
Abstract
Activity-dependent synaptic plasticity phenomena such as long-term potentiation and long-term depression are candidate mechanisms for storing information in the brain. Regulation of synaptic plasticity is critical for healthy cognition and learning and this is provided in part by metaplasticity, which can act to maintain synaptic transmission within a dynamic range and potentially prevent excitotoxicity. Metaplasticity mechanisms also allow neurons to integrate plasticity-associated signals over time. Interestingly, astrocytes appear to be critical for certain forms of synaptic plasticity and metaplasticity mechanisms. Synaptic dysfunction is increasingly viewed as an early feature of AD that is correlated with the severity of cognitive decline, and the development of these pathologies is correlated with a rise in reactive astrocytes. This review focuses on the contributions of astrocytes to synaptic plasticity and metaplasticity in normal tissue, and addresses whether astroglial pathology may lead to aberrant engagement of these mechanisms in neurological diseases such as Alzheimer’s disease.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
It has widely been accepted that memories are stored at least in part as changes in the strength of the synaptic connections between neurons, i.e., via synaptic plasticity. Metaplasticity regulates synaptic plasticity across time and among other things can help keep synaptic weights within a dynamic range by modifying the thresholds for long-term potentiation (LTP) and long-term depression (LTD) (Abraham 2008). Mounting evidence also suggests that the communication between neurons and astrocytes is critical for healthy brain functions. Notably, in this context, astrocytes can respond to neural activity and release gliotransmitters which feedback to neurons and regulate LTP and LTD, in part through generating metaplasticity (Gordon et al. 2009; Jones et al. 2013). Moreover, several studies have demonstrated the active participation of astrocytes in memory formation (Gibbs et al. 2008; Zorec et al. 2015).
Under neurodegenerative disease conditions, such as Alzheimer’s disease (AD), synaptic alterations in the hippocampus and association cortices are an early feature and correlate with the severity of cognitive decline (DeKosky et al. 1996). Astrocytes become activated under these conditions, undertaking amyloid-β (Aβ) clearance and degradation through reactive astrogliosis which is a complex, multistage pathological response. Astrocytes thus provide nutritional support and stability to neurons and isolate them from the amyloid plaque deposition in the brain (Rossner et al. 2005). However, signaling in astrocytes and calcium homeostasis is disrupted in transgenic mouse models of AD (Kuchibhotla et al. 2009; Takano et al. 2006), and such pathological astrocyte function might also be directly involved in the early stages of neuronal pathophysiology in AD. This may also occur in other neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), where astrocytes contribute to degeneration and death of motor neurons (Vargas and Johnson 2010; Haidet-Phillips et al. 2011).
This review addresses the astrocytic regulation of synaptic plasticity, including metaplasticity, focusing primarily in the hippocampus. We will also discuss whether the normal regulation of synaptic function becomes aberrant during pathological states, potentially contributing to the synaptic deficits in these pathologies, with a focus on Alzheimer’s disease. We will also consider whether astrocytes or gliotransmitters are potential targets for treating AD or other neurological disorders.
Astrocyte communication and signaling
Astrocytes encase many synaptic contacts and thus help to ensure normal neuronal excitability by maintaining extracellular ion homeostasis through clearing glutamate and potassium ions from the regions around synapses. Astrocytes, by synthesizing glutamine which is utilized by neurons to form glutamate, also contribute significantly to neuronal metabolic homeostasis. Importantly, astrocytes are well equipped to communicate bidirectionally with neurons at tripartite synapses (Araque et al. 1999). In the hippocampus, one astrocyte is in close vicinity to approximately 100 neurons (Agulhon et al. 2008; Hamilton and Attwell 2010) and can connect to thousands of synapses (Bushong et al. 2002). It is well established that neurotransmitters released from the neurons can induce elevations in astrocytic calcium levels. For example, activation of pyramidal neurons in the hippocampus induces elevations in astrocytic Ca2+ levels by stimulation of astrocytic metabotropic glutamate receptors (Porter and McCarthy 1996). This activation state can be communicated to neighboring astrocytes via gap junctions or release of extracellular signaling molecules (Giaume et al. 2010). In retina and the embryonic ventricular zone, for example, induction of intracellular calcium signals in astrocytes leads to Ca2+ waves that spread across astrocytic networks (Weissman et al. 2004). In turn, either electrical or mechanical stimulation of a single astrocyte raises their intracellular calcium levels causing extrasynaptic glutamate release and resulting in elevated calcium levels in neighboring neurons (Charles et al. 1991; Parpura et al. 1994). Through release of a variety of gliotransmitters such as glutamate, d-serine, ATP, GABA, tumor necrosis factor-α (TNF-α), and endocannabinoids, astrocytes are also capable of regulating synaptic plasticity (see Fig. 1).
Although the molecular mechanisms underlying release of these gliotransmitters from astrocyte are not well understood, both Ca2+-dependent exocytosis and Ca2+-independent mechanisms have been reported. Studies report that Ca2+-dependent release of gliotransmitter is based on Ca2+ and SNARE protein-dependent mechanisms (Bezzi et al. 2004; Araque et al. 2000) and through astrocytic vesicular compartments (Bezzi et al. 2004; Mothet et al. 2005) or lysosomal exocytosis (Jaiswal et al. 2007; Zhang et al. 2007). Such release can be triggered by activation of astrocytic G-protein coupled receptors (GPCRs) that couple by Gq to phospholipase C, the hydrolysis of which generates the second messenger IP3 which binds with the Type 2 IP3 receptor in the membrane of the astrocytic smooth endoplasmic reticulum to open Ca2+ channels and raise intracellular free calcium levels (Parpura and Zorec 2010; Pascual et al. 2005). Surprisingly, calcium-dependent entry of astrocytic mitochondria into neighboring neurons is also possible and this entry is believed to be vital for enhancing neuronal survival signals (Hayakawa et al. 2016). In certain cases, Ca2+-independent mechanisms for the release of gliotransmitters appear to coexist with Ca2+-dependent mechanisms. Such mechanisms include release through astrocytic hemi-channels (Ye et al. 2003), release by vesicles (Parpura et al. 1994), backward transport of Na+- dependent glutamate uptake (Anderson and Swanson 2000; Nicholls and Attwell 1990), through the channel pore of P2X7 receptors (Duan et al. 2003), and by volume regulated anion/Cl− channels (Kimelberg et al. 2006; Ye et al. 2009). Ca2+-independent mechanisms are known to occur in cultured astrocytic cells, but their relevance in vivo remains inconclusive.
In summary, astrocytes are in close contact with neurons, regulating neuronal function at the synaptic and network levels and thus are well placed to respond to neural network activity, and impose a significant impact on that activity under both physiological and pathological conditions.
Astrocytes and the regulation of synaptic transmission
Numerous studies have demonstrated that gliotransmitters released from astrocytes can modulate synaptic transmission. Astrocytic glutamate release modulates brain networks in multiple brain regions in situ including hippocampus, thalamus, etc. Slow inward currents are evoked by astrocytic glutamate in CA1 hippocampal pyramidal neurons by activating extrasynaptic NMDARs (Araque et al. 1998; Fellin et al. 2004; Angulo et al. 2004; Perea and Araque 2005; De Pitta and Brunel 2016). Astrocytic glutamate has been shown to synchronously excite hippocampal pyramidal neurons, indicating that gliotransmission may contribute to the coordinated neuronal firing patterns of neurons (Angulo et al. 2004). Moreover, activation of astrocytic CB1 receptors potentiates synaptic transmission via calcium-dependent release of glutamate (Navarrete and Araque 2010). ATP released from astrocytes during neuronal activity is able to influence synaptic transmission by acting on P2X or P2Y receptors (Palygin et al. 2010). Alternatively, ATP can be converted to adenosine by ectonucleotidases to act on pre-synaptic adenosine receptors (A1/A2) to increase (A2) or decrease (A1) transmitter release (Guthrie et al. 1999; Palygin et al. 2010). In the hypothalamus, the afferent activity-induced activation of astrocytes causes an enhancement in the amplitude of synaptic currents at glutamate synapses that depend on the release of glial ATP (Gordon et al. 2009). At CA3-CA1 synapses, astrocyte-derived ATP causes a quick and short-lasting form of pre-synaptic depression (Zhang et al. 2003). However, astrocytes can also regulate inhibitory synaptic transmission (Kang et al. 1998). Astrocytes may release ATP to stimulate interneuron excitability by acting on P2Y1 receptors and thereby potentiate GABAergic synaptic transmission (Bowser and Khakh 2004). Conversely, glutamate release from astrocytes in CA1 can promote pre-synaptic inhibition of inhibitory interneurons via activation of Group II/III mGluRs, although activation of neuronal kainate receptors by astrocyte-derived glutamate can counterbalance this inhibitory state and enhance inhibitory neurotransmission (Liu et al. 2004). In a nutshell, astrocytic-mediated gliotransmitter signaling regulates diverse synaptic and network functions under normal physiological conditions.
Astrocytic contribution to synaptic plasticity
The activation of mGluR and muscarinic cholinergic receptors generates LTP in the hippocampus in vivo and in vitro, an effect that requires simultaneous postsynaptic activity and astrocyte glutamate release and hence activation of neuronal mGluRs (Navarrete et al. 2012). Stimulation of single astrocytes in the hippocampus can cause glutamate release that elicits slow inward NMDAR-mediated currents, enhances the probability of glutamate release, and induces Group I mGluR-mediated LTP (Perea and Araque 2007). Conversely, the high affinity glutamate transporter-1 (GLT-1) coexists with aquaporin (AQP4) and reduced GLT-1 levels in AQP4-null mice results in decreased glutamate uptake by astrocytes and impaired LTP (Zeng et al. 2007).
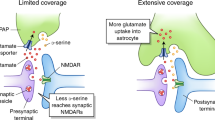
Besides glutamate, d-serine, traditionally considered to be released from astrocytes, enables LTP induction in cultures and acute hippocampal slices (Henneberger et al. 2010; Yang et al. 2003) and is essential for NMDA receptor-dependent synaptic plasticity in the supraoptic nucleus (Panatier et al. 2006). For example, local LTP induction at Schaffer collateral synapses in CA1 was abolished by clamping intracellular Ca2+ levels within a single nearby astrocyte. This LTP was rescued by bath application of d-serine (Henneberger et al. 2010). Similarly, blocking glial cell activation with fluoroacetate (a metabolic inhibitor) blocked LTP in the prefrontal cortex by attenuating the amount of D-serine present extrasynaptically (Fossat et al. 2012). d-Serine is a product of serine racemase, an enzyme first found in astrocytes (Schell et al. 1995) but later discovered to be primarily found in neurons (Kartvelishvily et al. 2006; Miya et al. 2008). In contrast to the traditional view therefore, it appears that it is l-serine that is synthesized and released by astrocytes and this is used by serine racemase in neurons to synthesize d-serine, which then modulates synaptic plasticity (Wolosker et al. 2016). In accord with this model, neuronal serine racemase conditional knockout mice display a significant reduction in LTP and reduced NMDAR currents, whereas astrocytic serine racemase conditional knockout mice show neither effect (Benneyworth et al. 2012). Taken together, these data indicate a key contribution by astrocytes to the D-serine regulation of LTP, but that this is an indirect role via the production and release of the precursor L-serine.
ATP release has also been implicated in hippocampal LTP (Wieraszko and Ehrlich 1994). ATP released from astrocytes, when converted to adenosine via ectonucleotidases, tonically suppressed synaptic transmission and subsequently enhanced the dynamic range for long-term potentiation and associated transient heterosynaptic depression in CA1 (Pascual et al. 2005). Glial ATP, when converted to adenosine, can also generate heterosynaptic LTD, thus amplifying the effect of the homosynaptically induced LTP, akin to lateral inhibition (Chen et al. 2013).
Another key set of gliotransmitters is cytokines. For example, TNF-α, possibly released from astrocytes in developing visual cortex, is essential for experience-dependent change in synaptic transmission (Kaneko et al. 2008). Mice deficient in TNF-α exhibited not only the normal loss of deprived eye responses following monocular visual deprivation, but also a lack of subsequent enhancement in open eye responses (Kaneko et al. 2008). Interestingly, acute application of TNF-α increased surface expression of AMPARs in both hippocampal culture and acute slices, resulting in increased synaptic strength and modulation of neuronal activity (Beattie et al. 2002). Even in the presence of tetrodotoxin, TNF-α upregulates postsynaptic AMPAR surface expression in culture, suggesting that astrocyte-derived TNF-α may be a crucial signal for synaptic up-scaling (Stellwagen and Malenka 2006). Similarly, interleukins can modulate synaptic plasticity in the brain. Treatment of hippocampal slices with recombinant human interleukin-6 (IL-6) impaired LTP in the CA1 (Li et al. 1997) and there was impaired LTP in a mouse model over-expressing IL-6 release from astrocytes in the dentate gyrus (Bellinger et al. 1995). Similarly, human recombinant IL-2 (Tancredi et al. 1990) and IL-1β inhibit hippocampal LTP (Bellinger et al. 1993), while IL-10 antagonizes the LTP inhibition by IL-1β (Kelly et al. 2001). Besides interleukins, intracerebroventricular injection of interferon gamma (IFN-ϒ) (Maher et al. 2006) and bath application of IFN-α (Mendoza-Fernandez et al. 2000) attenuates LTP in rat hippocampus. Thus, it is clear that cytokines play an active role in modulating synaptic plasticity, and although astrocytes (as well as microglia) are capable of releasing cytokines, the conditions under which astrocytes are stimulated to do so in vivo remain unclear.
Astrocytes and metaplasticity
How are LTP and LTD balanced in the healthy brain? One contributing mechanism is metaplasticity, whereby neural activity results in a change in neural state that alters the cell’s response to subsequent plasticity-inducing events (Abraham and Bear 1996). Metaplasticity mechanisms are capable of regulating events across time and space (from minutes to hours) to maintain synaptic plasticity and neuronal homeostasis inside the brain. Classical examples of metaplasticity are the ability of transient NMDAR activation (by weak HFS or by LFS) to inhibit subsequent LTP induction in hippocampal CA1, an effect lasting for less than an hour (Coan et al. 1989; Fujii et al. 1991), or the activation of Group I mGluRs to facilitate and prolong LTP (Raymond et al. 2000). Metaplasticity-like mechanisms have also been reported in human cortex (Bocci et al. 2014; Muller-Dahlhaus and Ziemann 2015), Xenopus (Dunfield and Haas 2009), and Aplysia (Fischer et al. 1997).
Metaplasticity phenomena can be broadly divided into homosynaptic and heterosynaptic subtypes. Homosynaptic metaplasticity is expressed at synapses that participate in the initial bout of priming activity, while heterosynaptic metaplasticity occurs when an episodic priming event at one set of synapses regulates subsequent plasticity at synapses in either nearby dendritic compartments or throughout the cell (Abraham 2008). In one recently studied example of heterosynaptic metaplasticity in hippocampal area CA1, high-frequency priming stimulation of one set of synapses caused a cell-wide shift in plasticity thresholds, such that subsequent LTD was enhanced and LTP was impaired (Hulme et al. 2012; Wang and Wagner 1999). This phenomenon appears to be mediated via astrocytes as the effect did not require postsynaptic neuronal depolarization, but involves release of calcium from intracellular stores by IP3, the opening of connexin-43 channels or hemi-channels located on astrocytes, the release and extracellular conversion of ATP to adenosine, and subsequent activation of adenosine type 2 receptors (A2Rs) (Hulme et al. 2012; Jones et al. 2013). Moreover, the stratum radiatum astrocytes were widely activated by priming stimulation in the stratum oriens, an effect that was almost completely blocked by an inhibitor of gap junctions and hemi-channels (Hulme et al. 2014). This range of evidence strongly supports the potential role of astrocytes in mediating at this type of heterosynaptic metaplasticity, although which glial transmitter may be exerting the plasticity regulation remains to be determined. Other examples of astrocyte-mediated metaplasticity have recently been reviewed (Jones 2015).
Astrocytic signaling in disease conditions
Neurodegenerative diseases are characterized by synaptic dysfunction, apoptotic cell death, and neuroinflammation, and are typically associated with severe cognitive decline and/or motor deficits. Accumulating evidence suggests a role for dysfunction in astrocyte-neuron signaling as an important contributor to pathology for most neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD) and Huntington’s disease (HD), and ALS. There is in particular substantial evidence for a role of astrocytes in AD and this section will focus mainly on how astrocytes are affected in AD and may contribute to the disease pathophysiology.
Three-dimensional reconstructions of amyloid plaques demonstrated that astrocytes are well placed to play a major role in amyloid-β (Aβ) degradation (Wegiel et al. 2000). Astrocytes degrade Aβ deposition by two ways—either by phagocytosis or by Aβ degrading proteases. There is substantial evidence that adult mouse astrocytes can phagocytose and degrade Aβ deposits in vitro (Wyss-Coray et al. 2003). Similarly, Aβ degrading proteases such as neprilysin display ability to degrade both Aβ1–40 and Aβ1–42 (Iwata et al. 2000; Leal et al. 2006). However, aberrant activity by astrocytes in disease conditions might also aggravate the pathophysiology. Abnormal astrocytic calcium signaling is seen in various pathological states including epilepsy, AD, stroke, and traumatic brain injury (Rodriguez et al. 2009). Calcium homeostasis and signaling in astrocytes are also disrupted in transgenic mouse models of AD (Kuchibhotla et al. 2009; Takano et al. 2006) and in astrocytes cultured with Aβ peptides (Haughey and Mattson 2003). Indeed, high levels of extracellular Aβ induce sporadic Ca2+ signals in astrocytes but not in isolated neurons, which may be due to differences in their membrane lipid composition (Abramov et al. 2003). The aberrant astrocytic calcium signaling begins near Aβ plaques and spreads for long distances through the cortex, suggesting that the astrocytic calcium signaling can be widespread. The ability of Aβ to dysregulate astrocytic calcium signaling in vitro and in vivo suggests the potential for astrocytes to contribute to the early stages of pathogenesis in AD (Kuchibhotla et al. 2009). Astrocytic networks in aged transgenic mice are aberrantly coupled by submissive intercellular diffusion/spread through gap junctions (Kuchibhotla et al. 2009) particularly in the neocortex (Peters et al. 2009).
Differences of pre-synaptic function in adult AD mice (APP/PS1) can arise from an altered calcium dynamic caused by the FAD-linked mutation in presenilin 1, which is known to enhance Ca2+ release from the endoplasmic reticulum (Megill et al. 2015). Hyperactive spontaneous short-lived astrocytic calcium waves, independent of neural activity and seen in many transgenic mice models, might influence neuronal signaling at synapses (Takano et al. 2006). Although the amyloid plaques have been one of the cardinal neuropathological feature of AD, it is now recognised that soluble Aβ oligomers are themselves toxic and lead to synaptic deterioration early in the disease process (Coleman et al. 2004). Of particular interest here, soluble Aβ oligomers contribute to impairments in astrocytic metabolism (Tarczyluk et al. 2015) and enhanced astrogliosis (Carrero et al. 2012). Atrophic astrocytes appear in the entorhinal cortex, hippocampus, and prefrontal cortex of 3xTg AD mice even before occurrence of extracellular β-amyloid depositions (Olabarria et al. 2010; Yeh et al. 2011) indicating a possible response to soluble Aβ oligomers.
Another neuropathological feature is reactive astrocytosis, during which astrocytes proliferate in response to ischaemia, injury, or disease, and show hypertrophy of the cell soma and increased gene expression of glial fibrillary acidic protein (GFAP) (Wisniewski and Wegiel 1991). Interestingly, hyperactive calcium signaling occurs in astrocytes that surround plaques, while atrophy occurs further away from plaques (Rodriguez et al. 2009), indicating that there is a complex alteration of astrocytic function in AD. Moreover, morphological atrophy of astrocytes results in reduced ability to envelop synapses which may lead to thinning and reduced numbers of synaptic contacts and thus decline in synaptic function (Coleman et al. 2004; Terry 2000). Reactive astrocytes along with microglia release proinflammatory cytokines, including interleukin-1β, interferon-γ, cyclooxygenase-2 (COX-2), interleukin-6, and TNF-α (Benzing et al. 1999; McGeer and McGeer 2010), noted above as being potent inhibitors of LTP. It is interesting, therefore, that although murine AD models differ in the temporal profile of astrocytosis and the onset of LTP deficits, the development of these features goes hand in hand (Table 1). While not conclusive, this correlation suggests that astrocytosis may be a major contributor to the LTP deficits and associated cognitive decline.
Early impairment of microvasculature may be another functional outcome of altered astrocyte calcium signaling in AD as astrocytic control of the microvasculature is altered in 3xTg, Dutch/Iowa mutants, and Tg2576 mouse models of AD mice at early stages of the disease, well prior to Aβ production, and loss of synapses (Takano et al. 2007). Moreover, astrocytes are well known to regulate glycogen processing, but development of AD induces progressive loss of glucose usage in the brain, contributing to a nutritional collapse that may have dire consequences on neuronal survival and synaptic plasticity (Allaman et al. 2010). These additional features of the AD brain add support to the concept that dysfunctional astrocytic signaling contributes to altered synaptic plasticity and thus cognition in AD.
Aberrant metaplasticity in disease condition
As discussed above, strong afferent activity can cause widespread-synchronous calcium elevations in astrocytes that drive the release of gliotransmitters to dampen LTP and enhance LTD throughout a neural network. The fact that spontaneous astrocytic and neuronal calcium waves are reported in AD models (Chakroborty et al. 2012; Kuchibhotla et al. 2009; Mattson and Chan 2003) is suggestive of endogenous hyper-activation of brain cells due to the disease and thus an excessive release of gliotransmitters. This raises the possibility that there may be an associated aberrant constitutive engagement of inhibitory metaplasticity mechanisms that underpin the impairment of LTP that is common to most mouse models of AD (Jones 2015).
The possible aberrant engagement of metaplasticity mechanisms in AD models was first supported by the finding that NMDAR-mediated inhibition of LTP was absent in APP23 mice, even before the onset of extracellular plaques (Balducci et al. 2010), suggesting that this mechanism was already active and mediating LTP inhibition basally. This could explain in part the therapeutic efficacy of memantine in AD. Memantine is a partial NMDAR antagonist that can rescue the attenuated LTP induction/expression due to application of low concentrations of NMDA in CA1 (Frankiewicz and Parsons 1999; Izumi et al. 1992; Zajaczkowski et al. 1997; Zorumski and Izumi 2012). The fact that cognitive deficits, like the altered synaptic metaplasticity, occur before the onset of plaques in the APP23 mice (Balducci et al. 2010) supports the important role played by soluble Aβ oligomers in synaptic dysfunction, and is consistent with the fact that the oligomers can enhance activation of the GluN2B-containing NMDARs that are targets of astrocytically released glutamate, thereby inhibiting LTP and favoring LTD (Li et al. 2011).
Targeting astrocytes may have therapeutic potential
Given the established role of astrocytes in regulating synaptic activity and metaplasticity, aberrations in astrocyte signaling may contribute to the progression of impairments in synaptic plasticity corresponding to cognitive decline in AD. In particular, it is worth paying attention to possible aberrant engagement of the inhibitory metaplasticity mechanisms in AD models. Understanding the mechanisms by which astrocyte regulates plasticity and generates metaplasticity in diseased states may suggest new therapeutic targets that are so urgently needed in this field. For example, an enhanced level of TNF-α (as produced by reactive astrocytes and no doubt activated microglia) not only causes impairments in LTP but also triggers a cascade of neuronal dysfunction and neurotoxicity as well as contributing to altered APP processing and plaque formation. Thus, antiTNF-α therapy has gained much attention in treating AD symptoms. Clinically, infliximab treatment can improve cognitive impairment and regulate Aβ1-42/p-tau levels in the cerebrospinal fluid (Shi et al. 2011). Other clinical antiTNF-α therapy such as and pentoxifylline (Sha and Callahan 2003) has been promising in treating severe cognitive decline in AD. Hence, targeting the TNF-α protein, its receptor or TNF-α converting enzyme (TACE) might be possible therapeutic interventions for the treatment of AD, although an early clinical trial with etanercept was inconclusive (Butchart et al. 2015). Thus the optimal agent, dose, and means of administration remain to be investigated further (Clark and Vissel 2016). In addition, considering the role played by p38 MAP kinase and NF-кB in TNF-α production in AD patients, inhibition of these molecules may be useful in treating neurodegeneration. Treatments against other inflammatory cytokines should also be considered, as well as the upregulation of the LTP pro-acting D-serine (Zou et al. 2016). Recent studies indicate that exogenous ATP may also be a therapeutic target as it helps in restoring LTP and protects dendritic loss as seen in AD (Jung et al. 2012). Finally, astrocytes themselves could be the direct target of gene therapies, as over-expression of the master autophagy/lysosome gene transcription factor EB in astrocytes facilitated Aβ clearance and reduced plaque formation in a mouse model of AD (Xiao et al. 2015). In summary, although anti-Aβ treatments have received the bulk of attention as Alzheimer’s treatment, the direct or indirect effects of aberrant astrocyte activity may also offer attractive targets as treatment options for rescuing at least the impairment in plasticity-associated memory and cognition.

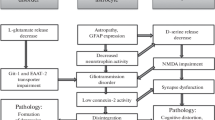
Astroglial regulation of plasticity in healthy vs diseased states. In the healthy state, activity-mediated release of gliotransmitters (such as d-serine, ATP, glutamate, TNF-α etc) from astrocytes regulates (+) LTP and metaplasticity, balancing synaptic plasticity with neuronal homeostasis. In AD, astrocytes are less able to sense the neuronal activity and withdraw their processes from synapses due to the presence of extrasynaptic amyloid plaque and neuroinflammation. Furthermore, reactive astrocytes and microglia increase the constitutive release of gliotransmitters (such as glutamate, GABA, S100β, IL-1β, TNF-α, etc.). This may induce aberrant metaplasticity states which result in impairments in LTP and neuronal homeostasis
References
Abraham WC (2008) Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci 9:387
Abraham WC, Bear MF (1996) Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci 19:126–130
Abramov AY, Canevari L, Duchen MR (2003) Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci 23:5088–5095
Agulhon C, Petravicz J, McMullen AB, Sweger EJ, Minton SK, Taves SR, McCarthy KD (2008) What is the role of astrocyte calcium in neurophysiology? Neuron 59:932–946
Allaman I, Gavillet M, Belanger M, Laroche T, Viertl D, Lashuel HA, Magistretti PJ (2010) Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J Neurosci 30:3326–3338
Anderson CM, Swanson RA (2000) Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32:1–14
Angulo MC, Kozlov AS, Charpak S, Audinat E (2004) Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci 24:6920–6927
Apelt J, Schliebs R (2001) Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res 894:21–30
Araque A, Parpura V, Sanzgiri RP, Haydon PG (1998) Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci 10:2129–2142
Araque A, Parpura V, Sanzgiri RP, Haydon PG (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22:208–215
Araque A, Li N, Doyle RT, Haydon PG (2000) SNARE protein-dependent glutamate release from astrocytes. J Neurosci 20:666–673
Aytan N, Choi JK, Carreras I, Brinkmann V, Kowall NW, Jenkins BG, Dedeoglu A (2016) Fingolimod modulates multiple neuroinflammatory markers in a mouse model of Alzheimer’s disease. Sci Rep 6:24939
Balducci C, Tonini R, Zianni E, Nazzaro C, Fiordaliso F, Salio M, Forloni G (2010) Cognitive deficits associated with alteration of synaptic metaplasticity precede plaque deposition in AbetaPP23 transgenic mice. J Alzheimers Dis 21:1367–1381
Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Malenka RC (2002) Control of synaptic strength by glial TNFα. Science 295:2282–2285
Beauquis J, Pavia P, Pomilio C, Vinuesa A, Podlutskaya N, Galvan V, Saravia F (2013) Environmental enrichment prevents astroglial pathological changes in the hippocampus of APP transgenic mice, model of Alzheimer’s disease. Exp Neurol 239:28–37
Bellinger FP, Madamba S, Siggins GR (1993) Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res 628:227–234
Bellinger FP, Madamba SG, Campbell IL, Siggins GR (1995) Reduced long-term potentiation in the dentate gyrus of transgenic mice with cerebral overexpression of interleukin-6. Neurosci Lett 198:95–98
Benneyworth MA, Li Y, Basu AC, Bolshakov VY, Coyle JT (2012) Cell selective conditional null mutations of serine racemase demonstrate a predominate localization in cortical glutamatergic neurons. Cell Mol Neurobiol 32:613–624
Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR (1999) Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging 20:581–589
Bezzi P, Gundersen V, Galbete JL, Seifert G, Steinhäuser C, Pilati E, Volterra A (2004) Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat Neurosci 7:613–620
Bocci T, Caleo M, Tognazzi S, Francini N, Briscese L, Maffei L, Sartucci F (2014) Evidence for metaplasticity in the human visual cortex. J Neural Transm 121:221–231
Bowser DN, Khakh BS (2004) ATP excites interneurons and astrocytes to increase synaptic inhibition in neuronal networks. J Neurosci 24:8606–8620
Bushong EA, Martone ME, Jones YZ, Ellisman MH (2002) Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 22:183–192
Butchart J, Brook L, Hopkins V, Teeling J, Puntener U, Culliford D, Holmes C (2015) Etanercept in Alzheimer disease: a randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 84:2161–2168
Carrero I, Gonzalo MR, Martin B, Sanz-Anquela JM, Arevalo-Serrano J, Gonzalo-Ruiz A (2012) Oligomers of beta-amyloid protein (Abeta1-42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1beta, tumour necrosis factor-alpha, and a nuclear factor kappa-B mechanism in the rat brain. Exp Neurol 236:215–227
Caruso D, Barron AM, Brown MA, Abbiati F, Carrero P, Pike CJ, Melcangi RC (2013) Age-related changes in neuroactive steroid levels in 3xTg-AD mice. Neurobiol Aging 34:1080–1089
Chakroborty S, Kim J, Schneider C, Jacobson C, Molgo J, Stutzmann GE (2012) Early presynaptic and postsynaptic calcium signaling abnormalities mask underlying synaptic depression in presymptomatic Alzheimer’s disease mice. J Neurosci 32:8341–8353
Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Hsiao, K K (1999) Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 2:271–276
Charles AC, Merrill JE, Dirksen ER, Sanderson MJ (1991) Intercellular signaling in glial cells: calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron 6:983–992
Chen J, Tan Z, Zeng L, Zhang X, He Y, Gao W, Duan S (2013) Heterosynaptic long-term depression mediated by ATP released from astrocytes. Glia 61:178–191
Clark IA, Vissel B (2016) Excess cerebral TNF causing glutamate excitotoxicity rationalizes treatment of neurodegenerative diseases and neurogenic pain by anti-TNF agents. J Neuroinflammation 13:236
Coan EJ, Irving AJ, Collingridge GL (1989) Low-frequency activation of the NMDA receptor system can prevent the induction of LTP. Neurosci Lett 105:205–210
Coleman P, Kurlan R, Crook R, Werner J, Hardy J (2004) A new presenilin Alzheimer’s disease case confirms the helical alignment of pathogenic mutations in transmembrane domain 5. Neurosci Lett 364:139–140
Crouzin N, Baranger K, Cavalier M, Marchalant Y, Cohen-Solal C, Roman FS,.Vignes M (2013) Area-specific alterations of synaptic plasticity in the 5XFAD mouse model of Alzheimer’s disease: dissociation between somatosensory cortex and hippocampus. PLoS One 8:e74667
De Pitta M, Brunel N (2016) Modulation of synaptic plasticity by glutamatergic gliotransmission: a modeling study. Neural Plast 2016:7607924. doi:10.1155/2016/7607924
DeKosky ST, Scheff SW, Styren SD (1996) Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration 5:417–421
Dewachter I, Reverse D, Caluwaerts N, Ris L, Kuiperi C, Van den Haute C, Van Leuven F (2002) Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J Neurosci 22:3445–3453
Duan S, Anderson CM, Keung EC, Chen Y, Chen Y, Swanson RA (2003) P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J Neurosci 23:1320–1328
Dunfield D, Haas K (2009) Metaplasticity governs natural experience-driven plasticity of nascent embryonic brain circuits. Neuron 64:240–250
Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G (2004) Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43:729–743
Fischer TM, Blazis DE, Priver NA, Carew TJ (1997) Metaplasticity at identified inhibitory synapses in Aplysia. Nature 389:860–865
Fossat P, Turpin TR, Sacchi S, Dulong J, Shi T, Rivet JM, Mothet JP (2012) Glial D-serine gates NMDA receptors at excitatory synapses in prefrontal cortex. Cereb Cortex 22:595–606
Frankiewicz T, Parsons CG (1999) Memantine restores long term potentiation impaired by tonic N-methyl-D-aspartate (NMDA) receptor activation following reduction of Mg2+ in hippocampal slices. Neuropharmacology 38:1253–1259
Fujii S, Saito K, Miyakawa H, Ito K, Kato H (1991) Reversal of long-term potentiation (depotentiation) induced by tetanus stimulation of the input to CA1 neurons of guinea pig hippocampal slices. Brain Res 555:112–122
Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C et al (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373:523–527
Giacchino J, Criado JR, Games D, Henriksen S (2000) In vivo synaptic transmission in young and aged amyloid precursor protein transgenic mice. Brain Res 876:185–190
Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N (2010) Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci 11:87–99
Gibbs ME, Hutchinson D, Hertz L (2008) Astrocytic involvement in learning and memory consolidation. Neurosci Biobehav Rev 32:927–944
Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O (2004) Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest 114:1624–1634
Gordon GR, Iremonger KJ, Kantevari S, Ellis-Davies GC, MacVicar BA, Bains JS (2009) Astrocyte-mediated distributed plasticity at hypothalamic glutamate synapses. Neuron 64:391–403
Guthrie PB, Knappenberger J, Segal M, Bennett MV, Charles AC, Kater SB (1999) ATP released from astrocytes mediates glial calcium waves. J Neurosci 19:520–528
Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Kaspar BK (2011) Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 29:824–828
Hamilton NB, Attwell D (2010) Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci 11:227–238
Harris JA, Devidze N, Halabisky B, Lo I, Thwin MT, Yu GQ, Mucke L (2010) Many neuronal and behavioral impairments in transgenic mouse models of Alzheimer’s disease are independent of caspase cleavage of the amyloid precursor protein. J Neurosci 30:372–381
Haughey NJ, Mattson MP (2003) Alzheimer’s amyloid beta-peptide enhances ATP/gap junction-mediated calcium-wave propagation in astrocytes. Neuromol Med 3:173–180
Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Lo EH (2016) Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535:551–555
Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, Landreth GE (2005) Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain 128:1442–1453
Henneberger C, Papouin T, Oliet SH, Rusakov DA (2010) Long-term potentiation depends on release of d-serine from astrocytes. Nature 463:232–236
Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274:99–102
Hulme SR, Jones OD, Ireland DR, Abraham WC (2012) Calcium-dependent but action potential-independent BCM-like metaplasticity in the hippocampus. J Neurosci 32:6785–6794
Hulme SR, Jones OD, Raymond CR, Sah P, Abraham WC (2014) Mechanisms of heterosynaptic metaplasticity. Philos Trans R Soc Lond B Biol Sci 369:20130148
Huttenrauch M, Baches S, Gerth J, Bayer TA, Weggen S, Wirths O (2015) Neprilysin deficiency alters the neuropathological and behavioral phenotype in the 5XFAD mouse model of Alzheimer’s disease. J Alzheimers Dis 44:1291–1302
Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Saido TC (2000) Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med 6:143–150
Izumi Y, Clifford DB, Zorumski CF (1992) Low concentrations of N-methyl-d-aspartate inhibit the induction of long-term potentiation in rat hippocampal slices. Neurosci Lett 137:245–248
Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Bloom FE (2006) Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 103:5161–5166
Jaiswal JK, Fix M, Takano T, Nedergaard M, Simon SM (2007) Resolving vesicle fusion from lysis to monitor calcium-triggered lysosomal exocytosis in astrocytes. Proc Natl Acad Sci USA 104:14151–14156
Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Lee CJ (2014) GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med 20:886–896
Jones OD (2015) Astrocyte-mediated metaplasticity in the hippocampus: Help or hindrance? Neuroscience 309:113–124
Jones OD, Hulme SR, Abraham WC (2013) Purinergic receptor- and gap junction-mediated intercellular signalling as a mechanism of heterosynaptic metaplasticity. Neurobiol Learn Mem 105:31–39
Jung JH, An K, Kwon OB, Kim HS, Kim JH (2011) Pathway-specific alteration of synaptic plasticity in Tg2576 mice. Mol Cells 32:197–201
Jung ES, An K, Hong HS, Kim JH, Mook-Jung I (2012) Astrocyte-originated ATP protects Abeta(1–42)-induced impairment of synaptic plasticity. J Neurosci 32:3081–3087
Kamphuis W, Mamber C, Moeton M, Kooijman L, Sluijs JA, Jansen AH, Hol EM (2012) GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS One 7:e42823
Kaneko M, Stellwagen D, Malenka RC, Stryker MP (2008) Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron 58:673–680
Kang J, Jiang L, Goldman SA, Nedergaard M (1998) Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci 1:683–692
Kartvelishvily E, Shleper M, Balan L, Dumin E, Wolosker H (2006) Neuron-derived D-serine release provides a novel means to activate N-methyl-D-aspartate receptors. J Biol Chem 281:14151–14162
Kelly A, Lynch A, Vereker E, Nolan Y, Queenan P, Whittaker E, Lynch MA (2001) The anti-inflammatory cytokine, interleukin (IL)-10, blocks the inhibitory effect of IL-1 beta on long term potentiation. A role for JNK. J Biol Chem 276:45564–45572
Kimelberg HK, MacVicar BA, Sontheimer H (2006) Anion channels in astrocytes: biophysics, pharmacology, and function. Glia 54:747–757
Kimura R, Ohno M (2009) Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol Dis 33:229–235
Kimura R, Devi L, Ohno M (2010) Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer’s disease transgenic mice. J Neurochem 113:248–261
Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ (2009) Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323:1211–1215
Kulijewicz-Nawrot M, Verkhratsky A, Chvatal A, Sykova E, Rodriguez JJ (2012) Astrocytic cytoskeletal atrophy in the medial prefrontal cortex of a triple transgenic mouse model of Alzheimer’s disease. J Anat 221:252–262
Larson J, Lynch G, Games D, Seubert P (1999) Alterations in synaptic transmission and long-term potentiation in hippocampal slices from young and aged PDAPP mice. Brain Res 840:23–35
Leal MC, Dorfman VB, Gamba AF, Frangione B, Wisniewski T, Castano EM, Morelli L (2006) Plaque-associated overexpression of insulin-degrading enzyme in the cerebral cortex of aged transgenic tg2576 mice with Alzheimer pathology. J Neuropathol Exp Neurol 65:976–987
Li AJ, Katafuchi T, Oda S, Hori T, Oomura Y (1997) Interleukin-6 inhibits long-term potentiation in rat hippocampal slices. Brain Res 748:30–38
Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ (2011) Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 31:6627–6638
Liu QS, Xu Q, Arcuino G, Kang J, Nedergaard M (2004) Astrocyte-mediated activation of neuronal kainate receptors. Proc Natl Acad Sci USA 101:3172–3177
Ma T, Hoeffer CA, Capetillo-Zarate E, Yu F, Wong H, Lin MT, Gouras GK (2010) Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS One 5(9):e12845. doi:10.1371/journal.pone.0012845
Maher FO, Clarke RM, Kelly A, Nally RE, Lynch MA (2006) Interaction between interferon gamma and insulin-like growth factor-1 in hippocampus impacts on the ability of rats to sustain long-term potentiation. J Neurochem 96:1560–1571
Mattson MP, Chan SL (2003) Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium 34:385–397
McGeer EG, McGeer PL (2010) Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J Alzheimers Dis 19:355–361
Megill A, Tran T, Eldred K, Lee NJ, Wong PC, Hoe HS, Lee HK (2015) Defective age-dependent metaplasticity in a mouse model of Alzheimer’s disease. J Neurosci 35:11346–11357
Mendoza-Fernandez V, Andrew RD, Barajas-Lopez C (2000) Interferon-alpha inhibits long-term potentiation and unmasks a long-term depression in the rat hippocampus. Brain Res 885:14–24
Miya K, Inoue R, Takata Y, Abe M, Natsume R, Sakimura K, Mori H (2008) Serine racemase is predominantly localized in neurons in mouse brain. J Comp Neurol 510:641–654
Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Van Leuven F (1999) Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem 274:6483–6492
Mothet J-P, Pollegioni L, Ouanounou G, Martineau M, Fossier P, Baux G (2005) Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter d-serine. Proc Natl Acad Sci USA 102:5606–5611
Muller-Dahlhaus F, Ziemann U (2015) Metaplasticity in human cortex. Neuroscientist 21:185–202
Navarrete M, Araque A (2010) Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 68:113–126
Navarrete M, Perea G, Fernandez de Sevilla D, Gomez-Gonzalo M, Nunez A, Martin ED, Araque A (2012) Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol 10:e1001259
Nicholls D, Attwell D (1990) The release and uptake of excitatory amino acids. Trends Pharmacol Sci 11:462–468
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Vassar R (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26:10129–10140
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, LaFerla FM (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39:409–421
Olabarria M, Noristani HN, Verkhratsky A, Rodriguez JJ (2010) Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58:831–838
Palygin O, Lalo U, Verkhratsky A, Pankratov Y (2010) Ionotropic NMDA and P2 × 1/5 receptors mediate synaptically induced Ca2+ signalling in cortical astrocytes. Cell Calcium 48:225–231
Panatier A, Theodosis DT, Mothet J-P, Touquet B, Pollegioni L, Poulain DA, Oliet SH (2006) Glia-derived d-serine controls NMDA receptor activity and synaptic memory. Cell 125:775–784
Parpura V, Zorec R (2010) Gliotransmission: exocytotic release from astrocytes. Brain Res Rev 63:83–92
Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG (1994) Glutamate-mediated astrocyte–neuron signalling. Nature 369:744–747
Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Haydon PG (2005) Astrocytic purinergic signaling coordinates synaptic networks. Science 310:113–116
Perea G, Araque A (2005) Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J Neurosci 25:2192–2203
Perea G, Araque A (2007) Astrocytes potentiate transmitter release at single hippocampal synapses. Science 317:1083–1086
Peters O, Schipke CG, Philipps A, Haas B, Pannasch U, Wang LP, Kettenmann H (2009) Astrocyte function is modified by Alzheimer’s disease-like pathology in aged mice. J Alzheimers Dis 18:177–189
Poisnel G, Herard AS, Bourrin NET, Volk A, Kober F, Dhenain, M (2012) Increased regional cerebral glucose uptake in an APP/PS1 model of Alzheimer’s disease. Neurobiol Aging 33:1995–2005
Porter JT, McCarthy KD (1996) Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci 16:5073–5081
Raymond CR, Thompson VL, Tate WP, Abraham WC (2000) Metabotropic glutamate receptors trigger homosynaptic protein synthesis to prolong long-term potentiation. J Neurosci 20:969–976
Rodriguez JJ, Olabarria M, Chvatal A, Verkhratsky A (2009) Astroglia in dementia and Alzheimer’s disease. Cell Death Differ 16:378–385
Rodriguez-Vieitez E, Ni R, Gulyas B, Toth M, Haggkvist J, Halldin C, Nordberg A (2015) Astrocytosis precedes amyloid plaque deposition in Alzheimer APPswe transgenic mouse brain: a correlative positron emission tomography and in vitro imaging study. Eur J Nucl Med Mol Imaging 42:1119–1132
Rossner S, Apelt J, Schliebs R, Perez-Polo JR, Bigl V (2001) Neuronal and glial beta-secretase (BACE) protein expression in transgenic Tg2576 mice with amyloid plaque pathology. J Neurosci Res 64:437–446
Rossner S, Lange-Dohna C, Zeitschel U, Perez-Polo JR (2005) Alzheimer’s disease beta-secretase BACE1 is not a neuron-specific enzyme. J Neurochem 92:226–234
Ruan L, Kang Z, Pei G, Le Y (2009) Amyloid deposition and inflammation in APPswe/PS1dE9 mouse model of Alzheimer’s disease. Curr Alzheimer Res 6:531–540
Saganich MJ, Schroeder BE, Galvan V, Bredesen DE, Koo EH, Heinemann SF (2006) Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J Neurosci 26:13428–13436
Sancheti H, Akopian G, Yin F, Brinton RD, Walsh JP, Cadenas E (2013) Age-dependent modulation of synaptic plasticity and insulin mimetic effect of lipoic acid on a mouse model of Alzheimer’s disease. PLoS One 8:69830
Sancheti H, Patil I, Kanamori K, Brinton R, Zhang W, Lin AL, Cadenas E (2014) Hypermetabolic state in the 7-month-old triple transgenic mouse model of Alzheimer’s disease and the effect of lipoic acid: a 13 C-NMR study. J Cereb Blood Flow Metab 34:1749–1760
Schell MJ, Molliver ME, Snyder SH (1995) D-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc Natl Acad Sci USA 92:3948–3952
Sha MC, Callahan CM (2003) The efficacy of pentoxifylline in the treatment of vascular dementia: a systematic review. Alzheimer Dis Assoc Disord 17:46–54
Shi JQ, Wang BR, Jiang WW, Chen J, Zhu YW, Zhong LL, Xu J (2011) Cognitive improvement with intrathecal administration of infliximab in a woman with Alzheimer’s disease. J Am Geriatr Soc 59:1142–1144
Stellwagen D, Malenka RC (2006) Synaptic scaling mediated by glial TNF-alpha. Nature 440:1054–1059
Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M (2006) Astrocyte-mediated control of cerebral blood flow. Nat Neurosci 9:260–267
Takano T, Han X, Deane R, Zlokovic B, Nedergaard M (2007) Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer’s disease. Ann N Y Acad Sci 1097:40–50
Tancredi V, Zona C, Velotti F, Eusebi F, Santoni A (1990) Interleukin-2 suppresses established long-term potentiation and inhibits its induction in the rat hippocampus. Brain Res 525:149–151
Tarczyluk MA, Nagel DA, Rhein Parri H, Tse EH, Brown JE, Coleman MD, Hill EJ (2015) Amyloid beta 1–42 induces hypometabolism in human stem cell-derived neuron and astrocyte networks. J Cereb Blood Flow Metab 35:1348–1357
Terai K, Iwai A, Kawabata S, Sasamata M, Miyata K, Yamaguchi T (2001) Apolipoprotein E deposition and astrogliosis are associated with maturation of beta-amyloid plaques in betaAPPswe transgenic mouse: implications for the pathogenesis of Alzheimer’s disease. Brain Res 900:48–56
Terry RD (2000) Cell death or synaptic loss in Alzheimer disease. J Neuropathol Exp Neurol 59:1118–1119
Trinchese F, Liu S, Battaglia F, Walter S, Mathews PM, Arancio O (2004) Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann Neurol 55:801–814
Vargas MR, Johnson JA (2010) Astrogliosis in amyotrophic lateral sclerosis: role and therapeutic potential of astrocytes. Neurother 7:471–481
Volianskis A, Kostner R, Molgaard M, Hass S, Jensen MS (2010) Episodic memory deficits are not related to altered glutamatergic synaptic transmission and plasticity in the CA1 hippocampus of the APPswe/PS1deltaE9-deleted transgenic mice model of ss-amyloidosis. Neurobiol Aging 31:1173–1187
Wang H, Wagner JJ (1999) Priming-induced shift in synaptic plasticity in the rat hippocampus. J Neurophysiol 82:2024–2028
Wegiel J, Wang KC, Tarnawski M, Lach B (2000) Microglia cells are the driving force in fibrillar plaque formation, whereas astrocytes are a leading factor in plague degradation. Acta Neuropathol 100:356–364
Weissman TA, Riquelme PA, Ivic L, Flint AC, Kriegstein AR (2004) Calcium waves propagate through radial glial cells and modulate proliferation in the developing neocortex. Neuron 43:647–661
Wieraszko A, Ehrlich YH (1994) On the role of extracellular ATP in the induction of long-term potentiation in the hippocampus. J Neurochem 63:1731–1738
Wisniewski HM, Wegiel J (1991) Spatial relationships between astrocytes and classical plaque components. Neurobiol Aging 12:593–600
Wolosker H, Balu DT, Coyle JT (2016) The rise and fall of the d-Serine-mediated gliotransmission hypothesis. Trends Neurosci 39:712–721
Wright AL, Zinn R, Hohensinn B, Konen LM, Beynon SB, Tan RP, Vissel B (2013) Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS One 8:e59586
Wu Z, Guo Z, Gearing M, Chen G (2014) Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s disease model. Nat Commun 5:4159
Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Husemann J (2003) Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 9:453–457
Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Lee JM (2015) Neuronal-targeted TFEB accelerates lysosomal degradation of APP, reducing Abeta generation and amyloid plaque pathogenesis. J Neurosci 35:12137–12151
Yang, G, Chen, Zhang, Shen, Wu, Duan (2003) Contribution of astrocytes to hippocampal long-term potentiation through release of d-serine. Proc Natl Acad Sci U S A 100:15194–15199
Ye Z-C, Wyeth MS, Baltan-Tekkok S, Ransom BR (2003) Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci 23:3588–3596
Ye ZC, Oberheim N, Kettenmann H, Ransom BR (2009) Pharmacological “cross-inhibition” of connexin hemichannels and swelling activated anion channels. Glia 57:258–269
Yeh CY, Vadhwana B, Verkhratsky A, Rodriguez JJ (2011) Early astrocytic atrophy in the entorhinal cortex of a triple transgenic animal model of Alzheimer’s disease. ASN Neuro 3:271–279
Zajaczkowski W, Frankiewicz T, Parsons CG, Danysz W (1997) Uncompetitive NMDA receptor antagonists attenuate NMDA-induced impairment of passive avoidance learning and LTP. Neuropharmacology 36:961–971
Zeng XN, Sun XL, Gao L, Fan Y, Ding JH, Hu G (2007) Aquaporin-4 deficiency down-regulates glutamate uptake and GLT-1 expression in astrocytes. Mol Cell Neurosci 34:34–39
Zhang JM, Wang HK, Ye CG, Ge W, Chen Y, Jiang ZI et al (2003) ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron 40:971–982
Zhang Z, Chen G, Zhou W, Song A, Xu T, Luo Q et al (2007) Regulated ATP release from astrocytes through lysosome exocytosis. Nat Cell Biol 9:945–953
Zorec R, Horvat A, Vardjan N, Verkhratsky A (2015) Memory formation shaped by astroglia. Front Integ Neurosc 9:56
Zorumski CF, Izumi Y (2012) NMDA receptors and metaplasticity: mechanisms and possible roles in neuropsychiatric disorders. Neurosci Biobehav Rev 36:989–1000
Zou C, Crux S, Marinesco S, Montagna E, Sgobio C, Shi Y, Herms J (2016) Amyloid precursor protein maintains constitutive and adaptive plasticity of dendritic spines in adult brain by regulating D-serine homeostasis. Embo J 35:2213–2222
Acknowledgements
This work was supported by grants from the Neurological Foundation of N.Z. and the Health Research Council of N.Z. A Singh was supported by a University of Otago doctoral scholarship. The authors thank Dr. O Jones for helpful comments and discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Singh, A., Abraham, W.C. Astrocytes and synaptic plasticity in health and disease. Exp Brain Res 235, 1645–1655 (2017). https://doi.org/10.1007/s00221-017-4928-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00221-017-4928-1