi.org/10.1111/j.1600-0447.1994.tb01534.x.
Dawson E, Powell JF, Sham PC, Nöthen M, Crocq MA, Propping P, et al. An association study of a neurotrophin-3 (NT-3) gene polymorphism with schizophrenia. Acta Psychiatr Scand. 1995;92(6):425–8.
Hattori M, Kunugi H, Akahane A, Tanaka H, Ishida S, Hirose T, et al. Novel polymorphisms in the promoter region of the neurotrophin-3 gene and their associations with schizophrenia. Am J Med Genet. 2002;114(3):304–9.
Sheldrick A, Camara S, Ilieva M, Riederer P, Michel TM. Brain-derived neurotrophic factor (BDNF) and neurotrophin 3 (NT3) levels in post-mortem brain tissue from patients with depression compared to healthy individuals-a proof of concept study. Eur Psychiatry. 2017;46:65–71.
Keshri N, Nandeesha H, Rajappa M, Menon V. Matrix metalloproteinase-9 increases the risk of cognitive impairment in schizophrenia. Nord J Psychiatry. 2021;75(2):130–4.
Arabska J, Łucka A, Strzelecki D, Wysokiński A. In schizophrenia serum level of neurotrophin-3 (NT-3) is increased only if depressive symptoms are present. Neurosci Lett. 2018;25(684):152–5.
Wysokiński A. Serum levels of brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3) in depressed patients with schizophrenia. Nord J Psychiatry. 2016;70(4):267–71.
Saruta J, Iida M, Kondo Y, To M, Hayashi T, Hori M, et al. Chronic stress induces neurotrophin-3 in rat submandibular gland. Yonsei Med J. 2012;53(6):1085–92.
Poduslo JF, Curran GL. Permeability at the blood-brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3. BDNF Mol Brain Res. 1996;36(2):280–6.
Pan W, Banks WA, Kastin AJ. Permeability of the blood-brain barrier to neurotrophins. Brain Res. 1998;788(1):87–94.
Di Carlo P, Punzi G, Ursini G. Brain-derived neurotrophic factor and schizophrenia. Psychiatr Genet. 2019;29(5):200–10.
Favalli G, Li J, Belmonte-de-Abreu P, Wong AHC, Daskalakis ZJ. The role of BDNF in the pathophysiology and treatment of schizophrenia. J Psychiatr Res. 2012;46(1):1–11.
Peng S, Li W, Lv L, Zhang Z, Zhan X. BDNF as a biomarker in diagnosis and evaluation of treatment for schizophrenia and depression. Discov Med. 2018;26(143):127–36.
Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry. 2003;60(8):804–15.
Fu X, Wang J, Du J, Sun J, Baranova A, Zhang F. BDNF gene’s role in schizophrenia: from risk allele to methylation implications. Front Psychiatry. 2020;15(11):564277. https://doi.org/10.3389/fpsyt.2020.564277.
Harrisberger F, Smieskova R, Schmidt A, Lenz C, Walter A, Wittfeld K, et al. BDNF Val66Met polymorphism and hippocampal volume in neuropsychiatric disorders: a systematic review and meta-analysis. Neurosci Biobehav Rev. 2015;55:107–18.
Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112(2):257–69.
Skibinska M, Groszewska A, Kapelski P, Rajewska-Rager A, Pawlak J, Dmitrzak-Weglarz M, et al. Val66Met functional polymorphism and serum protein level of brain-derived neurotrophic factor (BDNF) in acute episode of schizophrenia and depression. Pharmacol Rep. 2018;70(1):55–9.
Tang X, Zhou C, Gao J, Duan W, Yu M, Xiao W, et al. Serum BDNF and GDNF in Chinese male patients with deficit schizophrenia and their relationships with neurocognitive dysfunction. BMC Psychiatry. 2019;19(1):254.
Yang Y, Liu Y, Wang G, Hei G, Wang X, Li R, et al. Brain-derived neurotrophic factor is associated with cognitive impairments in first-episode and chronic schizophrenia. Psychiatry Res. 2019;273:528–36.
Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci. 2001;24:1217–81.
McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22(1):295–318.
Schinder AF, Poo M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000;23(12):639–45.
Neugebauer K, Hammans C, Wensing T, Kumar V, Grodd W, Mevissen L, Sternkopf MA, Novakovic A, Abel T, Habel U, Nickl-Jockschat T. Nerve growth factor serum levels are associated with regional gray matter volume differences in schizophrenia patients. Front Psychiatry. 2019;26(10):275. https://doi.org/10.3389/fpsyt.2019.00275.
Bora E, Fornito A, Radua J, Walterfang M, Seal M, Wood SJ, et al. Neuroanatomical abnormalities in schizophrenia: a multimodal voxelwise meta-analysis and meta-regression analysis. Schizophr Res. 2011;127(1):46–57.
Nakamura M, Nestor PG, Levitt JJ, Cohen AS, Kawashima T, Shenton ME, et al. Orbitofrontal volume deficit in schizophrenia and thought disorder. Brain. 2008;131(1):180–95.
Xiong P, Zeng Y, Zhu Z, Tan D, Xu F, Lu J, et al. Reduced NGF serum levels and abnormal P300 event-related potential in first episode schizophrenia. Schizophr Res. 2010;119(1):34–9.
Zakharyan R, Atshemyan S, Gevorgyan A, Boyajyan A. Nerve growth factor and its receptor in schizophrenia. BBA Clin. 2014;20(1):24–9.
Parikh V, Evans DR, Khan MM, Mahadik SP. Nerve growth factor in never-medicated first-episode psychotic and medicated chronic schizophrenic patients: possible implications for treatment outcome. Schizophr Res. 2003;60(2–3):117–23.
Rønn LC, Hartz BP, Bock E. The neural cell adhesion molecule (NCAM) in development and plasticity of the nervous system. Exp Gerontol. 1998;33(7–8):853–64.
Togashi H, Sakisaka T, Takai Y. Cell adhesion molecules in the central nervous system. Cell Adh Migr. 2009;3(1):29–35.
Vukojevic V, Mastrandreas P, Arnold A, Peter F, Kolassa I-T, Wilker S, et al. Evolutionary conserved role of neural cell adhesion molecule-1 in memory. Transl Psychiatry. 2020;10(1):1–13.
Sytnyk V, Leshchyns’ Ka I, Nikonenko AG, Schachner M. NCAM promotes assembly and activity-dependent remodeling of the postsynaptic signaling complex. J cell Biol. 2006;174(7):1071–85.
Stork O, Welzl H, Wolfer D, Schuster T, Mantei N, Stork S, et al. Recovery of emotional behaviour in neural cell adhesion molecule (NCAM) null mutant mice through transgenic expression of NCAM180. Eur J Neurosci. 2000;12(9):3291–306.
Sullivan PF, Keefe RSE, Lange LA, Lange EM, Stroup TS, Lieberman J, et al. NCAM1 and neurocognition in schizophrenia. Biol Psychiat. 2007;61(7):902–10.
Keshri N, Nandeesha H, Rajappa M, Menon V. Relationship between neural cell adhesion molecule-1 and cognitive functioning in schizophrenia spectrum disorder. Ind J Clin Biochem. 2021; Available from: https://doi.org/10.1007/s12291-020-00937-y
An H, Zhou L, Yu Y, Fan H, Fan F, Tan S, et al. Serum NCAM levels and cognitive deficits in first episode schizophrenia patients versus health controls. Schizophr Res. 2018;192:457–8.
An H, Qin J, Fan H, Fan F, Tan S, Wang Z, et al. Decreased serum NCAM is positively correlated with hippocampal volumes and negatively correlated with positive symptoms in first-episode schizophrenia patients. J Psychiatr Res. 2020;1(131):108–13.
Huntley GW. Synaptic circuit remodelling by matrix metalloproteinases in health and disease. Nat Rev Neurosci. 2012;13(11):743–57.
Löffek S, Schilling O, Franzke C-W. Biological role of matrix metalloproteinases: a critical balance. Eur Respir J. 2011;38(1):191–208.
Lepeta K, Kaczmarek L. Matrix metalloproteinase-9 as a novel player in synaptic plasticity and schizophrenia. Schizophr Bull. 2015;41(5):1003–9.
Szklarczyk A, Lapinska J, Rylski M, McKay RDG, Kaczmarek L. Matrix metalloproteinase-9 undergoes expression and activation during dendritic remodeling in adult hippocampus. J Neurosci. 2002;22(3):920–30.
Rivera S, Khrestchatisky M, Kaczmarek L, Rosenberg GA, Jaworski DM. Metzincin proteases and their inhibitors: foes or friends in nervous system physiology? J Neurosci. 2010;30(46):15337–57.
Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6(12):931–44.
Bozdagi O, Nagy V, Kwei KT, Huntley GW. In vivo roles for matrix metalloproteinase-9 in mature hippocampal synaptic physiology and plasticity. J Neurophysiol. 2007;98(1):334–44.
Wright JW, Harding JW. Contributions of matrix metalloproteinases to neural plasticity, habituation, associative learning and drug addiction. Neural Plast. 2010;10(2009):e579382.
Seitz-Holland J, Seethaler M, Makris N, Rushmore J, Cho KIK, Rizzoni E, et al. The association of matrix metalloproteinase 9 (MMP9) with hippocampal volume in schizophrenia: a preliminary MRI study. Neuropsychopharmacology. 2022;47(2):524–30.
Zaidel DW, Esiri MM, Harrison PJ. Size, shape, and orientation of neurons in the left and right hippocampus: investigation of normal asymmetries and alterations in schizophrenia. Am J Psychiatry. 1997;154(6):812–8.
Walker MA, Highley JR, Esiri MM, McDonald B, Roberts HC, Evans SP, et al. Estimated neuronal populations and volumes of the hippocampus and its subfields in schizophrenia. Am J Psychiatry. 2002;159(5):821–8.
Strzelecki D, Kałużyńska O, Szyburska J, Wysokiński A. MMP-9 serum levels in schizophrenic patients during treatment augmentation with sarcosine (results of the PULSAR study). Int J Mol Sci. 2016;17(7):1075.
Dwir D, Giangreco B, Xin L, Tenenbaum L, Cabungcal J-H, Steullet P, et al. MMP9/RAGE pathway overactivation mediates redox dysregulation and neuroinflammation, leading to inhibitory/excitatory imbalance: a reverse translation study in schizophrenia patients. Mol Psychiatry. 2020;25(11):2889–904.
Devanarayanan S, Nandeesha H, Kattimani S, Sarkar S. Relationship between matrix metalloproteinase-9 and oxidative stress in drug-free male schizophrenia: a case control study. Clin Chem Lab Med (CCLM). 2016;54(3):447–52.
Lepeta K, Purzycka KJ, Pachulska-Wieczorek K, Mitjans M, Begemann M, Vafadari B, Kaczmarek L. A normal genetic variation modulates synaptic MMP-9 protein levels and the severity of schizophrenia symptoms. EMBO Mol Med. 2017;9(8):1100–16.
Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest. 1996;98(11):2572–9. https://doi.org/10.1172/JCI119076.
Tromp A, Mowry B, Giacomotto J. Neurexins in autism and schizophrenia-a review of patient mutations, mouse models and potential future directions. Mol Psychiatry. 2021;26(3):747–60.
Kirov G, Gumus D, Chen W, Norton N, Georgieva L, Sari M, et al. Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum Mol Genet. 2008;17(3):458–65.
Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, et al. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature. 2003;423(6943):939–48.
Etherton MR, Blaiss CA, Powell CM, Südhof TC. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci USA. 2009;106(42):17998–8003.
Owczarek S, Bang ML, Berezin V. Neurexin-neuroligin synaptic complex regulates schizophrenia-related DISC1/kal-7/Rac1 “signalosome.” Neural Plast. 2015;2015:167308.
Kirov G, Grozeva D, Norton N, Ivanov D, Mantripragada KK, Holmans P, et al. Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum Mol Genet. 2009;18(8):1497–503.
Kirov G, Rujescu D, Ingason A, Collier DA, O’Donovan MC, Owen MJ. Neurexin 1 (NRXN1) deletions in schizophrenia. Schizophr Bull. 2009;35(5):851–4.
Rujescu D, Ingason A, Cichon S, Pietiläinen OPH, Barnes MR, Toulopoulou T, et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum Mol Genet. 2009;18(5):988–96.
Ishizuka K, Yoshida T, Kawabata T, Imai A, Mori H, Kimura H, et al. Functional characterization of rare NRXN1 variants identified in autism spectrum disorders and schizophrenia. J Neurodev Disord. 2020;12(1):25.
Calabrese F, Molteni R, Racagni G, Riva MA. Neuronal plasticity: a link between stress and mood disorders. Psychoneuroendocrinology. 2009;1(34):S208–16.
Murakami S, Imbe H, Morikawa Y, Kubo C, Senba E. Chronic stress, as well as acute stress, reduces BDNF mRNA expression in the rat hippocampus but less robustly. Neurosci Res. 2005;53(2):129–39.
Kim D-M, Leem Y-H. Chronic stress-induced memory deficits are reversed by regular exercise via AMPK-mediated BDNF induction. Neuroscience. 2016;2(324):271–85.
Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci. 1995;15(3 Pt 1):1768–77.
Aguayo FI, Pacheco AA, García-Rojo GJ, Pizarro-Bauerle JA, Doberti AV, Tejos M, et al. Matrix metalloproteinase 9 displays a particular time response to acute stress: variation in its levels and activity distribution in rat hippocampus. ACS Chem Neurosci. 2018;9(5):945–56.
Smith MA, Makino S, Altemus M, Michelson D, Hong SK, Kvetnansky R, et al. Stress and antidepressants differentially regulate neurotrophin 3 mRNA expression in the locus coeruleus. Proc Natl Acad Sci USA. 1995;92(19):8788–92.
Di Filippo M, Sarchielli P, Picconi B, Calabresi P. Neuroinflammation and synaptic plasticity: theoretical basis for a novel, immune-centred, therapeutic approach to neurological disorders. Trends Pharmacol Sci. 2008;29(8):402–12.
Di Filippo M, Chiasserini D, Gardoni F, Viviani B, Tozzi A, Giampà C, et al. Effects of central and peripheral inflammation on hippocampal synaptic plasticity. Neurobiol Dis. 2013;52:229–36.
De Picker LJ, Morrens M, Chance SA, Boche D. Microglia and brain plasticity in acute psychosis and schizophrenia illness course: a meta-review. Front Psych. 2017;8:238.
Allimuthu P, Nandeesha H, Chinniyappan R, Bhardwaz B, Blessed RJ. Relationship of brain-derived neurotrophic factor with interleukin-23, testosterone and disease severity in schizophrenia. Indian J Clin Biochem. 2021;36(3):365–9.
Jesudas BR, Nandeesha H, Menon V, Allimuthu P. Relationship of elevated neural cell adhesion molecule 1 with interleukin-10 and disease severity in bipolar disorder. Asian J Psychiatr. 2020;47:101849.
Chenniappan R, Nandeesha H, Kattimani S, Nanjaiah ND. Interleukin-17 and interleukin-10 association with disease progression in schizophrenia. Ann Neurosci. 2020;27(1):24–8.
Gruol DL. IL-6 regulation of synaptic function in the CNS. Neuropharmacology. 2015;1(96):42–54.
Author information
Authors and Affiliations
Contributions
Both HN and NK equally contributed to this review with conception of the study, literature review, analysis, drafting, critical revision and approval of the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Keshri, N., Nandeesha, H. Dysregulation of Synaptic Plasticity Markers in Schizophrenia. Ind J Clin Biochem 38, 4–12 (2023). https://doi.org/10.1007/s12291-022-01068-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12291-022-01068-2